
Abstract
The primary goal of this study is the enhancement of the mechanical properties of polyurethane (PU) coating via introducing a hybrid sandwich-like nanofiller composed of layered double hydroxide (LDH) grown on graphene oxide. To increase the nanofiller compatibility with the PU matrix, its surface was functionalized with an amino-functional silane coupling agent(APTES). Characterization of the particles with Raman, TGA, EDX-mounted FESEM, XRD approved the prosperous synthesis and modification of the nanosheets. The untreated (LDH@GO) and silane-treated (LDHGO@Si) nanoplatelets at three loading levels (1, 2, and 3 wt%) were incorporated and the stress-strain behavior of various nanocomposites films were evaluated. The stress-strain diagrams revealed that there is an optimum content (2wt.%) for both nanosheets that causes a rise in mechanical properties. While LDH@GO could improve the toughness of the PU by 95%, the treated counterpart (LDHGO@Si) resulted in a stronger impact, leading to a 184% rise concerning unfilled PU (from 1.27 to 3.6 GPa) in toughness thanks to its better dispersion, more extensive covalent and non-covalent interactions with PU chains. DMTA results also depicted that the treated nanosheets formed a reinforced network with increased Tg and crosslinking density that can efficiently delocalize the stress under tensile pressure, resulting in promoted energy dissipation and toughness.
Similar content being viewed by others


Introduction
New composite materials with outstanding mechanical properties can be designed by introducing a nanoscale reinforcing agent into different polymeric matrices1. Nowadays, utilizing polymeric nanocomposites, especially polyurethane-based ones, has presented attractive solutions where a high level of short- and long-term mechanical integrity is a high demand2,3,4.
Protective polymeric coating systems are usually applied on various substrates to protect them against different environmental attackers during coating service life. A coating system, depending on its practical application, can be threatened by various factors from its surroundings. Weathering (i.e. radiations like sunlight, humidity or water, biological substance, thermal shocks, corrosive species, etc.) and mechanical (traffic, abrasion, scratch, stone-chipping, etc.) are among the main aggressive parameters that can cause gradual degradation of the coating integrity and its protective function during its service life5,6,7,8,9. Thus, coating systems must be fortified to tolerate such aggressions. The most innovative approach to promote the mechanical performance of an organic coating is to introduce an intrinsically high modulus filler into the organic media10,11,12,13,14,15.
The literature survey concluded that the prosperity of such a particle-reinforcement strategy is highly correlated to the extent and the strength of the interfacial interactions that are formed at the filler-matrix interface area16. Not always, but usually, the greater sites of interactions and the stronger covalent/non-covalent bonds between the filler surface and the polymeric media result in a more resistant substance against mechanical tensions. Nano-sized fillers with high aspect ratios have been desired fillers for mechanical properties enhancement17,18,19,20,21,22.
Layered double hydroxides (LDHs) have been recently considered capable functional fillers for different purposes, especially for the mechanical enhancement of polymeric nanocomposites. –––They, with the general chemical formula of [M2+ (1 –X) M3+ X (OH)2]X+ (An−)x/n.mH2O, possess tunable composition due to the relative proportions for both di- and trivalent metal cations (M2+ and M3+), and interlayer anions(A)23. Such elemental composition has led to the positively charged outer layers that are neutralized with anions occupying the interlayer spaces. They are typically synthesized via three conventional methods; layered- by-layered assembling, sol-gel, and co-precipitation24,25,26,27,28. Among these, the co-precipitation method has attracted interest due to its easy procedure. LDH-based compounds, thanks to their inorganic character, lamellar shape, and high aspect ratio have been utilized in many applications including where there is a high demand for mechanical enhancement of polymeric composites29,30.
Graphene is one of the strongest, lightest, and thinnest materials already recognized by man. As structurally known, it is a single thin layer of Sp2 hybridized carbon atoms that are 2-dimensionally arrayed in a densely packed honeycomb crystal lattice31,32. The unique covalently-tightened carbon-carbon bonds have endowed graphene with impressive mechanical attributes (Young’ modulus and ultimate strength of 1 Tera Pascal and 130 Giga Pascal, respectively). These incredible characteristics have enabled graphene, and its derivatives, to be implemented in high-performance polymeric nanocomposite production and development, as reported particularly as a reinforcing nanofiller in a wide range of polymeric matrices33,34,35,36,37,38. Adak et al. used graphene in a urethane matrix39. It was concluded that the strong physical interactions of PU/functionalized graphene, along with the high level of dispersion of graphene into the PU matrix, were the primary reasons for achieving a reinforced nanocomposite, exhibiting significant improvement in mechanical and long-term performances compared to the unfilled PU films.
Since both inorganic and organic nanoparticles, including LDHs and graphene derivatives, intrinsically tend to aggregate or agglomerate in the organic matrices, their surface modification, aiming at reaching a stable dispersion within the end-user matrix, is a favorable topic for scientific research40,41,42. Surface decoration and modification with functional silane coupling agents are among the most frequent strategies that have been implemented for this purpose11,12,43.
Polyurethane (PU) coatings are used in protective and functional coatings where durability and toughness are of the utmost concern. Conventional PU materials may not in all cases offer the necessary toughness and physical stability in severe applications. One of the most effective means of overcoming such limitations is using nanoscale fillers in the synthesis of reinforced PU nanocomposites. More intriguing are 2D nanomaterials such as graphene oxide (GO) and layered double hydroxides (LDHs), which have extremely high aspect ratios and huge surface areas and are thus fantastically efficient polymer reinforcing materials44. Though both GO and LDH are individually highly active as polymer nanofillers, both of these two nanomaterials have the inherent shortcoming that cancels their reinforcement effects.
LDHs also restack easily in loosely dispersed tactoids, leading to interfacial adhesion weakness to the polymer44. GO sheets also restack easily due to strong π–π interactions. GO–LDH hybridization can overcome the aforementioned disadvantages and create dispersion stability and interfacial compatibility. For example, Long et al. demonstrated that strongly coupled graphene oxide–FeNi LDH hybrids possess superior structure strength and dispersion stability that remarkably enhances their reinforcing efficiency45. Recent studies confirm synergistic enhancements obtained with GO–LDH hybrid fillers in polymer matrices. Zhu et al. achieved a record-high tensile strength and elongation in a GO–PU composite by engineering strong hydrogen bonding interactions46.
Yang et al. demonstrated that coupling Zn–Cr LDHs with graphene significantly improved the dispersion, interfacial charge transfer, and structural integrity of the hybrid system, suggesting strong potential for mechanical and functional enhancement in polymer matrices47.
Zhang et al. prepared GO–LDH hybrids to dramatically enhance gas barrier property and mechanical toughness in styrene–butadiene rubber composites48. These hybrids created interpenetrating networks of fillers and hence helped improve fracture toughness as well as prevent gas permeability. Cong et al. also demonstrated that GO-functionalized CaAl–LDH hybrids greatly enhanced tensile fracture strength, elongation at break, and chemical stability of fluoroelastomer composites through improved dispersion and interface interaction44. On the basis of such research, the current work extends further in the addition of LDH@GO hybrid nanosheets into thermosetting PU coatings as well as additional surface modification with silane coupling agents for further enhancement of filler–polymer adhesion. The hierarchical approach is likely to bring further advancements in mechanical toughness, fracture resistance, and weatherability with new reference points in future high-performance polyurethane coatings.
