
Abstract
Species boundaries in scleractinian corals remain highly elusive due to conflicting patterns between morphological and molecular phylogenies, often caused by morphological plasticity, occurrence of cryptic species, incomplete lineage sorting or introgressive hybridization. Here, we use an integrated systematics approach, which combines reduced representation genome sequencing (nextRAD), micro-morphometric characterization, SEM analyses and compilation of life history traits, to infer phylogenetic relationships among closely related species in the Caribbean coral genus Madracis. In total, we analyzed 235 Madracis specimens from Curaçao and Bermuda collected from 10–90 m depth. Sequence- and SNP-based analyses for 115 samples generated unprecedented species resolution in Madracis, greatly supporting the morphology-based taxonomy of the current, accepted Caribbean species M. senaria, M. decactis, M. formosa, M. carmabi and M. mirabilis (M. auretenra). The exception was M. pharensis, in which we found evidence for three separate lineages, and for which we found signatures of admixture and introgression. These three M. pharensis lineages showed distinct depth distributions (thus classified as shallow, deep and very deep) and were partially distinguishable on the basis of fine microstructural elements of the collumella, septa and coenosteum. Further taxonomic comparisons are needed to formalize these putative cryptic species. Overall, our integrated systematics approach further resolves species relationships in the Caribbean genus Madracis, supports the morphological descriptions for most of the recognized species, but also reveals the existence of cryptic diversity in groups marked by high admixture, thus suggesting hybridization as a driving force in coral species diversity.
Similar content being viewed by others

Introduction
Reef-building corals face unprecedented local and global stressors, yet their conservation is of utmost importance to keep coral reefs functional throughout the Anthropocene1,2. Underpinning any present and future coral conservation efforts is a solid understanding of species relationships, as well as establishing species classifications that represent real ecological and evolutionary entities3,4. However, phylogenetic studies in corals have traditionally been hindered by few morphological characters available for species delimitation5,6,7,8 as well as by widespread conflicting patterns between molecular and morphological data7,8,9. These inconsistencies arise from a variety of factors, such as phenotypic plasticity10,11, genetic mixing via introgressive hybridization12,13,14, morphological stasis or homoplasy resulting in cryptic species15,16,17, or incomplete lineage sorting and sharing of polymorphisms in recently diverged taxa18. Moreover, phylogenetic studies have been hindered by the slow evolution of the mitochondrial genome in anthozoans that makes the application of classical barcoding gene regions (e.g., cox1) of limited use in scleractinian corals19,20,21,22.
Next generation sequencing (NGS) data have been used to solve phylogenetic questions at unprecedented resolution using reduced-representation genome sequencing approaches23. In particular, restriction-site associated DNA sequencing (RADseq24,25), a technique allowing the regions adjacent to restriction endonuclease sites to be surveyed with deep coverage by high-throughput sequencing, produces a panel of markers to describe patterns of population structure, delimit species boundaries and study phylogenetic relationships24,26,27. Such developments were useful for solving questions at different evolutionary scales, from very divergent taxa such as in beetles skinks28, to recently diverged taxa such as in cichlids29. In soft corals, RADseq has been used, for example, to generate fine-tuned evolutionary interpretations in two diverse groups of deep-sea octocorals (genera Chrysogorgia and Paramuricea)30,31, to infer the presence of cryptic species within the Paragorgia and Heliopora genera32,33, or to disentangle the confounding effects of recent speciation and hybrid speciation in the genus Sinularia34. Likewise, RADseq techniques have recently clarified evolutionary relationships in scleractinian corals recognized for traditional phylogenetic difficulties35. For instance, in the genus Pocillopora, RADseq was used to show that introgression may play a role in generating polyphyletic morphospecies13. A larger follow-up study including seven Pocillopora species found reciprocal monophyly among all species (i.e., no evidence for introgressive hybridization), except for the most derived sister species in the group, P. damicornis and P. acuta36. Rosser et al.37 compared genome-wide to previously available single (nuclear and mitochondrial) gene phylogenies to dispute introgressive hybridization as cause for molecular incongruences in the Acropora genus38. Dimond et al.39 used RADseq to find a strong genetic basis for branching Porites morphospecies in Belize. Forsman et al.14 used a combination of de novo assembly and reference mapping approaches to compare RADseq loci of Porites lobata and P. compressa in Hawai’i to find that these two species with contrasting morphologies appear to experience ongoing hybridization. The ubiquitous Indo-Pacific Pachyseris speciosa has recently been shown by a combination of nextRAD sequencing and an annotated reference genome to represent a cryptic species complex with substantial ecological and physiological differentiation despite ancient morphological stasis40. Coral phylogenomics is therefore contributing greatly to capturing the hidden diversity and delimiting coral species, critical steps not only warranted for efficient conservation efforts, but also to answer important open questions concerning the evolutionary ecology of corals and how ecological drivers contribute to shape species interactions and boundaries.
