
Abstract
Mosquitoes are important vectors of a wide range of viruses and serve as pivotal vectors with complex ecological functions that facilitate the transmission of these pathogens between hosts, such as humans, through biting behavior. In the mosquito surveillance conducted in Weifang and Yantai cities of Shandong Province, genetic material of a novel virus, temporarily named Fangshan bunya-like virus, was detected. From 3013 female mosquitoes, genetic material was detected in 9 pools (infection rate: 0.30%) using qRT-PCR and nested PCR. Analysis of the distances between pairs revealed a significant level of nucleotide similarity among these amplified sequences. Phylogenetic analysis placed Fangshan bunya-like virus within the Mobuvirus genus, Phenuiviridae family, clustering with Pyongtaek Culex Bunyavirus and indicating a common ancestor. The genomic characteristics and phylogenetic classification of Fangshan bunya-like virus offered valuable insights into the diversity and evolution of Phenuiviridae among mosquito populations, despite the challenge of not having isolated the live virus.
Similar content being viewed by others

Introduction
Arboviruses are a group of viruses transmitted between humans and animals by blood-sucking arthropods, which bite susceptible vertebrate hosts and cause human and animal diseases1. Mosquitoes, as one of the main vectors, pose a significant threat to human health due to their ability to carry various viruses. It is widely recognized that mosquitoes play an indispensable role in the transmission and evolution of viruses2. With advancing research on mosquito ecological biology, it has become evident that an increasing number of mosquito-borne viruses have emerged as pivotal factors in the emergence of new infectious diseases. For example, mosquito-borne viruses such as Banna virus3 and West Nile Virus4 have become significant public health concerns within the public health.
In the year 2017, the ICTV, which is the global authority on the classification and nomenclature of viruses, upgraded the taxonomic status of the Bunyaviridae family to the order Bunyavirales. This upgrade was accompanied by a significant expansion of its taxonomic range, with the incorporation of several new families that account for both non-segmented and segmented RNA viruses impacting a diverse range of life forms including animals, fungi, vegetation, and protozoa. Moving forward to April 2024, the order Bunyavirales was further promoted to the class Bunyaviricetes. The class Bunyaviricetes now encompasses two significant orders, namely Ellioviricolae and Hareolae, and is estimated to harbor a vast array of approximately 600 viral species, cementing its status as the most expansive taxon within the broader category of arboviruses. Bunyaviruses can be transmitted through various arthropod vectors, with the majority being transmitted by mosquitoes, such as La Crosse encephalitis virus5 and Jamestown Canyon virus6. Tick-borne viruses are also part of bunyaviruses, with severe fever with thrombocytopenia virus (Bandavirus) and Tacheng tick virus 1 being representative viruses. Traditionally, most classical bunyaviruses were three-segmented negative-stranded RNA viruses. These segments encoded important viral components such as RNA-dependent RNA polymerase (RdRp), Glycoprotein (GP), and Nucleoprotein (NP). However, there were a few exceptions where only two RNA segments (L and S) encoded the necessary viral components7,8,9. Vectors that transmit bunyaviruses can infect vertebrates, arthropods and/or plants, making bunyaviruses a diverse group with a wide range of potential hosts10. The discovery of new viruses within the class Bunyaviricetes has presented unique challenges to the field of public health. These emerging viruses bring with them unknown transmission kinetics, pathogenicity, and the potential to adapt to new hosts and environments. Understanding and addressing these challenges is crucial for effective public health management and response. In recent years, mosquito-borne viral diseases had shown increasingly complex epidemiological patterns in China and neighboring countries11. Dengue fever, Japanese encephalitis, chikungunya, and Zika virus had periodically emerged across multiple regions in Asia12. With accelerating climate change and urbanization, the geographical distribution of these diseases had continued to expand13. Countries have implemented comprehensive control strategies, including vector surveillance, vaccine development, early case detection, and community-based vector control. However, the emergence of novel mosquito-borne viruses had posed new challenges to existing control systems, making enhanced viral surveillance and pathogen identification crucial for preventing future outbreaks.
In the present study, we described the genome sequence, morphological features, and phylogenetic relationships of a novel Bunyavirus discovered in China in 2020. Based on the principle of territoriality and the structural features of the virus sequence, we named it Fangshan bunya-like virus14. Phylogenetic analysis suggested that this virus was evolutionarily conserved and clustered with the yongtaek Culex Bunyavirus and Narangue virus. However, the pathogenicity of the virus and its impact on the public health require further research.