In this paper, we intend to fabricate a composite filler using two already-explained 2D nanoparticles (graphene oxide and LDH nanosheets) and study its effectiveness in boosting the mechanical properties of a typical PU matrix. Thus, at first, graphene oxide nanoplatelets will be synthesized. Then, it will be decorated with the Zn–Al–LDH nanosheets via an in-situ approach. The gradual growth of LDH on the graphene oxide surface can function not only as a barrier to physically hinder the graphene oxide aggregation but also contribute (via sandwich–like filler assembly) to promote the mechanical performance of the matrix at which they are intended to be incorporated. An additional surface modification process was also targeted on the LDH-decorated graphene oxide by amino-silane coupling agents to prevent the dual sheet-like particles from agglomeration and also increase the tendency (covalently and noncovalently) of such nanocomposite filler toward PU matrix backbone, promoting the nanosheets tendency toward surrounding PU media. The untreated synthesized nanofillers and their silane-treated counterparts were embedded in the PU. The influence of such embedment on the mechanical and structural properties was explored by tensile and dynamic mechanical thermal analysis (DMTA) techniques, respectively, and the toughening mechanisms were thoroughly studied by FE-SEM.
Experimental
Material
Steel plates (150 mm × 200 mm × 2 mm) were obtained from a domestic factory (Foolad Mobarakeh Steel Co., Iran). The polyurethane resin was prepared by stoichiometrically mixing a typical OH-functional acrylic polyol (AC753, OH content of 4.5%, solid content of 57.17 wt%, manufactured by PolyResin Co., Karaj, Iran), Desmodur N75 (a widely-used aliphatic polyisocyanate, Covestro, Germany) and a solvent solution including butyl acetate and xylene (50/50 w/w). All the reagents/reactants used for the synthesis of nanosheets were of analytical grade. Expandable graphite, Hydrochloric acid (37%), Potassium Permanganate, Sodium Nitrate, Sulfuric acid (98%), Sodium hydroxide, Hydrogen Peroxide (H2O2), Zinc Nitrate Hexahydrate, Sodium Chloride were purchased from Merck Co. 2-Amino Propyl Triethoxy Silane(APTES) for surface modification of nanoparticles was procured from Exir Co.(Iran). Aluminum nitrate Nonahydrate was sourced from Aldrich.
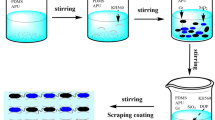
Graphene oxide synthesis and decoration with LDH nanoparticles (LDH@GO)
Starting from expandable graphite powder and through the modified Hummer’s approach, an aqueous graphene oxide colloidal suspension was prepared under the nitrogen atmosphere. Details of synthesis are given in the Ref12.
It was attempted to grow Zn–Al–NO3-LDH on the graphene oxide nanosheet through an in-situ co-precipitation procedure under N2 purging49. Double distilled water was utilized for the synthesis of all solutions to avoid contamination caused by undesired anions. Zinc Nitrate (0.8 M) and Aluminum Nitrate (0.6 M) solution were stirred to obtain aqueous suspension for 30 min. Following that, the sodium nitrate solution was gently dropped into the previous solution. Finally, the solution pH was attempted to remain constant at 12 ± 0.2 during the process by NaOH addition (2.4 M). The final particles were filtered, washed with water five times, and oven-dried at 80 °C for 24 h. The product in this stage was coded as “LDH@GO”.
Particle surface treatment (LDHGO@Si)
For the surface modification of the synthesized nanoparticle, the LDH@GO nanoparticles (5 g) were dispersed in ethanol (26.8 ml) for 5 min at 25 ± 1 °C by a homogenizer (Viggen, model 0310) at 10,000 rpm. 2.8 ml of aminopropyl triethoxy silane (APTES) was gently added to the dispersion and the temperature was adjusted between 80 and 90 °C under the reflux condition for 24 h. The resultant products were subsequently washed 6 times with ethanol: deionized water (60:40 by volume) and the precipitated sample was kept at 80 °C for 24 h. The resultant surface-modified nanosheet was coded as “LDHGO@Si”.
Coating preparation
A schematic representation of the whole coating system, including all layers applied in this study, is illustrated in Fig. 1. The steel panels were wiped out with an alkaline degreaser and deionized water, followed by immersion in the phosphate activator solution (50 °C for 3 min). The phosphate samples were then covered (with a dry film thickness of 15 ± 5 μm) by a commercial water-borne electrodeposition epoxy coating and exposed to a thermal curing at 180 °C for 20 min. The next layer (as a primer layer) was a polyester/melamine (2.5/1, binder/binder) system that was applied with a 50 μm-thick on the electrodeposition layer. The primer was thermally cured at 145 °C for 20 min. Afterward, a 40 μm-thick 2 K PU was employed to be coated as a solid basecoat.

A schematic representation of the multi-layer coating system.
Finally, the clearcoat, which was designed to have a similar chemistry to the basecoat (PU), was prepared by the addition of various loadings (1, 2, and 3%) of synthesized LDH@GO and LDHGO@Si nanoparticles as nanofillers. The nanofillers were embedded into polyol resin and homogenized for 20 min to achieve uniform dispersion. At a stoichiometric weight mixing ratio of 2:1(polyol: hardener), the isocyanate-based hardener was introduced into the dispersion. A solvent solution including butyl acetate and xylene (50/50 w/w) was used for viscosity adjustment when required. After application of the targeted clearcoats on the basecoat layer, the panels were cured at 80 °C for 20 min. The codings of the panels are given in Table 1. For the free film preparation, only the last layer (clear coat) was coated on the glass sheets. After their curing (similar to the clearcoats applied on the steel), coated glasses were then immersed for 1–2 min in the water. The free-standing films were easily detached from the substrate.
Tests and methods
The nanoparticles synthesized and further modified were characterized with different techniques. Using 785 nm laser excitation and utilizing a Renishaw inVia reflex Raman spectrometer (UK), Raman spectra were obtained over a wavenumber range of 500–3200 cm− 1. An X-ray 6000 (Shimadzu, Japan) instrument (Cu Ka radiation (\(\:\lambda\:=\) 0.1540 nm) at 40 kV with 30 mA current) was employed to collect the X-ray diffraction (XRD) patterns. The thermogravimetry behavior of samples was investigated by a TGA (STA 150 instrument). The TGA was run in a nitrogen atmosphere from 25 to 800 °C with a heating rate of 10 °C/min. Moreover, the samples’ surface morphology was studied using a Field emission scanning electron microscope (FESEM, SU8010; CRMD, USA).