The pocilloporid genus Madracis Milne Edwards and Haime, 1849 is archetypal of many of the difficulties associated with coral systematics. In the Caribbean, the genus Madracis includes six shallow water hermatypic species, distinguished based on their macro-morphology41,42,43, and whose biology and ecology have been fairly well studied44,45,46,47,48,49,50,51,52,53,54,55,56,57. Madracis taxonomic assignment to species level, however, remains considerably problematic because the topologies provided by genetic markers often do not yield monophyletic lineages44,58. From the six recognized morphospecies41,42,43, only one, the submassive M. senaria Wells, 1973b, seems to emerge as a “true” species based on ribosomal and nuclear genetic data44,58. The other species are characterized by extensive sharing of genetic variation, suggesting semi-permeable species boundaries and that introgressive hybridization has played an important role in the evolution of the genus58. Likewise, there is ongoing taxonomic debate, with controversy fueled by high levels of inter- and intraspecific colony variation. For instance, Fenner59 proposed that the encrusting/submassive depth-generalist M. pharensis (Heller, 1868) is actually an ecomorph of the morphologically plastic but typically knobby M. decactis (Lyman, 1859), a conclusion challenged by the overlapping light use strategies of the two morphospecies45. In several molecular phylogenetic studies, these species appear to form an intricate species complex with the deep-water branching M. formosa Wells, 1973, and the deep-water branching M. carmabi Vermeij, Diekmann and Bak 2003, the latter of which has been hypothesized to be a hybrid species43,44. Finally, M. mirabilis (Duchassaing & Michelotti 1860) (i.e., M. auretenra sensu Locke, Weil and Coates, 2007)60, a branching shallow-water specialist typically forming extensive beds in the shallows, is in some studies reportedly monophyletic44. Although the different Madracis species seem to occupy distinct depth niches across the steep reef slopes, their distributions are to a certain extent overlapping56. Such recurrent conflicts between morphology-based taxonomy and molecular phylogenetics as seen in Madracis, are common in other pocilloporiid genera such as Stylophora61,62, Seriatopora63 and Pocillopora64,65.
In addition, significant genetic divergence was found (using the mitochondrial nad5 intron) between shallow and deep populations within M. pharensis, with threshold at ca. 25 m depth58. These results have been confirmed by the mitochondrial ATP8 region, showing further evidence for a divergence in the M. pharensis populations even deeper into the lower mesophotic reef66. The consequence of these divergences for the taxonomic status of M. pharensis is still to be evaluated. More recently, the existence of significant genetic and morphological differences between the (obligate) zooxanthellate M. pharensis in the Caribbean and the (facultative zooxanthellate) M. pharensis in the Mediterranean was reported67, with the latter region being the type locality of the species68. This work suggested that the zooxanthellate Caribbean form, also named M. pharensis f. luciphila Wells, 1973 by some41,69, represents a different species (or group of species) whose identity and relationships with remaining Caribbean Madracis are still to be ascertained. While prior Madracis studies using a handful of molecular markers have generated useful information, many basic ecological and evolutionary questions in Madracis remain unresolved. Here, we apply an integrated systematics approach by combining the power of reduced representation genomic libraries (which we compare to a commonly used mitochondrial marker, ATP8) with corallite-level morphometrics and several ecological traits to determine species relationships in the Caribbean genus Madracis.