Materials and methods
Mosquito collecting and nucleic acid extraction
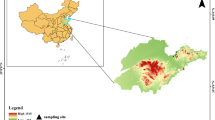
During June and July of 2020, CO2-baited light traps were used to collect mosquitoes in the sheep pens located in Yantai and Weifang cities, Shandong Province, China (Fig. 1). Specifically, samples were collected from five sites in Yantai (Site 1: 37.5462° N, 121.3816° E; Site 2: 37.5103° N, 121.4215° E; Site 3: 37.4891° N, 121.3574° E; Site 4: 37.5578° N, 121.2983° E; Site 5: 37.5042° N, 121.5107° E) and five sites in Weifang (Site 1: 36.7234° N, 119.1423° E; Site 2: 36.6986° N, 119.1876° E; Site 3: 36.7512° N, 119.0984° E; Site 4: 36.7103° N, 119.2254° E; Site 5: 36.6792° N, 119.1527° E). Five CO₂-baited light traps had been placed at each sampling site, with a minimum distance of 100 m between traps, and operated from 18:00 to 06:00 daily. During the sampling period, Yantai city had an average temperature of 24–28 °C, relative humidity of 65–75%, and predominantly hilly and coastal plain terrain; Weifang city had an average temperature of 23–27 °C, relative humidity of 70–80%, and primarily plain terrain. Sampling sites had been selected around sheep pens, which typically have high mosquito density and multiple potential host animals, facilitating the study of potential zoonotic viruses. Morphological identification was performed using a stereomicroscope, following taxonomic keys including Illustrated keys to the mosquitoes of Thailand and Guidelines for morphological identification of mosquitoes. Samples were categorized based on external features (wing scales, proboscis morphology), species, and sexes15,16,17. Male mosquitoes were discarded. Females were maintained at room temperature for 6–8 h to allow partial digestion of blood meals, thereby reducing potential PCR inhibition caused by blood components. This period was determined to be sufficient for our detection protocols while maintaining viral RNA integrity. Subsequently, the mosquitoes were divided into groups of no more than 50 mosquitoes each and placed in microcentrifuge tubes. Using a tissue homogenizer (Qiagen, Germany), the mosquitoes were homogenized in 1000 µL of DMEM. The clarified supernatant from the mosquito homogenates was then extracted for RNA using TIANamp RNA extraction kit (Tiangen, China) following the manufacturer’s instructions after being mixed and centrifuged at 4 °C and 10,000 g for 5 min. Nucleic acids are used immediately or stored at -40 °C.

Geographical location of mosquitoes capturing sites: Yantai and Weifang city, within the Shandong Province. Black flag mark the mosquito collection sites.
Library construction
RNA from each mosquito pool was aspirated 5 µl and mixed into two large assay tubes for RNA library construction. Nucleic acid concentration quantification and rRNA removal were performed using the FastSelect rRNA Kit (Vazyme, China). Specifically, samples with RNA concentrations below 10 ng/µL were not subjected to rRNA removal, while samples above 10 ng/µL were subjected to rRNA removal. Subsequently, the NGS library was constructed using the VAHTS® Universal V8 RNA-seq Library Prep Kit (Vazyme, China). Finally, the constructed libraries were sent to Beijing Geinga Medical Laboratory Co., Ltd. for quantitative quality control by Agilent 2100 Bioanalyzer and qPCR, and the library was rolled and replicated to form nanorods and loaded onto the sequencing chip, and then subjected to high throughput sequencing.
Sequence alignment
Overall, the DNBSEQ-T7 sequencer (BGI) and the double-end sequencing method (2 × 150 bp) was selected for sequencing in this study. The raw data of next-generation sequence (NGS) were analyzed by CLC Genomics Workbench version 23.0.2 (Qiagen) for quality control (QC) and trimming of low-quality sequencing data. In this study, the identification of mosquito-filtered reads was conducted through a series of bioinformatics analyses. Initially, the reads were processed using STAR v2.5.0a to align against host rRNAs and genome sequences. Subsequently, non-host reads were subjected to de novo assembly using MEGAHIT v1.1.2 to match against assembled contigs. The resulting contigs were then compared against viral nucleotides and proteins through blastn and blastx algorithms, employing an E-value threshold of 1E−2 to ascertain viral sequences. Finally, a further validation step was performed by comparing these contigs against the nonredundant nucleotide database (as of May 2024) using blastn with an E-value cutoff of 1E–10 to eliminate any potential false positives. To ensure the authenticity of detected viral sequences and minimize false positives, we implemented additional validation steps. After initial sequence alignment, we evaluated each nucleotide position for quality scores (Phred score > 30), minimum coverage depth (> 10×), and potential host genomic contamination. Sequences with ambiguous alignments or high similarity to host genomic material were flagged for further inspection. Additionally, we calculated the error probability at each nucleotide position and established a stringent threshold (p < 0.001) to distinguish true viral sequences from potential sequencing artifacts. All the software tools used in this study were run with default parameters unless otherwise specified. The adaptor sequences and low-quality bases (Phred score < 20) were trimmed from the raw reads. For host sequence removal, the clean reads were mapped to the mosquito reference genome (Aedes aegypti, AaegL5) using STAR v2.5.0a with default settings. The resulting high-quality non-host reads were then de novo assembled using MEGAHIT v1.1.2, with the k-mer size set from 21 to 149.
Detection of Fangshan bunya-like virus in mosquitos
The detection of the Fangshan bunya-like virus was conducted through qRT-PCR utilizing the Qiagen One-Step RT-PCR Kit and virus-specific primer-probe sets. The procedure was executed in strict adherence to the manufacturer’s guidelines. The reaction took place in a 25 µL volume, comprising 5 µL of buffer, 1 µL of dNTP, 1 µL of enzyme mix, 11.75 µL of RNase-free water, 0.5 µL each of upstream and downstream primers (refer to Table 1), 0.25 µL of the probe, and 5 µL of RNA. The primers were subjected to BLAST alignment analysis on the National Center for Biotechnology Information (NCBI) database to confirm their uniqueness and specificity against other sequences. The qRT-PCR protocol involved an initial step at 50 °C for 30 min followed by 95 °C for 15 min. Subsequently, 40 cycles were performed at 94 °C for 30 s, 55 °C for 30 s, and 72 °C for 1 min, with a final extension at 72 °C for 10 min. A cycle threshold (Ct) value of 40 cycles was set to determine a positive sample. The primer-probe sets utilized in this investigation were meticulously crafted and assessed using Primer Express v3.0 from Applied Biosystems in the USA. Initially, alignments were conducted with Clustal W (BioEdit v7.0.9) using the complete genomic sequences of Fangshan bunya-like virus segment L sourced from the GenBank database. Regions displaying significant conservation were selected following visual inspection. The primer-probe pairs were meticulously designed and evaluated using Primer Express v3.0, and a BLAST analysis was conducted to verify the specificity of the primer-probe set. Negative controls carried through all protocols showed that the system was not contaminated for the presence of Fangshan bunya-like virus. Furthermore, the qRT-PCR-positive samples underwent sequencing post nested PCR amplification. Nested PCR was executed with Fangshan bunya-like virus-specific primers to amplify the positive samples (refer to Table 1). The products of nested PCR amplification were purified through 1% agarose gel electrophoresis and visualized using an imaging system (ChemiDoc Touch, USA). High-quality amplification products were forwarded to the Qingdao Sangon Biotech Company for Sanger sequencing.