The mechanical and thermo-mechanical aspects of variously modified polyurethane nanocomposite films were analyzed by tensile and dynamical mechanical thermal analyzing (DMTA) techniques, respectively. Tensile testing was conducted according to ASTM D882-12 employing a Tinius Olsen Universal Tensile Tester. The film dimension was 45 mm × 100 mm with a thickness of around 30 μm. Before the test, samples were stored at 23 ± 2 °C and 50 ± 10% relative humidity for 24 h. The experiment was run at a tensile speed of 10 mm/min.
DMTA test was carried out using a NETZSCH DMA 242. The details of the DMTA experiment are tension mode, frequency of 1 Hz, temperature range of 25–150 °C, with a rate of 5 °C/min.
Results and discussion
Characterization
Transmission electron microscopy (TEM) analysis was employed to investigate the morphology of the synthesized LDH@GO nanoplatform in greater detail, and the results have been compiled in Fig. 2. The images clearly reveal the two-dimensional sheet-like structure of graphene oxide, which appears as a transparent background. According to previous studies, graphene oxide consists of layered sheets which, despite exhibiting wrinkles, maintain a clean and smooth surface50. Detailed analysis of the TEM images of the LHD@GO sample reveals the presence of an additional phase beyond the graphene oxide sheets. Flake-like structures with dimensions ranging from 40 to 90 nm were observed overlaying the graphene oxide layers, representing plate-like LHD particles. The obtained results show good agreement with similar studies, indicating that the graphene oxide sheets are effectively covered51. However, the growth of LDH particles in the bulk phase cannot be considered entirely unavoidable.

Morphological characterization of the synthesized GO@LDH structure using TEM analysis.
Raman spectroscopy provides crucial insights into chemical structures. The presence of the graphitic structure of graphene oxide in LDH@GO and LDHGO@Si composites has been confirmed by Raman spectroscopy, a well-known technique for identifying sp2 and sp3 hybridized carbon atoms. According to the findings of previous studies, the Raman spectrum of graphene oxide exhibits three characteristic peaks about 1060, 1381, and 1590 cm− 1, corresponding to the C–O–C bonds, the characteristic D and G bands, respectively52. The Raman test results for composites LDH@GO and LDHGO@Si are presented in Fig. 3. The nitrate anion exhibits only one mode (ν1), with the major peak of metal nitrate appearing at 1066 cm− 1. Notably, two main peaks are seen between 1000 and 2000 cm− 1. While that centered at 1361 cm− 1 is attributed to imperfections in the disordered graphitic carbon structure, known as the D-band, the peak at 1598 cm− 1 is assigned to the in-plane vibration of well-ordered SP2 bonded carbon (Raman-active E2g mode), termed the G-band. The ID/IG ratio is commonly utilized to assess the graphitization degree of carbonaceous materials and the average size of SP2 domains53. The graphitization ID/IG ratio for the GO powder was presented in our previously published paper. It was about 0.88 for the pure GO powder54. This value for the LDH@GO is about 1.03. Such increased graphitization ID/IG ratio for the LDH@GO could be due to the increment of the defect after the decoration of the GO sheets with the LDH structures, and the reduction of oxygenated functional groups (epoxide, hydroxyl, and carboxylic)55. When the graphitization degree of LDH@GO increases, the crystallinity degree of the material also increases, which could be due to the increment of defects in graphene oxide sheets due to the formation of complexes of zinc and aluminum metals present in the LDH structure with oxygen functional groups in GO sheets56. The result showed that the graphitization ID/IG value decreases for the LDHGO@Si. Such reduced ID/IG value for LDHGO@Si suggests the formation of carbon rings by the recovery of the fragment of dangling bonds through the carbonization process in the presence of the APTES molecules, and also the reduction of surface defects, which may result from the formation of a silane shell on the surface of the LDHGO sheets57.

Raman spectra for the LDH@GO and LDHGO@Si sheets.
XRD analysis is a valuable technique for evaluating a material’s crystalline structure (Fig. 4). The XRD diffraction patterns of the GO powder are presented in our previously published paper54. The pristine GO powder revealed a peak at 2θ value of 10.8° with an interlayer d-spacing of 8.1 Å, reflecting the basal peak of (001) carbon skeletal plane54. The LDH@GO XRD pattern showed characteristic peaks centered at 2θ of 11.25, 22.33, 33.98, 38.83, 45.82, and 60.55° (as presented in Table 2 as well) that are assigned to the (003), (006), (012), (015), (018), and (110) planes, respectively55.
The LDHGO@Si XRD pattern is similar to that of LDH@GO. It can be observed that the (003), (006), (012), (015), (018), and (110) planes are slightly shifted to the higher 2θ of 11.72, 22.47, 35.08, 39.44, 46.66, and 61.24°, respectively. According to the results, the characteristic peak for the GO sheets (at 2θ value of 10.8°) is shifted to the higher values of 2θ for the LDH@GO and LDHGO@Si powders. The basal peak of (001) plane for the GO sheets overlapped with the basal peak of (003) plane for the LDH@GO and LDHGO@Si powders that are located at 2θ values of 11.25°, and 11.72°, respectively. Moreover, the interlayered space value (d-spacing) was calculated using the Bragg equation55. Although the d-spacing of LDHGO@Si (003) is lower (7.54 Å) than that of LDH@GO (8.31 Å), the peak intensity is increased compared to LDH@GO. This phenomenon can be due to the bridging of the Si networks and the convergence of the layers.

XRD patterns for the LDH@GO and LDHGO@Si nanosheets.
As depicted in Fig. 5., the thermal stability of the synthesized powders was studied through TGA-DTG analysis. The TGA plots of the pristine GO powder was presented in our previously published paper54. The total weight loss values for GO, LDH@GO, and LDHGO@Si at 800 °C were approximately 75, 34.88, and 21.87%, respectively. According to the DTG plots, two significant weight loss stages were observed between 25 and 150 °C and 150–300 °C for both LDH@GO and LDHGO@Si samples. For LDH@GO, the first loss stage (T = 90 °C, weight loss ~ 10%) is ascribed to the evaporation of water/solvent molecules that have physically attached to both the external and internal surfaces of the LDH@GO. The next weight-loss stage (~ 25%) is explained by the decomposition of GO/LDH interactions and oxygenated functional groups (i.e. epoxy, hydroxyl, and carboxylic) of graphene oxide. The ash content of LDH@GO was approximately 65%.
Compared to LDH@GO, the TGA plot for LDHGO@Si demonstrated considerable thermal stability improvement for all weight loss stages studied here. The first stage for LDHGO@Si is approximately 5% lower than that of LDH@GO, suggesting that the more hydrophobic character of LDHGO@Si, compared to LDH@GO (which has plenty of oxygen-containing functionalities), which leads to lower water absorption and thus lower water content. Experiencing a weight loss of 14% during the second weight loss stage again proves the higher resistance of LDHGO@Si compared to the untreated nanosheets. Such improvement can be attributed to the inorganic parts of the APTES structure, which can create Si-O-Si networks on the surface of LDHGO@Si, thus forming a thermal shield that protects the interior graphene and LDH sheets at higher temperatures. The Si-O-Si can be formed during either initial surface modification or via thermally-triggered self-condensation of unreacted silanol functional groups as the temperature increases12.The ash content value of LDHGO@Si at 800 °C was around 78%, approximately 33% larger than that of LDH@GO, confirming the formation of inorganic Si-O-Si networks by APTES molecules on the surface of modified nanosheets.