Materials and methods
(a) Sample collection
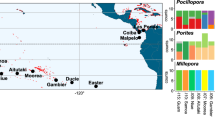
Madracis coral specimens were collected from three locations in Curaçao (in 2013, 2014 and 2017) and from seven locations in Bermuda (in 2013 and 2017) (see Fig. 1). Small coral fragments were collected using SCUBA diving (5-70 m depth) or manned submersible (>70 m depth) in Curaçao while using only SCUBA diving (5-60 m depth) in Bermuda. Subsamples (< 4 cm2) from each specimen were stored in salt-saturated buffer solution containing 20 % DMSO and 0.5 M EDTA. The remaining fragments were bleached for skeletal measurements58. Taxonomic assertion used morphological descriptions from Wells41,42 for M. mirabilis (i.e., M. auretenra Locke, Weil and Coates, 2007), M. senaria and M. formosa, and from Vermeij et al.43 for M. carmabi. Colony macromorphology following Fenner59 was used to characterize M. pharensis (i.e., M. pharensis f. luciphila) and M. decactis (i.e., encrusting and nodular colonies were classified as M. pharensis and M. decactis, respectively). Colonies were chosen as best representatives of the six recognized morphospecies and by attempting to cover their depth distributions. Intermediate phenotypes (between M. pharensis and M. decactis) were purposely avoided and will be targeted in future research. Throughout the manuscript we refer to M. auretenra as M. mirabilis and to M. pharensis f. luciphila as M. pharensis, as these are the commonly used names by most research groups.

Sampling sites (A) with numbers of specimens collected from each of six recognized Madracis morphospecies (B) for each location (Curaçao in the South Caribbean, and Bermuda in the West Atlantic), across a large depth range (0-90 m depth). Sampling depth is shown as five discrete depth categories with bubble area representative of sample size. For sample details and the application of the different methods across samples please see Table S1. Underwater photos by Pedro R. Frade.
DNA extraction, library preparation and sequencing
Genomic DNA (gDNA) was extracted using a modified salt-extraction method3, where Symbiodiniaceae contamination was reduced through additional centrifugation steps unless this resulted in too low gDNA yield. Quality and yield were assessed using a Qubit fluorometer and gel electrophoresis to select a subset of samples viable for downstream sequencing given overall yield and quality issues. Genomic DNA of the samples was used to prepare nextRAD genotyping-by-sequencing libraries (SNPsaurus, LLC), which uses selective PCR primers to amplify loci consistently between samples. Genomic DNA was fragmented and ligated with adapter sequences using Nextera reagent (Ilumina, Inc). These fragments were then amplified using primers matching the adapter and a 9-basepair selective sequence (“GTGTAGAGG”). This protocol was chosen to produce similar results to that of a SbfI restriction enzyme digest commonly used in regular RAD sequencing70, and which proved to be successful in sea anemones71 and octocorals30. Barcodes differing by at least 2 nt were used to differentiate pooled samples. The libraries were run on a single HiSeq 2500 (Ilumina, Inc) lane using 100 bp single-end chemistry and the manufacturer’s recommended protocol. Nextera adapters (with overlap of at least 5 bp) and low quality ends (PHRED < 20) were trimmed using TrimGalore (https://github.com/FelixKrueger/TrimGalore), before discarding reads that became less than 30 bp. Poorly performing samples (files below 10 MB in size) were removed from subsequent analyses.
Sequence clustering, variant calling, filtering and quality control
Obtained (demultiplexed) sequence data were analyzed using the iPyRAD pipeline v. 0.7.172, using a minimum clustering coverage of 6 at each base position (Mindepth), a clustering threshold of 85% (Wclust), a maximum of 10 sites with a PHRED quality below 33 (NQual) and a minimum number of 20 samples in a final locus (MinCov). One representative sequence was then extracted (using pyrad2fasta.py script) from each of the obtained loci and BLASTN-searched (https://www.ncbi.nlm.nih.gov) against the Symbiodiniaceae B1 (Breviolum minutum) nuclear73 and mitochondrial74 genomes, as well as against nextRAD sequencing data (generated using the same selective primers) of 15 isolated Symbiodiniaceae strains (including types B7, B13 and B15 in the Breviolum genus, commonly associating with Madracis spp.)3 (Bongaerts et al. unpublished). A threshold of 50 bp identity match and a stringent maximum E-value of 10−15 were used to remove potential Symbiodiniaceae loci and produce a conservative set of Madracis coral loci. Other potential contaminants were identified through BLASTN searches against non-redundant NCBI non-Cnidarian databases using a less stringent maximum E-value of 10−5.