Cell culture
In this investigation, the utilization of Vero cells, BHK-21 cells, and C3/36 cells was pivotal. The C6/36 cells were cultivated in SF-9 medium supplemented with 10% fetal bovine serum (FBS, Gibco) and 1% penicillin/streptomycin, maintained at 30 °C with 3% CO2. Conversely, Vero and BHK-21 cells were nurtured in DMEM supplemented with 10% fetal bovine serum (FBS, Gibco) and 1% penicillin/streptomycin, incubated at 37 °C with 5% CO2. These cell lines are securely stored within the laboratory facilities for ongoing research endeavors.
Virus isolation
In the study, mosquito homogenates from qRT-PCR positive pools underwent filtration using a 0.22 μm microporous filter. Subsequently, 100 µL of the filtrate from each pool was extracted and inoculated onto monolayer C6/36 cells in a six-well plate with varying perforations. The cells were then incubated at 30 °C with 3% CO2 for an hour. Following this initial incubation period, 2 mL of SF-9 medium supplemented with 10% fetal bovine serum and 1% streptomycin were added to each well. The cells were cultured under the same conditions for a duration of 7 to 10 days, with blind passaging through three successive generations. Throughout the culture period, the cell supernatant from each generation was collected, and daily observations were made to monitor the cytopathic effect (CPE). RNA was extracted from the collected supernatant samples, and quantitative reverse transcription polymerase chain reaction (qRT-PCR) was conducted to ascertain the presence of the Fangshan bunya-like virus.
Phylogenetic analysis
Genome sequences of representative viruses in different families of the class Bunyaviricetes were downloaded from the GenBank database. Sequences were subjected to MAFFT (v7.310) alignment under the G-INS-i option18. A full tree search for protein models was then performed using the ModelFinder19 implemented in IQ-TREE (v1.6.12)20. Then, phylogenetic inference using maximum likelihood (ML) was performed by IQ-TREE using the best model, supplying branch support with ultrafast bootstrap (UFboot) value optimized using a hill-climbing nearest neighbor interchange (NNI) search21. Finally, the FigTree (http://tree.bio.ed.ac.uk/software/figtree/) was used for viewing the acquired tree files and further construction of phylogenetic trees with manual modifications.
Data analysis
The pooled infection rate was determined by dividing the number of positive specimen pools by the total number of processed specimen pools. The identification of open reading frames (ORFs) and the conserved domains of the virus were conducted through the utilization of the NCBI ORF finder tool and CD-Search software available on the NCBI platform and protein model prediction was conducted using PyMOL. Statistical analyses were conducted using Prism software version 5.00. Fisher’s exact test was employed to assess the statistical significance of differences in the positive rates, with p-values below 0.05 considered as indicative of statistical significance. Furthermore, viral sequence alignment and pairwise divergence analyses were carried out using MegAlign, a component of the DNASTAR software package (Lasergene, USA).
Result
Trapping of mosquitoes and nucleic acid extraction
A total of 3,400 mosquitoes were collected from 10 sampling sites in Weifang and Yantai, of which 3,013 females were used for the study (Fig. 1). The female mosquitoes were distributed across sampling sites as follows: Weifang site 1 (432 mosquitoes), Weifang site 2 (488 mosquitoes), Weifang site 3 (452 mosquitoes), Weifang site 4 (511 mosquitoes), Weifang site 5 (488 mosquitoes), Yantai site 1 (127 mosquitoes), Yantai site 2 (135 mosquitoes), Yantai site 3 (122 mosquitoes), Yantai site 4 (118 mosquitoes), and Yantai site 5 (140 mosquitoes). Morphological identification was performed on the collected samples using a stereomicroscope, and they were then categorized based on their external features, species, and sexes. The female mosquitoes were further divided into 71 pools, with each pool containing fewer than 50 mosquitoes, grouped according to collection date, species, and sampling site. Specifically, the 71 pools were distributed as follows: Weifang sites (58 pools in total: 12 pools from site 1, 12 pools from site 2, 11 pools from site 3, 11 pools from site 4, and 12 pools from site 5) and Yantai sites (13 pools in total: 2 pools from site 1, 3 pools from site 2, 2 pools from site 3, 3 pools from site 4, and 3 pools from site 5). Morphological characterisation has shown that Culex tritaeniorhynchus species was recorded at 889, Culex quinquefasciatus species at 1408, Anopheles sinensis species at 103, Aedes albopictus species at 396, and Armigeres subalbatus mosquitoes at 217 (Table 2). Evidently, Cx. quinquefasciatus emerged as the predominant species among the mosquitoes gathered in this research, with Cx. tritaeniorhynchus following closely behind. The male mosquitoes were excluded, retaining only the females for experimental purposes. Furthermore, these mosquitoes were categorized into 71 pools based on the date, species, and collection site. Nucleic acid extraction was performed separately for each pool.