TGA and DTG plots for the LDH@GO and LDHGO@Si sheets.
The morphology of pristine GO was investigated in our previous study58. Graphene oxide has a sheet-like structure with two-dimensional layers. The graphene oxide sheets have a mirror-like graphenenic and clean surface sheets. The morphological investigations of the synthesized LDH@GO and LDHGO@Si composites were performed by FE-SEM, as presented in Fig. 6. FE-SEM images reveal smaller agglomerations and stacks of graphene oxide sheets covered with LDH particles. To further confirm the distribution of elements inside the LDH@GO and LDHGO@Si composites, elemental mapping was carried out utilizing Energy-dispersive X-ray spectroscopy (EDS). The EDS mapping results showed a homogeneous distribution of N (orange zone), oxygen (yellow zone), Zn (red zone), and Al (green zone) on the LDH@GO surface, while carbon (blue zone) derived from graphene is spread throughout the selected domain. Additionally, Si (light blue) is evenly distributed throughout LDHGO@Si. Furthermore, EDS data indicates the coexistence of Al, Zn, O, C, N, and Si elements in the resulting product. The decrease in Al and Zn values observed in the EDS results when Si–O–Si molecules decorate the surface of LDH@GO powders confirms the synthesis and modification route undertaken.

(a) SEM images of LDH@GO (b) SEM images of LDHGO@Si (c) EDX analysis of LDH@GO (d) EDX analysis of LDHGO@Si; (e) Elemental mapping of LDHGO@Si.
Physical and mechanical properties

Stress-strain diagrams of neat and different PU films filled with unmodified and modified fillers.
The stress-strain diagrams can be utilized to extract some mechanical parameters including toughness, Young’s Modulus, Maximum stress (strength), and strain at break. These parameters were calculated and presented in Table 3.
Loading LDH@GO into PU at any portion has promoted mechanical parameters, leading to a considerable enhancement in toughness. Besides that improvement, it is clear that the particle dosage has an optimum (2 wt% in the PU-LDH@GO-2), where the highest value in modulus, strength, and toughness are achieved, and after that such favorable factors decrease. For the PU-LDH@GO-2, the Young‘s modulus has increased from 5.54 to 7.94 GPa (43% growth ) and the strength from 30.61 to 47.18 MPa (54%), the strain at break from 0.73 to 0.88 (20%). Such desired changes have led to a 95% enhancement in toughness compared to the PU-Blank (from 1.27 to 2.48 GPa).
According to Fig. 7; Table 3, it is exhibited that the silane-modified nanosheets have also resulted in a positive impact on the PU composite mechanical properties. While there is also an optimum content for LDHGO@Si in boosting the mechanical properties (similar to LDH@GO), the high loading of modified filler ( PU-LDHGO@Si-3 sample) exhibits an inferior performance compared to the optimum sample(PU-LDHGO@Si-2 sample). Almost at all filler dosages (1, 2, and 3%), the LDHGO@Si nanoplatelets have promoted the mechanical parameters much more efficiently compared to the LDH@GO, confirming the great influence of silane modification on the composite performance. The best mechanical attributes have been achieved for the PU-LDHGO@Si-2, where Young’s modulus, maximum strength, maximum strain, and toughness have respectively experienced considerable enhancements as much as 33%, 60%, 78%, and 184% compared to the pristine PU. Such a desired enhancement in the mechanical behavior of PU nanocomposite can be attributed to the influential impact of silane surface modifier that facilitates the nanosheets distribution of modified nanofillers and increases the covalent and non-covalent interaction between the silane-modified filler and the polar PU backbone11,39,59,60.
Through inspection of the fractured surface morphology, insightful information about the toughening mechanism of incorporated nanosheets can be achieved. FE-SEM images of the fractured surface of unfilled and filled PU films are revealed in Fig. 8. Pristine PU revealed a relatively even surface with the least roughness, reflecting a typical fragile damage morphology for the 3D-crosslinked PU network. Upon incorporating 2% LDH@GO into the matrix (PU-LDH@GO-2), the fractured morphology changes distinguishably as coarser surface roughness accompanied by disordered crack lines are formed. Despite the obvious asperities, in most areas of the fractured surface, a smooth surface is still observed. Such morphology indicates that unmodified LDH@GO particles have not been homogeneously dispersed within the matrix. In contrast, PU-LDHGo@Si-2 shows relatively more ordered and finer formations of grooves which may indicate a crack deflection process that has happened. Additionally, precise observation in the smoother area exhibits a very fine roughness in the nanoscale can be detected that was absent in PU-Blank, and PU-LDH@GO-2 morphologies. Significant differences in modulus and Poisson’s ratio between the PU matrix and surface-treated nanoplatelets cause a vast stress concentration in the spatial peripheral of each nanosheet, eventually resulting in extensive energy dissipation61,62. Uniform dispersion and distribution of LDHGO@Si nanosheets and their suitable interfacial adhesion with the PU matrix have led to better strain delocalization and toughness enhancement.

FE-SEM images of cross-section of (a) PU-Blank, (b) PU-LDH@GO-2, and (c) PU-LDHGO@Si-2.
The network structure (including glass transition temperature (Tg), cross-linking density, and the level of homogeneity) of PU nanocomposites filled with various portions of LDH@GO and LDHGO@Si nanoparticles was studied by the DMTA technique. Figure 9 illustrates the DMTA diagrams of various films as temperature sweep of storage modulus and Tan δ.

E′ (storage modulus) and Tan delta plots for PU-Blank, PU-LDH@GO-2, and PU-LDHGO@Si-2.
As expected and similar to almost all polymeric networks, the storage modulus descends from a glassy state (below Tg) to the rubbery zone (above Tg) via a sharp turning point (around Tg). For thermosetting networks like PU, the rubbery zone is usually perceived as a plateau area (after Tg) due to the covalently fixed chemical structure of the 3-dimensionally cross-linked network. Crosslinking density, υe, can be derived from the below equation.
In the above equation, the storage modulus and temperature at the onset of the plateau (rubbery) zone are respectively utilized for E′ and T, and R-value is the molar gas constant63,64,65. The temperature at which the Tan δ reaches its maximum value is ascribe to Tg. Meanwhile, the Tan δ width (at its half height) has a close connection to the homogeneity of the PU network66,67. The variations in the crosslinking density, Tan δ peak width, and Tg data are presented in Table 4.
As seen in Table 4, the addition of both nanosheets (LDH@GO and LDHGO@Si) has increased the Tg, crosslinking density, and also the inhomogeneity in the coating chemical network. In such enhancements, the silane-modified nano platelet has caused greater impacts on the structural properties. For example, it is observed that Tg has raised from 89.4 °C for the unfilled PU to 100.7 °C and 103.3 °C for PU-LDH@GO-2 and PU-LDHGO@Si-2, respectively. In the case of crosslinking density, an increase of as much as 7% (from 7.84 to 8.45 mmol/cm3) And 45% (from 7.84 to 11.4 mmol/cm3) has occurred with the incorporation of unmodified and aminosilane-modified nanosheets, respectively. The homogeneity of the three-dimensional crosslinked network has considerably decreased which seems logical. Incorporating un- and aminosilane-modified nanosheets can affect the extent and the type of crosslinking reactions that may form during network curing, leading to establishing a more diverse and less homogenous network compared to the unfilled PU network.