The resulting VCF file containing SNP data was filtered using VCFtools v.0.1.1375 to remove singleton and doubleton (single occurrence in homozygote individual) alleles, as well as monomorphic SNPs. After filtering, all 115 individuals were kept, with 315,362 out of an initial 928,829 SNPs remaining. Given a slight overrepresentation of SNPs towards the end of reads, all SNPs located after position 95 bp were eliminated (using vcfreadtrim.py). Number of shared loci between all samples was assessed and individuals with low coverage across sites (i.e., with less than 5% of loci genotyped) were removed, leaving 95 out of 115 individuals in the analysis, and keeping 282,328 out of a possible 287,387 sites. In order to identify potential clones, the genotyping accuracy of technical replicates (duplicates, for 7 samples in total) was calculated (only considering SNPs genotyped for at least 50% of samples), and a 96% similarity threshold was then applied to remove potential clones and replicated samples (retaining the sample with the least missing data in each case). This resulted in 78 of 95 individuals and 268,839 of 282,328 sites after filtering.
SNP-based analyses
Overall genetic structure of the SNP dataset (based on 95 individuals, before filtering out potential clones and technical replicates) was visualized using a Neighbor-Joining (NJ) tree based on genetic distance with the ggtree package76 in R. The presence of major clusters as well as potential signatures of admixture were further explored using the Bayesian clustering STRUCTURE approach (using the multi-threading wrapper structure_mp) for a reduced dataset including only SNPs genotyped for at least 50% of samples (22,550 out of a possible 268,839 sites). Individual runs (up to k=8) were aggregated using CLUMPP77. For each of the 20 CLUMPP-aligned replicates, we computed the maximum assignment value for each cluster to assess the presence of “ghost clusters”. Samples were separated into clusters using a (minimum) mean ancestry assignment cut-off value of 0.8 and samples below this threshold were marked as unassigned. To assess the extent of within-clade divergence, three separate datasets representing three distinct clades/clusters were analysed through STRUCTURE following the same approach described above (up to k=5). Genetic structure in the same reduced dataset was further explored with Discriminant Analysis of Principal Components (DAPC) using the adegenet package in R78. The adegenet::find.clusters function, which uses Bayesian Information Criteria (BIC) to determine the optimal number of clusters, consistently indicated five clusters, though four and six clusters were also explored. Six principal components and four discriminant functions were retained for plotting and the adegenet::compoplot function was used to visualize the assignment probability of individuals to each cluster. Both STRUCTURE and DAPC are based on genetic variation alone and do not consider any previous contextual information on taxonomic or geographic origin.
Coalescent-based SNAPP analyses (v. 1.5.1) were performed in BEAST2 (v. 2.6.3)79 for SNPs genotyped in at least 50% of samples (22,550 sites). After converting VCF file to nexus format using vcf2phylip.py, BEAUTi was used to select models and prepare input data for BEAST (MCMC length of 100,000,000, pre-burnin of 10,000,000, samplefreq = 10,000). Because SNAPP are computationally costly analyses, initially only two samples per morphospecies were included (a total of 12 samples; 3,548 SNPs; including only unlinked biallelic SNPs with 0% missing data as required), and this number was increased to include putatively new Madracis lineages as suggested by the previous STRUCTURE and DAPC analyses (leading to two more runs of 14 and 17 samples, and 2,791 and 125 SNPs, respectively). Because no missing data are allowed in the analyses, the number of SNPs included was relatively low, and decreased as more individuals were included. This was particularly true for the run with 17 samples, which included three samples with high percentages of missing SNPs (only 11-24% genotyped). Default parameters were used for mutation rate, coalescent rate and ancestral population sizes. Log files (every 10,000 runs) were combined using Log Combiner and input into Tracer for assessment after a burnin of 10% (i.e., 10,000,000 out of a chain length of 100,000,000). Maximum clade credibility trees were generated with TreeAnnotator and then Densitree (included in the BEAST2 package), and tree topologies visualized in FigTree.