Genome-wide analysis of Fangshan bunya-like virus
In total, two libraries generated data of 9.06G and 14.87G bases, including approximately 30.20 million and 49.58 million 150 bp paired end readings. After removing the host sequence, we obtained 12,054 and 25,321 clean readings related to the Fangshan bunya like virus. The complete genome of Fangshan bunya-like virus was successfully obtained, which comprised of three segments corresponding with previously defined categories of L, M, and S bunyaviral genomic segment nomenclature. The L segment’s genome was characterized by a length of 7477 bp and a guanine-plus-cytosine (G + C) content of 45.15%. The M segment, on the other hand, had a genome length of 1821 bp with a G + C content of 45.09%. The S segment exhibited a genome length of 1,408 bp and a higher G + C content of 52.27% (Fig. 2).

Gene structure diagram of three segments of the Fangshan bunya-like virus.
For the 20-LZ-1 strain, the average read depth was 170× (range: 12.3×–542.1×), with 99.2% of nucleotides covered at ≥ 20×. The 20-WF-12 strain showed an average depth of 360× (range: 28.7×–894.3×), with 99.5% coverage at ≥ 20×. SNP detection identified 123 high-quality variants (QUAL ≥ 30, DP ≥ 10) across the three genomic segments, including 97 transitions (78.9%) and 26 transversions (21.1%). No indels were detected. The majority of SNPs were synonymous (89/123, 72.4%), with non-synonymous substitutions distributed evenly across the L (12), M (8), and S (14) segments (Appendix Fig. 6). The false positive analysis of detected viral sequences revealed high confidence in the obtained genomic data. The mean Phred quality score across all viral contigs was 38.7, corresponding to a base-calling accuracy of 99.98%. Coverage depth analysis showed consistent coverage (mean depth: 170–360×) across all segments, with no significant coverage drops that might indicate chimeric sequences. When comparing against host genomic sequences, all viral contigs showed < 80% nucleotide identity to any mosquito genomic regions, confirming their viral origin rather than host contamination. The probability of false positive detection was calculated to be < 0.0001 for each of the reported viral segments, providing high confidence in the authenticity of the Fangshan bunya-like virus sequences. All sequences in this study have been uploaded to NCBI. (GenBank accession no. OR192222.1-OR192224.1, OR160351.1-OR160353.1)
Detection of Fangshan bunya-like virus RNA in mosquitoes
The qRT-PCR testing revealed the presence of Fangshan bunya-like virus RNA in 9 out of 71 mosquito pools, indicating a detection rate of 12.68% for the viral genetic material. Specifically, 6 positive pools originated from Cx. quinquefasciatus, while the remaining 3 pools were from Cx. tritaeniorhynchus. The MIR (minimum infection rate per 100 mosquitoes) of Fangshan bunya-like virus was 0.34% (3/889) in Cx. quinquefasciatus and 0.43% (6/1,408) in Cx. tritaeniorhynchus. There is no statistically significant difference in the MIR between the two species of mosquitoes. Geographically, the positivity rates in Yantai and Weifang are 7.69% and 13.79%, respectively, with no statistically significant difference (Table 3). Based on the second-generation sequencing, the two samples with higher titers, 20-LZ-1 and 20-WF-12, were successfully obtained in full sequence. Positive and negative controls carried through all protocols showed that the system was both sensitive to the presence of Fangshan bunya-like virus and free from contamination. Nested PCR amplification of pools with positive qRT-PCR assays using two sets of primers, and the partial segment was successfully amplified and sequenced from 7 of 9 pools.
Structural analysis of virus encoded proteins
The unique genome structure of the class Bunyaviricetes provided the basis for a deeper understanding of their biology and pathogenesis. Using Alphafold, we generated an accurate 3D model of the protein encoded by the L fragment, which predicted a sequence of 3426 amino acids. Functional domain prediction using the UniProt database revealed that the L fragment contains two structural domains: RdRp and the GsiA superfamily. RdRp plays a crucial role in viral RNA replication and transcription, with its core structural features being highly conserved among viruses within the same genus. The GsiA superfamily protein typically comprises a catalytic domain and a central domain, with key functions including the facilitation of adenylyltransferase import, enhancement of RdRp activity, and augmentation of deconjugase activity. Subcellular localization prediction using CELLO v.2.5 suggests that the protein is primarily localized in the cytoplasmic region, offering insights into the virus’s function within the host cell (Fig. 3).

3D cartoon representation of the L fragment (A), M fragment (B) and S fragment (C) structure of the Fangshan bunya-like virus generated by Alphafold. (A) L fragment: RdRp in yellow (aa 1925–2226) and GsiA super family in red (aa 401–471). (B) M fragment: Transmembrane domain in green (aa 105–127) and glycoprotein in red (aa 344–567). (C) S fragment: Nucleoprotein in red (aa 133–411). The scale bar represents the number of protein residues considered in the model predictions, with the color gradient from green to white reflecting the confidence level of the predictions. In the figure, green represents a high degree of confidence, corresponding to a smaller prediction error, while white indicates low confidence and a larger prediction error.
The protein encoded by the M fragment was predicted to contain 567 amino acids. Structural domain analysis indicated that the sequence contains both transmembrane and glycoprotein structural domains. Glycoproteins and membranes play crucial roles in the viral life cycle, being involved not only in the assembly and maturation processes but also in host cell recognition and viral invasion. Moreover, the protein sequence exhibits distinct transmembrane characteristics. Subcellular localization prediction indicated that the protein is primarily localized in the cytoplasm or outer membrane.