The increase in crosslinking density in the presence of LDH@GO and LDHGO@Si is assigned to the capability of such functional particles to participate in chemical reactions occurring during curing. The former via its hydroxyl groups and the latter via its hydroxyl and amine functional groups can react with the hardener isocyanate groups. Besides functional hydroxyl groups of resin, such nano platelets provide numerous (due to their nano-scale dimension and high aspect ratio) additional sites for the curing reaction with the hardener, forming a more reinforced and cross-linked network for the nanocomposite coatings. The presence of nanosheets within the PU matrix can physically and chemically tighten the network, resulting in a decrease in the network free volume, and consequently, a Tg increase. As previously stated, the aminosilane-modified nanofiller (LDHGO@Si) has a more profound influence on the structural properties and also mechanical performance compared to the untreated LDH@GO. This can be attributed to the dispersion quality and also the additional amine functional groups available on the surface of the modified nanosheet. Aminosilane modification provides a hybrid organic/inorganic interface for the nanosheets to increase their interaction (physically and chemically) with the PU backbone, leading to better dispersion quality in comparison with the unmodified particles. Additionally, the LDHGO@Si has additional functional groups (amine) that abundantly are accessible to interact (physically and chemically) with the surrounding matrix. Such surplus capability has resulted in a much more reinforcing and interactive role for the modified nanofiller that has led to much superior performance in comparison with the LDH@GO- and un-filled PU coating.
The increase in crosslinking density, as approved by DMTA results, can also be supported by the changes in coating surface attributes. Water contact angle (WCA), surface hardness, and surface topology were studied to investigate these changes after incorporating various particles. Table 5 presents the WCA and hardness values for all coating samples.
The WCA values reveal that embedding the particles has increased WCA. The influence of such addition is deeper for the surface-treated particles. Increased WCA implies the reduced surface polarity of the coating. The hardness also represents a general ascending trend with respect to the PU-Blank, indicating the higher hardness for the filled samples, especially those loaded with the higher particle content and those filled with surface-treated LDHGO@Si nanosheets. Both WCA and hardness results are aligned with the DMTA findings. Increased crosslinking density of the filled samples, as approved with DMTA, was the main reason for the increased hardness and WCA (due to the more efficient curing process in the presence of nanosheets, leading to the reduced unreacted functional group and consequently reduced polarity).
Additionally, surface topology of various coatings was observed to study the influence of particle inclusion on the surface morphology. Figure 10 depicts AFM micrographs of various coatings.

AFM topology micrographs of unfilled and filld coatings.
It is observed that while PU-Blank presents an almost uniformly smooth surface without any small or coarse asperities (there is only a texture probably attributed to the trace of solvent evaporation), PU-LDHGO-2 and PU-LDHGO@Si samples exhibit a rougher surface with some small and large asperities scattered on the surface which are attributed to the particle inclusion. Comparing these two AFM micrographs demonstrates that the coating loaded with untreated particles (PU-LDHGO-2) exhibits several big asperities with no or few small asperities. On the other hand, for the coating filled with silane-treated particles (PU-LDHGO@Si-2), the number of small asperities has increased and the number of larger asperities (that are assigned to the particle aggregation) has diminished compared to PU-LDHGO-2. These changes in topology can directly indicate a better dispersion and finer particle distribution of treated nanosheets within the PU matrix in comparison with the untreated LDHGO nanosheets.
Discussing the precise impact of filler orientation on mechanical properties requires explicit evidence obtained from high-resolution electron microscopic techniques, which have not been targeted in this study. However, there is implicit evidence induced from AFM topological studies and improved mechanical properties in the presence of nanosheets, specifically the treated ones, that the GOLDH@Si nanosheets seem to have much more exfoliated and parallel-oriented arrangement in comparison with the untreated GO@LDH nanosheets. The particle exfoliation and parallel orientation provide not only a much more extensive and powerful interface with the surrounding matrix, but also obtain a much larger and more efficient stress transfer from the low-modulus PU matrix toward high-modulus nanofillers, resulting in considerable enhancement in toughness of nanocomposites.
It was attempted to investigate the possibility of any correlation between our data. To this end, the correlation (direct relationship) between structural (DMTA) and mechanical (Tensile) properties was examined. Interesting relationships were found with a high correlation factor (Fig. 11). The best results were found to be between crosslinking density (in mmol/cm^3), peak width (or inhomogeneity in °C), and Tg ( in °C) .

The correlation of crosslinking density, peak width, and Tg.
These equations confirm that as the crosslinking density increases with the addition of LDH@GO and LDHGO@Si, the peak width (inhomogeneity) and also the toughness of the coatings increase. It is an interesting finding and can be logically explained here. PU-Blank has a 3-D thermosetting network composed of crosslinks which are formed as a consequence of reactions between functional hydroxyl groups of polyol with NCO functional groups of hardener. The addition of LDH@GO nanoparticles into such PU chemistry adds new functional groups (such as inorganic hydroxyl from the LDH and the oxygenated functional groups available on the GO surface) that will participate in the curing reactions, forming new crosslinks with a wide range of molecular length and mass, increasing the network inhomogenity that would result in more dense and divergent network formation, needing higher energy to reach a certain chain mobility (Higher Tg) and greater energy absorption and dissipation upon tension (higher toughness). In a similar manner, for the PU samples filled with LDHGO@Si nanosheets, the higher crosslinking, and consequently, the greater toughness and inhomogeneity can be accounted for by the extra functional groups (silanol and amine groups) available on the treated filler surface, besides all functional groups mentioned for LDH@GO. These additional groups can form an even more extensive and more varied network, leading to higher crosslinking density, inhomogeneity, and toughness compared to the PU-LDH@GO.
Conclusion
This paper reported the fabrication of a sandwich-like hybrid nanofiller via growing layer double hydroxide on GO nanoplatelets. The hybrid was then functionalized with an APTES silane coupling agent. The results obtained from different characterization techniques (Raman spectroscopy, XRD, TGA, and FESEM) approved the success of nanofiller fabrication and silane functionalization. Both pristine (LDH@GO) and functionalized (LDHGO@Si) fillers were embedded into a typical PU coating at different dosages (1, 2, and 3 wt%) to explore the impact of unmodified and modified filler content on the mechanical properties of resultant PU nanocomposites. The tensile test results proved that the hybrid nanofiller (in both untreated and APTES-treated forms) has positively affected the mechanical properties especially the toughness of PU nanocomposite. It was concluded that the best mechanical response occurred at 2 wt% of the nanofiller content. Such impact was much more profound for the functionalized LDHGO@Si compared to the untreated LDH@GO. Such toughness was the consequence of favorable enhancement in Young modulud, Strain at break and maximum stress (strength). Structural parameters derived from DMTA demonstrated that the particles have reinforced the PU nanocomposites via physical and chemical interactions between the nanofiller surface with the PU backbone, leading to the increased Tg and crosslinking density. Additionally, toughening mechanism studies by FESEM revealed that such particle-reinforced nanocomposite triggers extensive stress delocalization, leading to energy dissipation and toughness enhancement.