Sequence-based analyses
To reconstruct phylogenetic relationships, the filtered loci dataset (78 individuals; only samples with >50% loci genotyped) was trimmed to 95 bp, and three files were generated including only loci genotyped for at least 5%, 25% and 50% of the individuals, respectively. Different thresholds for the number of loci were used to accommodate the effect of missing data in the observed genetic patterns. Resulting loci were concatenated into FASTA files to generate maximum likelihood trees using RAxML v. 8.2.XX80 available on the CIPRES portal81, with a GTRGAMMA nucleotide substitution model (and estimated proportion of invariable sites), using RAxML-HPC-BlackBox’s rapid bootstrapping algorithm on XSEDE. Three samples representing putatively new lineages were also included despite a low percentage of genotyped loci (11-24%). To obtain outgroup data for phylogenetic inference, a single reference sequence was used for each RAD locus and mapped against the genome of Acropora digitifera82 using BWA-MEM. Generated loci files were then concatenated and used as an outgroup. ML trees were based on all available loci and ran on RAxML-HPC-BlackBox as described above (68 individuals plus outgroup; only samples with >50% loci genotyped).
Bayesian inference was performed in BEAST (v. 1.10.4) using as initial source the same concatenated FASTA file including all loci and the outgroup, but only one sample per Madracis morphospecies (as data would otherwise not upload to BEAUTi). A chain-length of 100,000,000 (logged every 10,000 iterations) and a 10% burn-in were used for Bayesian inference using GTR, estimated base frequencies and gamma+ invariant sites as site heterogeneity model (plus speciation Yule process and random starting tree). Three log files and trees output by BEAST had high acceptance ratios and were combined using LogCombiner, with the resulting log file passed on to Tracer. Maximum clade credibility tree was built in TreeAnnotator and further visualized using FigTree.
Hybridization tests
To assess gene flow across the different recognized and putatively new Madracis species, the D-statistic (ABBA-BABA)83 was calculated in Dsuite84, based on the VCF file. Given a set of biallelic SNPs across four target taxa, Dsuite tests the probability that the frequencies of ancestral and derived alleles are explained by recent contact between one of the derived lineages and an ancestral lineage, rather than the null hypothesis of no gene flow. D-statistics were calculated for (the whole) and several parts (clusters) of the Madracis tree, based on the topology generated by RAxML approach with the A. digitifera as outgroup (i.e., here defining the ancestral allele). Dsuite outputs results of three complementary analyses: 1) in the “BBAA” output, where each trio of lineages (one ancestral and two derived) is ordered assuming that the correct tree is the one that minimizes any significant ABBA or BABA pattern; 2) in “Dmin” there is no attempt to infer the true tree, but rather the trio is ordered to minimize any difference between frequency of ABBA vs that of BABA; and 3) in the output “tree”, where the test is calculated on top of the provided topology (in Newick format).
Symbiodiniaceae reads
Using the set of loci-representative sequences (before filtering and quality control), loci mapped to the Symbiodiniaceae B1 nuclear genome73 by BWA-MEM85 were extracted to create a database of symbiont loci. These were further filtered to contain only loci genotyped for at least 70% of samples and containing at least 5 SNPs. A set of random loci were analyzed after converting to FASTA format in order to generate Symbiodiniaceae trees with clustalx (https://doi.org/10.1093/bioinformatics/btm404). Two loci that maximized the separation of expected Symbiodiniaceae lineage according to Madracis host species, were chosen to retrieve Symbiodiniaceae type information from the RADseq data. In order to validate this choice, a known fingerprinting assay (ITS2 rRNA, using Denaturing Gradient Gel Electrophoresis56) was run for 37 representative samples, which included all six recognized Madracis species. This validation provided an exact match (fingerprinting vs RAD data) in 20 out of 21 comparisons, plus 13 samples with missing results for at least one of the assays (no comparison possible), and three cases where the ITS2 DGGE assay seem to indicate co-dominance of the two lineages (whereas RADseq clustering indicated only one of the symbionts). Even when excluding the three cases of ITS2 co-dominance, the accuracy of >80% between expected and observed Symbiodiniaceae genotypes allowed us to use the RADseq data to retrieve symbiont information.