In the predicted protein model encoding the S fragment, the coat protein is a crucial component of the viral particle. Structural domain predictions suggest that the region spanning amino acids 133 to 411 may contain the nucleoprotein structural domain. This region binds to the viral RNA genome, forming the ribonucleoprotein (RNP) complex, which is essential for viral genome stability, replication, and packaging. Subcellular localization predictions suggest that the nucleoprotein is predominantly found in the cytoplasm. The nucleotide sequences of the three fragments of the two strains of viruses 20-LZ-1 and 20-WF-12 obtained in this study were partially mutated, but the translated amino acid sequences were mainly synonymous mutations (Appendix Fig. 1). Sequence comparison analysis revealed high similarity between Fangshan bunya-like virus and Pyongtaek Culex Bunyavirus. The three representative viral proteins encoded by Fangshan bunya-like virus, RdRp, NP and GP have the highest homology with the related proteins of Pyongtaek Culex Bunyavirus, with amino acid sequence similarities of 95.82%, 86%, and 87.91%, respectively. At the nucleotide level, the L, S, and M segments showed identity percentages of 85.30%, 82.33%, and 87.03% with corresponding segments of Pyongtaek Culex Bunyavirus. These high identity values in both amino acid and nucleotide sequences provide strong evidence that Fangshan bunya-like virus is a novel Bunyavirus closely related to Pyongtaek Culex Bunyavirus, likely representing a distinct genetic variant within the same evolutionary lineage. In addition, amino acid sequence comparison of Fangshan bunya-like virus and its similar family of viruses showed some of their amino acid substitutions (Appendix Figs. 3, 4 and 5).
Virus isolation
In the present study, we focused on the isolation of Fangshan bunya-like virus from nucleic acid positive samples and observed its growth characteristics in C6/36, BHK-21, and Vero cells. In the meantime, the presence of viral RNA in the supernatants of cells was detected using qRT-PCR with specific primers and the CPE of the cell monolayer was monitored. Unfortunately, the virus was not isolated from any positive mosquito samples, and none of the three cell lines showed any morphological changes.
Notably, after inoculation with 20-LZ-1 and 20-WF-12 homogenate, the virus genome was detectable in the first two passages in all three cell types, and the virus copy number is gradually decreasing. Subsequently, the virus could not be traced after three passages. Overall, the current data can only prove that viruses are not suitable for growth in these three cell lines, and does not provide evidence of the virus entering cells for replication.
Phylogenetic analyses
The class Bunyaviricetes is the largest known family of RNA viruses, with approximately 600 viruses identified. The genomes of the vast majority of bunyaviruses are composed of three segments of single-stranded negative-sense RNA22. This segmented character increases the likelihood of genetic reassortment among the members of the virus family. Therefore, we chose to analyze the evolutionary relationship of Fangshan bunya-like virus by comparing the similarity of the three proteins encoded by this virus. Clearly, the top BLAST hit (i.e., highest e-value) for RdRp, glycoprotein and nucleoprotein with relatively clear virological classification status was all Pyongtaek Culex Bunyavirus, from the order Bunyavirale. Next, we analyzed the three proteins of representative strains from all families under Bunyavirale, and discovered that Fangshan bunya-like virus and Pyongtaek Culex Bunyavirus formed a monophyletic group nested within the diversity of the family Phenuiviridae, which suggests that these viral segments had a single origin from the Phenuiviridae (Figs. 4, 5, 6). Meanwhile, phylogenetic trees based on three proteins all indicated that Fangshan bunya-like virus had the closest relationship with members of the genus Mobuvirus, so we speculated that Fangshan bunya-like virus was a new member of this genus.

Phylogenetic analysis of the RdRp sequences of Fangshan bunya-like virus identified from mosquito in this study (marked black with a circle. The protein substitution model used for this fragment is: LG + F + R7. Bootstrap values are shown at the branches. GenBank accession numbers are listed for each strain/isolate/amplicon. Descriptions of the sequences published in GenBank were reproduced without changes.

Phylogenetic analysis of the GP sequences of Fangshan bunya-like virus identified from mosquito in this study (marked black with a circle. The protein substitution model used for this fragment is: Blosum62 + F + R4. Bootstrap values are shown at the branches. GenBank accession numbers are listed for each strain/isolate/amplicon. Descriptions of the sequences published in GenBank were reproduced without changes.

Phylogenetic analysis of the NP sequences of Fangshan bunya-like virus identified from mosquito in this study (marked black with a circle. The protein substitution model used for this fragment is: LG + F + I + G4. Bootstrap values are shown at the branches. GenBank accession numbers are listed for each strain/isolate/amplicon. Descriptions of the sequences published in GenBank were reproduced without changes.
Furthermore, the partial S segment (about 1073 bp) of Fangshan bunya-like virus was amplified and sequenced from 7 of 9 qRT-PCR positive samples. A pairwise distance analysis demonstrated that the 7 sequences had a nucleotide identity of 98.1–99.9% with each other. A phylogenetic tree was constructed based on the 7 nucleotide sequences, along with the corresponding sequences of the Phenuiviridae. As shown in Fig. 7, all of the Fangshan bunya-like virus strains that were identified in this study clustered with each other, and showed a closer evolutionary relationship with Narangue virus of the genus Mobuvirus.

Phylogenetic analysis of 7 partial sequences of Fangshan bunya-like virus obtained in this study with other reference sequences from GenBank. The phylogenetic tree was built based on about 1000 bp using nucleic acid substitution model: HKY + F + G4. The numbers above the branches indicate bootstrap values. Black circle indicated Fangshan bunya-like virus sequences identified in the current study.