Data availability
All data generated or analysed during this study are included in this published article.
References
Rafiee, M. A. et al. Enhanced mechanical properties of nanocomposites at low graphene content. ACS Nano. 3 https://doi.org/10.1021/nn9010472 (2009).
Ghosh, B. & Urban, M. W. Self-repairing oxetane-substituted Chitosan polyurethane networks. Science 323, 1458–1460. https://doi.org/10.1126/science.1167391 (2009).
Mirchandani, G., Waghoo, G., Parmar, R., Haseebuddin, S. & Ghosh, S. K. Oligomeric Silsesquioxane reinforced polyurethane with enhanced coating performance. Prog. Org. Coat. 65, 444–449. https://doi.org/10.1016/j.porgcoat.2009.03.009 (2009).
Kim, H., Miura, Y. & Macosko, C. W. Graphene/polyurethane nanocomposites for improved gas barrier and electrical conductivity. Chem. Mater. 22, 3441–3450. https://doi.org/10.1021/cm100477v (2010).
Yari, H., Mohseni, M. & Ramezanzadeh, B. A mechanistic study of degradation of a typical automotive clearcoat caused by bird droppings. J. Coat. Technol. Res. https://doi.org/10.1007/s11998-010-9273-2 (2011).
Ramezanzadeh, B., Mohseni, M., Mohammad Rabea, A. & Yari, H. Attributing the resistance against simulated tree gum of an acrylic/melamine film loaded with an active silicone additive to its surface free energy. Int. J. Adhes. Adhes. 31, 775–783 (2011).
Razin, A. A., Yari, H. & Ramezanzadeh, B. Stone-chipping and adhesion deterioration of automotive coating systems caused by outdoor weathering of underneath layers. J. Ind. Eng. Chem. 31, 291–300. https://doi.org/10.1016/j.jiec.2015.07.001 (2015).
Alizadeh Razin, A., Ramezanzadeh, B. & Yari, H. Detecting and estimating the extent of automotive coating delamination and damage indexes after stone chipping using electrochemical impedance spectroscopy. Prog. Org. Coat. https://doi.org/10.1016/j.porgcoat.2015.11.023 (2015).
Pakravan, H. R. & Yari, H. The influence of nanostructured UV-blockers on mechanical properties of carbon fiber epoxy composites during accelerated weathering condition. Polym. Adv. Technol. 29, 970–981. https://doi.org/10.1002/pat.4208 (2018).
Wetzel, B., Haupert, F. & Qiu Zhang, M. Epoxy nanocomposites with high mechanical and tribological performance. Compos. Sci. Technol. 63, 2055–2067. https://doi.org/10.1016/S0266-3538(03)00115-5 (2003).
Lotfi, M., Azizi, A., Yari, H. & Ganjaee Sari, M. Aminosilane-co-graphene oxide/epoxy nanocomposite coating: an approach towards toughness and viscoelastic properties enhancement. Prog. Org. Coat. 151, 106050. https://doi.org/10.1016/j.porgcoat.2020.106050 (2021).
Lotfi, M., Yari, H., Sari, M. G. & Azizi, A. Fabrication of a highly hard yet tough epoxy nanocomposite coating by incorporating graphene oxide nanosheets dually modified with amino silane coupling agent and hyperbranched polyester-amide. Prog. Org. Coat. 162, 106570. https://doi.org/10.1016/j.porgcoat.2021.106570 (2022).
Song, H-J., Zhang, Z-Z. & Men, X-H. The tribological behaviors of the polyurethane coating filled with nano-SiO2 under different lubrication conditions. Compos. Part. Appl. Sci. Manuf. 39, 188–194. https://doi.org/10.1016/j.compositesa.2007.11.003 (2008).
Yari, H., Mohseni, M. & Messori, M. Toughened acrylic/melamine thermosetting clear coats using POSS molecules: mechanical and morphological studies. Polym. (United Kingdom). https://doi.org/10.1016/j.polymer.2015.02.040 (2015).
Yari, H., Mohseni, M., Messori, M. & Ranjbar, Z. Tribological properties and scratch healing of a typical automotive nano clearcoat modified by a polyhedral oligomeric Silsesquioxane compound. Eur. Polym. J. 60, 79–91. https://doi.org/10.1016/j.eurpolymj.2014.08.023 (2014).
Rostami, M., Ranjbar, Z. & Mohseni, M. Investigating the interfacial interaction of different aminosilane treated nano silicas with a polyurethane coating. Appl. Surf. Sci. 257, 899–904. https://doi.org/10.1016/j.apsusc.2010.07.087 (2010).
Chen, J., Gao, X. & Song, W. Effect of various carbon nanofillers and different filler aspect ratios on the thermal conductivity of epoxy matrix nanocomposites. Results Phys. 15, 102771. https://doi.org/10.1016/j.rinp.2019.102771 (2019).
Pandele, A. M. et al. Non-covalent functionalization of GO for improved mechanical performances of pectin composite films. Compos. Part. Appl. Sci. Manuf. 103, 188–195. https://doi.org/10.1016/j.compositesa.2017.10.005 (2017).
Idumah, C. I. & Obele, C. M. Understanding interfacial influence on properties of polymer nanocomposites. Surf. Interfaces. 22, 100879. https://doi.org/10.1016/j.surfin.2020.100879 (2021).
Kuan, H. T. N., Tan, M. Y., Shen, Y. & Yahya, M. Y. Mechanical properties of particulate organic natural filler-reinforced polymer composite: A review. Compos. Adv. Mater. 30, 263498332110075. https://doi.org/10.1177/26349833211007502 (2021).
Asmatulu, R., Nguyen, P. & Asmatulu, E. Nanotechnology safety in the automotive industry. vol. #volume#. 1st ed. © 2013 Elsevier B.V. All rights reserved. https://doi.org/10.1016/B978-0-444-59438-9.00005-9 (2013).
Hu, Y. et al. In-situ hybridization of an epoxy resin using polyurethane and MXene nanoplatelets for thermally stable nanocomposites with improved strength and toughness. Polym. (Guildf). 302, 127065. https://doi.org/10.1016/J.POLYMER.2024.127065 (2024).
Alibakhshi, E., Ghasemi, E., Mahdavian, M., Ramezanzadeh, B. & Farashi, S. Evaluation of the active corrosion protection and self-healing properties of a silane sol-gel coating doped with Zn-Al-PO43- layered double hydroxide nanoparticles. Eur. Corros. Congr EUROCORR 2016. 4, 2870–2877 (2016).
Wang, Q. & Ohare, D. Recent advances in the synthesis and application of layered double hydroxide (LDH) nanosheets. Chem. Rev. https://doi.org/10.1021/cr200434v (2012).