Discussion
Mosquitoes serve as vectors of a wide range of viral pathogens, leading to zoonotic diseases characterized by complex and unpredictable epidemiological dynamics, thus posing a significant threat to human and animal health. These viruses were typically endemic in tropical and subtropical regions, but the geographical distribution of these viruses is shifting with climate change and mosquito spread23. In recent years, our understanding and ability to manage mosquito-borne viruses improved significantly with the development of high-throughput sequencing technologies24. Metagenomic approaches have been employed to analyze a multitude of mosquito species approaches to identify medically important viruses and their core viral groups25,26. In this study, we investigated the development of the genome structure and phylogeny of a new member of the family Phenuiviridae, which was the first virus identified in Cx. tritaeniorhynchus and Cx. quinquefasciatus mosquitoes. Notably, Cx. tritaeniorhynchus and Cx. quinquefasciatus mosquitoes have been documented as vectors for other arboviruses in Asia, including Japanese encephalitis virus27, West Nile virus28, and Batai virus29. These species exhibit broad vector competence, highlighting the potential public health risk of Fangshan bunya-like virus.
The observed minimal infection rate (MIR) was determined to be 0.30%, indicative of a modest yet significant presence of the virus within the sampled vectors. The detection of Fangshan bunya-like virus genetic material in two distinct mosquito species suggests the potential for vector promiscuity, with implications for the virus’s transmission dynamics and potential public health risk. Similar to some arboviruses that infect a wide range of vectors, the ability of Fangshan bunya-like virus to infect a wide range of mosquitoes means that it has the potential to spread and be maintained more widely in mosquito communities30,31. The lack of statistically significant differences in detection rates between the two mosquito species suggests that the viral genetic material exhibits no significant preference for specific species. The virus was detected in only one pool of the mosquitoes collected from Yantai, while in Weifang, it was detected in eight pools. There is no difference in the positivity rate between the two regions. This not only indicated that the virus was widely present within Shandong Province, but also suggested that the prevalence of the virus was roughly the same across different regions. While we detected Fangshan bunya-like virus genetic material in both Cx. tritaeniorhynchus and Cx. quinquefasciatus, it is important to note that detection of viral RNA alone does not confirm vector competence. The presence of viral RNA may indicate that the mosquitoes had fed on an infected host or that the virus may be capable of infecting mosquito cells. However, comprehensive vector competence studies involving experimental infections, assessment of viral replication within mosquito tissues, and transmission experiments are necessary to definitively establish these mosquito species as competent vectors for Fangshan bunya-like virus. Such studies would provide valuable insights into the virus’s transmission dynamics and potential public health significance.
Comparing the genome structures of Fangshan bunya-like virus with other viruses in the Phenuiviridae family revealed that despite differences in specific nucleotide sequences, they exhibited a high degree of similarity in terms of function and structure. The conservation of the genome structures of Segment-L, Segment-M, and Segment-S ensured the virus’s efficient replication and transmission capabilities32. The structural conservation of these segments reflected the dependency of the family Phenuiviridae viruses on these key proteins during the evolutionary process, which also ensured their survival and adaptability in various host environments. However, Fangshan bunya-like virus exhibits significant structural differences from members of the family Phenuiviridae within the class Bunyaviricetes. Firstly, the Segment-M of the Fangshan bunya-like virus is relatively short, potentially impacting the size and characteristics of the glycoprotein precursors Gn and Gc that it encodes33. Secondly, different from the common situation with bunyaviruses, the genome of the Fangshan bunya-like virus did not currently exhibit the non-structural protein NSS (Non-structural protein S), which typically plays a role in disrupting the host’s immune response in other bunyaviruses34. Lastly, the non-coding region of the Fangshan bunya-like virus has not yet been found to contain sequences capable of forming a “panhandle” structure, a conserved feature in many RNA viruses that is associated with viral replication and stability35. Our study presents a putative coding sequence, and we have not yet determined the complete genomes of the Fangshan bunya-like virus. Further research, including the completion of RACE analyses, is necessary to fully understand the replication mechanisms, host interactions, and the adaptability and transmission modes of this virus. We look forward to contributing to the ongoing body of research in this area.
We successfully sequenced the complete genomes of two distinct strains of the same virus, revealing a high degree of sequence identity across their three genomic segments (L, M, and S). Our pairwise sequence comparison, conducted using the ClustalW algorithm, demonstrated that the two strains, despite being from geographically distinct host populations, exhibit a very high nucleotide sequence identity. This high identity is reflected in the amino acid sequences, which show a remarkable conservation with identities. While a small number of nucleotide mutations were observed, our analysis confirmed that these mutations are predominantly synonymous, leading to no significant alterations in the amino acid sequences of the encoded proteins. This is further supported by the structural predictions based on the translated protein sequences, which confirmed that these genetic variations do not impact the integrity of the proteins’ secondary or tertiary structures. Our findings underscore the remarkable conservation of the viral genome and its encoded functional elements, suggesting limited genetic drift potential within the viral population. This study once again emphasizes the importance of whole-genome sequencing in understanding the evolutionary dynamics and structural constraints of viral populations.
This study clarified the phylogenetic status of Fangshan bunya-like virus within the vast genus of bunyaviruses, thereby revealing its familial affiliations. Through detailed comparisons of three key proteins encoded by the virus, it unveiled the genetic relationship between this virus and other viruses within the family Phenuiviridae. Our analysis, based on the conserved RdRp sequence, definitively placed Fangshan bunya-like virus within the family Phenuiviridae, highlighting its close evolutionary ties with XiangYun bunya-arena-like virus 5 and Pyongtaek Culex Bunyavirus (Appendix Fig. 2). Consistency observed in the systematic evolutionary analysis of the G protein and N protein sequences further confirmed this connection. These three groups of viruses constituted a robustly supported clade in phylogenetic analysis, indicating a shared evolutionary history and potential similar biological characteristics. Further investigation of the genomic sequences of the isolates 20-LZ-1 and 20-WF-12 revealed a high degree of genomic homology between these two strains of Fangshan bunya-like virus isolated from different geographical locations. This finding does not automatically suggest a recent transmission or spread of a common viral lineage. Further research is required to substantiate any claims regarding the transmission of a common lineage. Moreover, the genetic consistency among the seven isolates of Fangshan bunya-like virus indicated limited genetic variation within the RNA genomes of these viral strains, possibly due to a low mutation rate during viral replication or strong natural selection pressures36. This level of genetic conservation among these isolates may reflect a robust adaptive strategy or a constrained evolutionary pathway37.
Despite the initial detection of Fangshan bunya-like virus in the viral supernatant of the first two generations, it could not be successfully isolated from the cells. This posed an intriguing challenge and warranted further investigation. The transient presence of the virus in these cell lines suggested potential limitations in the viral replication machinery or compatibility with the cellular environment. It was plausible that the virus, while capable of initial infection, did not sustain its replication cycle in either cell type, possibly due to intrinsic antiviral responses or inadequate provision of necessary cofactors for viral assembly and release. Future work will concentrate on optimizing conditions for viral isolation, including refining cell culture techniques, adjusting passage strategies, and exploring the molecular mechanisms of virus-host cell interactions, with the aim of improving the success rate of viral isolation and gaining a deeper understanding of its replication patterns in different cell lines. Additionally, investigating virus-vector interactions, such as viral persistence in mosquito tissues, transmission barriers, and potential effects on vector behavior and fitness, will be crucial for understanding the ecological role of this virus and its transmission dynamics in natural settings.
In conclusion, our study presented the first identification and characterization of the Fangshan bunya-like virus, a novel member of the family Phenuiviridae, from mosquito populations in Shandong Province. The high genomic similarity across sequences and their close phylogenetic relation to known viruses emphasized the value of continuous mosquito surveillance. Our findings have identified a new member of the Phenuiviridae family, enriching the known diversity of the class Bunyaviricetes. However, further research is needed to fully characterize its pathogenicity in animal models and assess potential zoonotic risks to humans.
Data availability
The data underlying this article are available in NCBI and can be accessed with the following accessions: OR192222.1, OR192223.1, OR192224.1, OR160351.1, OR160352.1, OR160353.1. (https://www.ncbi.nlm.nih.gov/nuccore/OR192222.1)
References
Young, P. R. & Arboviruses A family on the move. Adv. Exp. Med. Biol. 1062, 1–10. https://doi.org/10.1007/978-981-10-8727-1_1 (2018).
Mayanja, M. N. et al. Mosquito-borne arboviruses in uganda: History, transmission and burden. J. Gen. Virol. https://doi.org/10.1099/jgv.0.001615 (2021).
Li, J. et al. Evolutionary analysis of a newly isolated Banna virus strain from yunnan, China. Arch. Virol. 167, 1221–1223. https://doi.org/10.1007/s00705-022-05403-z (2022).
Saxena, V. et al. West nile virus. Clin. Lab. Med. 37, 243–252. https://doi.org/10.1016/j.cll.2017.01.001 (2017).
Khan, U. M. et al. La Crosse Encephalitis. StatPearls. Treasure Island (FL) Ineligible Companies. Disclosure: Aashrai Gudlavalleti Declares No Relevant Financial Relationships with Ineligible Companies: StatPearls Publishing Copyright © 2024 (StatPearls Publishing LLC., 2024).
Schneider, E. F. et al. Jamestown Canyon virus in collected mosquitoes, Maine, United States, 2017–2019. Emerg. Infect. Dis. 28, 2330–2333. https://doi.org/10.3201/eid2811.212382 (2022).
Byford, O. et al. Lymphocytic choriomeningitis arenavirus utilises intercellular connections for cell to cell spread. Sci. Rep. 14 https://doi.org/10.1038/s41598-024-79397-w (2024).
Földvári, G. et al. Genomic characterization of Volzhskoe tick virus (Bunyaviricetes) from a hyalomma marginatum tick, Hungary. Sci. Rep. 14 https://doi.org/10.1038/s41598-024-69776-8 (2024).
Simo Tchetgna, H. et al. Molecular characterization of a new highly divergent Mobala related arenavirus isolated from Praomys sp. rodents. Sci. Rep. 11, 10188. https://doi.org/10.1038/s41598-021-88046-5 (2021).
Boshra, H. An overview of the infectious cycle of bunyaviruses. Viruses 14 https://doi.org/10.3390/v14102139 (2022).
Ni, H. et al. Epidemiological characteristics and transmission dynamics of dengue fever in China.
Weaver, S. C. et al. Zika, chikungunya, and other emerging Vector-Borne viral diseases. Annu. Rev. Med. 69, 395–408. https://doi.org/10.1146/annurev-med-050715-105122 (2018).
Wang, Y. et al. Projection of dengue fever transmissibility under climate change in South and Southeast Asian countries. PLoS Negl. Trop. Dis. 18, e0012158. https://doi.org/10.1371/journal.pntd.0012158 (2024).
Dong, X. et al. A novel bunyavirus discovered in Oriental shrimp (Penaeus chinensis). Front. Microbiol. 12, 751112. https://doi.org/10.3389/fmicb.2021.751112 (2021).
Rattanarithikul, R. et al. Illustrated keys to the mosquitoes of thailand. VI. Tribe Aedini. Southeast. Asian J. Trop. Med. Public. Health. 41 (Suppl 1), 1–225 (2010).
Yaghoobi-Ershadi, M. R. et al. Morphological studies on adult mosquitoes (Diptera: Culicidae) and first report of the potential Zika virus vector Aedes (Stegomyia) unilineatus (Theobald, 1906) in Iran. Bull. Soc. Pathol. Exot. 110, 116–121. https://doi.org/10.1007/s13149-016-0530-1 (2017).