Kardar, P. & Amini, R. Studying the active corrosion inhibition effect of the Ce3+/2-Mercaptobenzothiazole loaded NaY Zeolite/Zn-Al LDH based containers in a silane coating. Prog. Color. Color. Coat. 15, 1–9. https://doi.org/10.30509/PCCC.2022.81675 (2022).
Wu, L. et al. Corrosion resistance of the GO/ZIF-8 hybrid loading benzotriazole as a multifunctional composite Filler-Modified MgAlY layered double hydroxide coating. Langmuir 38, 10338–10350. https://doi.org/10.1021/ACS.LANGMUIR.2C01915/ (2022).
Wang, Y. et al. Superhydrophobic and self-healing Mg-Al layered double hydroxide/silane composite coatings on the Mg alloy surface with a long-term anti-corrosion lifetime. Langmuir 37, 8129–8138. https://doi.org/10.1021/ACS.LANGMUIR.1C00678/SUPPL_FILE/LA1C00678_SI_002.MP4 (2021).
Jafari, M., Ganjali, F., Eivazzadeh-Keihan, R., Maleki, A. & Geranmayeh, S. Recent advances in applications of graphene-layered double hydroxide nanocomposites in supercapacitors and batteries. FlatChem 45, 100658. https://doi.org/10.1016/J.FLATC.2024.100658 (2024).
Morlat, S., Mailhot, B., Gonzalez, D. & Gardette, J. L. Photo-oxidation of polypropylene/montmorillonite nanocomposites. 1. Influence of nanoclay and compatibilizing agent. Chem. Mater. 16, 377–383. https://doi.org/10.1021/cm031079k (2004).
Morlat-Therias, S., Mailhot, B., Gonzalez, D. & Gardette, J. L. Photooxidation of polypropylene/montmorillonite nanocomposites. 2. Interactions with antioxidants. Chem. Mater. 17, 1072–1078. https://doi.org/10.1021/cm040172l (2005).
Huang, X. et al. Graphene-based materials: synthesis, characterization, properties, and applications. Small. 7. https://doi.org/10.1002/smll.201002009 (2011).
Sadegh, N., Dehcheshmeh, I. M. & Sadegh, F. Review of zeolitic imidazolate framework/graphene oxide: A synergy of synthesis, properties and function for multifaceted applications in nanotechnology. FlatChem. 44, 100618. https://doi.org/10.1016/J.FLATC.2024.100618 (2024).
Amrollahi, S., Ramezanzadeh, B., Yari, H., Ramezanzadeh, M. & Mahdavian, M. In-situ growth of ceria nanoparticles on graphene oxide nanoplatelets to be used as a multifunctional (UV shield/radical scavenger/anticorrosive) hybrid compound for exterior coatings. Prog. Org. Coat. https://doi.org/10.1016/j.porgcoat.2019.105241 (2019).
Amrollahi, S., Ramezanzadeh, B., Yari, H., Ramezanzadeh, M. & Mahdavian, M. Synthesis of polyaniline-modified graphene oxide for obtaining a high performance epoxy nanocomposite film with excellent UV blocking/anti-oxidant/ anti-corrosion capabilities. Compos. Part. B Eng. https://doi.org/10.1016/j.compositesb.2019.05.015 (2019).
Mahmudzadeh, M., Yari, H., Ramezanzadeh, B. & Mahdavian, M. Urtica dioica extract as a facile green reductant of graphene oxide for UV resistant and corrosion protective polyurethane coating fabrication. J. Ind. Eng. Chem. 78, 125–136. https://doi.org/10.1016/j.jiec.2019.06.026 (2019).
Dagdag, O. & Kim, H. Recent advances for poly(cyclotriphosphazene) functionalized graphene oxide composites: synthesis, properties and applications. J. Ind. Eng. Chem. 136, 89–122. https://doi.org/10.1016/J.JIEC.2024.02.052 (2024).
Saadatmandi, S., Asghari, M. & Ramezanzadeh, B. Effective epoxy composite coating mechanical/fracture toughness properties improvement by incorporation of graphene oxide nano-platforms reduced by a green/biocompataible reductant. J. Ind. Eng. Chem. 75, 271–284. https://doi.org/10.1016/J.JIEC.2019.03.038 (2019).
Bandeira de Souza, Z. S. et al. Influence of graphene functionalization on the curing kinetics, dynamical mechanical properties and morphology of epoxy nanocomposites. Polym. (Guildf). 320, 128067. https://doi.org/10.1016/J.POLYMER.2025.128067 (2025).
Adak, B., Joshi, M. & Butola, B. S. Polyurethane/functionalized-graphene nanocomposite films with enhanced weather resistance and gas barrier properties. Compos. Part. B Eng. https://doi.org/10.1016/j.compositesb.2019.107303 (2019).
Zhang, M. et al. In-situ defect repairing in hydroxyapatite/phytic acid hybrid coatings on AZ31 magnesium alloy by hydrothermal treatment. J. Alloys Compd. 658, 649–656. https://doi.org/10.1016/j.jallcom.2015.10.282 (2016).
Guo, L. et al. Layered double hydroxide coatings on magnesium alloys: A review. J. Mater. Sci. Technol. 34, 1455–1466. https://doi.org/10.1016/j.jmst.2018.03.003 (2018).
Kamiyama, N. et al. Effect of treatment time in the Mg(OH)2/Mg–Al LDH composite film formed on Mg alloy AZ31 by steam coating on the corrosion resistance. Surf. Coat. Technol. 286, 172–177. https://doi.org/10.1016/j.surfcoat.2015.11.051 (2016).
Ma, P. C., Kim, J. K. & Tang, B. Z. Functionalization of carbon nanotubes using a silane coupling agent. Carbon N. Y. 44, 3232–3238. https://doi.org/10.1016/j.carbon.2006.06.032 (2006).
Cong, C. et al. Effect of graphene oxide-modified CaAl-layered double hydroxides on the carbon dioxide permeation properties of fluoroelastomers. Polym. (Basel). 15, 4151. https://doi.org/10.3390/POLYM15204151/S1 (2023).
Long, X. et al. A strongly coupled graphene and FeNi double hydroxide hybrid as an excellent electrocatalyst for the oxygen evolution reaction. Angew Chemie. 126, 7714–7718. https://doi.org/10.1002/ANGE.201402822 (2014).
Zhu, X., Zhang, W., Lu, G., Zhao, H. & Wang, L. Ultrahigh mechanical strength and robust Room-Temperature Self-Healing properties of a Polyurethane-Graphene oxide network resulting from multiple dynamic bonds. ACS Nano. 16, 16724–16735. https://doi.org/10.1021/ACSNANO.2C06264/SUPPL_FILE (2022).
Lan, M., Fan, G., Yang, L. & Li, F. Significantly enhanced visible-light-induced photocatalytic performance of hybrid zn-cr layered double hydroxide/graphene nanocomposite and the mechanism study. Ind. Eng. Chem. Res. 53, 12943–12952. https://doi.org/10.1021/IE501650G/SUPPL_FILE/IE501650G_SI_001.PDF (2014).