Gyawali, N. et al. A morphological identification key to the mosquito disease vectors of the Pacific. 64, e70003. (2025). https://doi.org/10.1111/aen.70003
Nakamura, T. et al. Parallelization of MAFFT for large-scale multiple sequence alignments. Bioinformatics 34, 2490–2492. https://doi.org/10.1093/bioinformatics/bty121 (2018).
Kalyaanamoorthy, S. et al. ModelFinder: Fast model selection for accurate phylogenetic estimates. Nat. Methods. 14, 587–589. https://doi.org/10.1038/nmeth.4285 (2017).
Nguyen, L. T. et al. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 32, 268–274. https://doi.org/10.1093/molbev/msu300 (2015).
Hoang, D. T. et al. UFBoot2: Improving the ultrafast bootstrap approximation. Mol. Biol. Evol. 35, 518–522. https://doi.org/10.1093/molbev/msx281 (2018).
Elliott, R. M. Emerging viruses: The Bunyaviridae. Mol. Med. 3, 572–577 (1997).
Xia, H. et al. Mosquito-associated viruses in China. Virol. Sin. 33, 5–20. https://doi.org/10.1007/s12250-018-0002-9 (2018).
Roossinck, M. J. Deep sequencing for discovery and evolutionary analysis of plant viruses. Virus Res. 239, 82–86. https://doi.org/10.1016/j.virusres.2016.11.019 (2017).
Fauver, J. R. et al. West African Anopheles Gambiae mosquitoes harbor a taxonomically diverse Virome including new insect-specific flaviviruses, mononegaviruses, and totiviruses. Virology 498, 288–299. https://doi.org/10.1016/j.virol.2016.07.031 (2016).
Fetters, A. M. et al. The pollen Virome of wild plants and its association with variation in floral traits and land use. Nat. Commun. 13, 523. https://doi.org/10.1038/s41467-022-28143-9 (2022).
Faizah, A. N. et al. Mosquito populations originating from nonendemic areas have the potential to transmit recently emerging Japanese encephalitis virus genotype IV. Emerg. Microbes Infect. 14, 2438661. https://doi.org/10.1080/22221751.2024.2438661 (2025).
Khan, S. A. et al. Detection of West nile virus in six mosquito species in synchrony with seroconversion among Sentinel chickens in India. Parasit. Vectors. 10 https://doi.org/10.1186/s13071-016-1948-9 (2017).
Sudeep, A. B. et al. Vector competence of certain culex and Aedes mosquitoes for the Chittoor virus, the Indian variant of the Batai virus. Can. J. Microbiol. 64, 581–588. https://doi.org/10.1139/cjm-2017-0514 (2018).
Mansfield, K. L. et al. Batai orthobunyavirus: An emerging Mosquito-Borne virus in Europe. Viruses 14 https://doi.org/10.3390/v14091868 (2022).
Mravcová, K. et al. Ťahyňa virus-A widespread, but neglected mosquito-borne virus in Europe. Zoonoses Public. Health. 70, 371–382. https://doi.org/10.1111/zph.13042 (2023).
Sun, M. H. et al. Highly adaptive phenuiviridae with biomedical importance in multiple fields. J. Med. Virol. 94, 2388–2401. https://doi.org/10.1002/jmv.27618 (2022).
Ribeiro, M. S. et al. High prevalence of a newly discovered Wutai mosquito phasivirus in mosquitoes from Rio de janeiro, Brazil. Insects. https://doi.org/10.3390/insects10050135 (2019).
Sizikova, T. E. et al. The molecular evolution of Dabie bandavirus (Phenuiviridae: Bandavirus: Dabie bandavirus), the agent of severe fever with thrombocytopenia syndrome. Vopr Virusol. 66, 409–416. https://doi.org/10.36233/0507-4088-68 (2022).
Klempa, B. et al. Sangassou virus, the first hantavirus isolate from Africa, displays genetic and functional properties distinct from those of other murinae-associated hantaviruses. J. Virol. 86, 3819–3827. https://doi.org/10.1128/jvi.05879-11 (2012).
Guterres, A. et al. New bunya-like viruses: Highlighting their relations. Infect. Genet. Evol. 49, 164–173. https://doi.org/10.1016/j.meegid.2017.01.019 (2017).
Simmonds, P. et al. Detection of genome-scale ordered RNA structure (GORS) in genomes of positive-stranded RNA viruses: Implications for virus evolution and host persistence. Rna 10, 1337–1351. https://doi.org/10.1261/rna.7640104 (2004).
Acknowledgements
We are deeply grateful to Mr. Zongdong Liu and Hongchen Fu for his contributions in sample collection.
Funding
This research was supported by the National Natural Science Foundation of China (82104053), Development Program for Youth Innovation Team in Colleges and Universities of Shandong Province (2022KJ263), the Science and Technology Development Program in Weifang (2020GX014). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Author information
Authors and Affiliations
Contributions
Y.Z. conceived and designed the study. Y.L. analyzed the data and interpreted some of the results. L.L. carried out formal analysis. Y.T. and M.R. collected the specimens. H.S. managed the overall coordination of the study. Y.G. wrote the original draft and the review. Y.Z. conducted the molecular experiments. All authors reviewed the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Zhang, Y., Liu, Y., Liu, L. et al. Genome of a novel Fangshan bunya-like virus identified in mosquitoes from Shandong Province, China. Sci Rep 15, 31356 (2025). https://doi.org/10.1038/s41598-025-16304-x
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-16304-x

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}