Zhang, X., Xu, Z., Sun, C., Zheng, L. & Wen, S. Enhanced gas barrier and mechanical properties of Styrene-Butadiene rubber composites by incorporating electrostatic Self-Assembled graphene oxide @ layered double hydroxide hybrids. ACS Omega. 9, 39846–39855. https://doi.org/10.1021/acsomega.4c05304 (2024).
Alibakhshi, E., Ghasemi, E., Mahdavian, M. & Ramezanzadeh, B. Corrosion inhibitor release from Zn-Al-[PO 4 3-]-[CO 3 2-] layered double hydroxide nanoparticles. Prog. Color. Color. Coat. 9, 233–248 (2016).
Goswami, R. N., Mourya, P., Saini, R., Khatri, O. P. & Ray, A. Polyaniline-wrapped nitrogen-doped graphene nanocomposites as protective functional fillers in epoxy coatings for remarkable enhancement of corrosion Inhibition performance. Prog. Org. Coat. 189, 108335. https://doi.org/10.1016/j.porgcoat.2024.108335 (2024).
Wang, Z. et al. Insights into the hydrophobic coating with integrated high-efficiency anti-corrosion, anti-biofouling and self-healing properties based on anti-bacterial nano LDH materials. Corros. Sci. 231, 111995. https://doi.org/10.1016/j.corsci.2024.111995 (2024).
Asif, M. et al. Development of Co-Al LDH/GO composite photocatalyst for enhanced degradation of textile pollutant under visible light irradiation. Results Phys. 42, 105997. https://doi.org/10.1016/J.RINP.2022.105997 (2022).
Javidparvar, A. A., Naderi, R. & Ramezanzadeh, B. Epoxy-polyamide nanocomposite coating with graphene oxide as cerium nanocontainer generating effective dual active/barrier corrosion protection. Compos. Part. B Eng. 172, 363–375. https://doi.org/10.1016/J.COMPOSITESB.2019.05.055 (2019).
Ramezanzadeh, M., Ramezanzadeh, B. & Mahdavian, M. Graphene skeletal nanotemplate coordinated with pH-Responsive porous Double-Ligand Metal-Organic frameworks (DL-MOFs) through ligand exchange theory for High-Performance smart coatings. Chem. Eng. J. 461, 141869. https://doi.org/10.1016/J.CEJ.2023.141869 (2023).
Haddadi, S. A., Ramezanzadeh, M., Haji Naghi Tehrani, M. E. & Ramezanzadeh, B. Sodium lignosulfonate-loaded ZnAl-layered double hydroxide decorated graphene oxide nanolayers; toward fabrication of sustainable nanocomposite for smart corrosion prevention. J. Clean. Prod. 374, 133980. https://doi.org/10.1016/J.JCLEPRO.2022.133980 (2022).
Rashed, S. H. et al. Preparation and characterization of layered-double hydroxides decorated on graphene oxide for dye removal from aqueous solution. J. Mater. Res. Technol. 17, 2782–2795. https://doi.org/10.1016/j.jmrt.2022.02.040 (2022).
Su, Y. et al. Sulfonated polyaniline assisted hierarchical assembly of graphene-LDH nanohybrid for enhanced anticorrosion performance of waterborne epoxy coatings. Chem. Eng. J. 426, 131269. https://doi.org/10.1016/J.CEJ.2021.131269 (2021).
Ramezanzadeh, M., Ramezanzadeh, B., Mahdavian, M. & Bahlakeh, G. Development of metal-organic framework (MOF) decorated graphene oxide nanoplatforms for anti-corrosion epoxy coatings. Carbon N. Y. 161, 231–251. https://doi.org/10.1016/j.carbon.2020.01.082 (2020).
Ahmadi, Y., Yadav, M. & Ahmad, S. Oleo-polyurethane-carbon nanocomposites: effects of in-situ polymerization and sustainable precursor on structure, mechanical, thermal, and antimicrobial surface-activity. Compos. Part. B Eng. 164, 683–692. https://doi.org/10.1016/j.compositesb.2019.01.078 (2019).
Theiler, G., Wachtendorf, V., Elert, A. & Weidner, S. Effects of UV radiation on the friction behavior of thermoplastic polyurethanes. Polym. Test. 70, 467–473. https://doi.org/10.1016/j.polymertesting.2018.08.006 (2018).
Meijer, H. E. H. & Govaert, L. E. Mechanical performance of polymer systems: the relation between structure and properties. Prog. Polym. Sci. 30, 915–938. https://doi.org/10.1016/J.PROGPOLYMSCI.2005.06.009 (2005).
Wu, H. & Fan, G. An overview of tailoring strain delocalization for strength-ductility synergy. Prog. Mater. Sci. 113, 100675. https://doi.org/10.1016/J.PMATSCI.2020.100675 (2020).
Palimi, M. J., Rostami, M., Mahdavian, M. & Ramezanzadeh, B. Surface modification of Fe 2 O 3 nanoparticles with 3-aminopropyltrimethoxysilane (APTMS): an attempt to investigate surface treatment on surface chemistry and mechanical properties of polyurethane/fe 2 O 3 nanocomposites. Appl. Surf. Sci. 320, 60–72. https://doi.org/10.1016/j.apsusc.2014.09.026 (2014).
Zheng, J. et al. Vitrimers: current research trends and their emerging applications. Mater. Today. 51, 586–625. https://doi.org/10.1016/J.MATTOD.2021.07.003 (2021).
Yari, H., Mohseni, M., Messori, M. & Ranjbar, Z. Tribological properties and scratch healing of a typical automotive nano clearcoat modified by a polyhedral oligomeric Silsesquioxane compound. Eur. Polym. J. 60 https://doi.org/10.1016/j.eurpolymj.2014.08.023 (2014).
Fantoni, A., Koch, T., Baudis, S. & Liska, R. Synthesis and characterization of homogeneous epoxy networks: development of a sustainable material platform using epoxy-Alcohol polyaddition. ACS Appl. Polym. Mater. 5, 731–742. https://doi.org/10.1021/ACSAPM.2C01728/ASSET (2023).
Bandzierz, K. et al. Influence of network structure on glass transition temperature of elastomers. Mater. (Basel Switzerland). https://doi.org/10.3390/MA9070607 (2016).
Author information
Authors and Affiliations
Contributions
A.A.: Formal analysis, methodology, data curation, writing- original draft preparation. H.Y.: Conceptualization, methodology, data curation, writing-reviewing and editing. M.R. Formal analysis, methodology, data curation, writing-original draft preparation. M.R.: Conceptualization, methodology, data curation, writing- reviewing and editing. M.J.: Conceptualization, methodology, data curation, writing- original draft preparation. B.R.: Conceptualization, writing- reviewing and editing.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Aslani, A., Yari, H., Rezaei, M. et al. Graphene oxide decoration with ZnAl LDH and further functionalization with APTES for enhancing the toughness of polyurethane coatings. Sci Rep 15, 30876 (2025). https://doi.org/10.1038/s41598-025-16495-3
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-16495-3