Sequencing of mitochondrial ATP8 gene
To extrapolate interpretations based on RADseq dataset to a broader number of Madracis samples for which skeletal vouchers are available, we sequenced a commonly used single mitochondrial marker, ATP862,63,65,66,67,86. This marker has been used to characterize the depth divergence within M. pharensis66 and allows for further evaluation of genome-wide data versus a traditional marker for delimiting species in the genus Madracis. A 1200 bp fragment, which spans a small portion of the nad5 region, the entire trnW and ATP8 regions, an intergenic spacer and a small portion of cox1, was amplified using the FNAD5-RCOI3 primer pair87 and PCR conditions and cycling protocol previously described86. PCR products were run on agarose gels, purified using ExoSAP-IT and sequenced in both the forward and reverse directions (ABI BigDye Terminator chemistry, Australian Genome Research Facility). Obtained chromatograms were analyzed using Codoncode Aligner (https://www.codoncode.com/aligner/) and sequences were aligned with MUSCLE (https://doi.org/10.1186/1471-2105-5-113) and blasted on GenBank, where all unique sequences were deposited. Phylogenetic analyses based on ML were performed in Geneious 5.6.5 (https://www.geneious.com) under the delayed transition setting and calculation of bootstrap values based on 1000 replicates. Outgroup sequences were retrieved from GenBank. The best-fit model of molecular evolution (with a HKYGAMMA nucleotide substitution model) was selected by hierarchical Bayesian information criterion (BIC) using Modeltest2 in the CIPRES platform. Samples available from a previous publication58 were also characterized with the ATP8 marker.
Morphometrics
Specimens collected in 2017 from Bermuda and Curaçao generated 93 skeletal vouchers deposited at the Natural History Museum in Vienna, which are available for further comparisons (inventorized as convolute objects NHMW-ZOO-EV-21523 to NHMW-ZOO-EV-21534; see Table S1 for a list).
Coral skeleton macro- and micro-morphological features were observed using light microscopy and scanning electron microscopy (SEM), respectively. Dried skeleton chips were imaged using cameras mounted on a stereomicroscope (Leica and Nikon) at constant magnification (63 x) to obtain images of the corallum surface. Image J software was used to take measurements of the inter columella distance (ID), defined as the linear distance between the centre of the columella and the center of an adjacent corallite, as well as of the calice diameter (CD). About 25 replicates of ID and CD were taken for each specimen (roughly five measurements in each of five distinct sections of the skeleton) at as many different random positions as possible along the available skeleton chip, and caution was taken to avoid corallites undergoing budding. Number of primary and secondary septa was reassessed confirming species identifications in the field. Specimen ID and CD of genetic lineages obtained from RADseq were included in a Linear Discriminant Analysis (LDA, function “lda” in package MASS88) and differences among Madracis species/groups were assessed by applying Permutational Multivariate Analysis of Variance (PERMANOVA) on LDA scores followed by pairwise tests with corrections for multiple comparisons. For SEM, fragments were examined with a Hitachi TM4000 SEM at the Museum of Tropical Queensland. The following skeletal features were visualized and described blindly (i.e., without information on species identification or depth origin of specimen): structure and shape of the columella, the septa and the coenosteum, septal fusion patterns, prevalence of septal spines, septa second cycle, septal step, pali, and boundary teeth. SEM was used to study phenotypic variation and cryptic diversity mostly (but not exclusively) within the M. pharensis phenotype/group. All SEM and optical microscopy images are available via MorphoBank89 through the following link: http://morphobank.org/permalink/?P5806
Results
Samples and RADseq sequencing
A total of 235 Madracis coral specimens were collected at Curaçao and Bermuda from depths between 10–90 m. For details on species identification, collection site, depth of origin, and datasets retrieved for each specimen, see Table S1 and Fig. 1. While all six recognized Madracis morphospecies were identified and sampled in Curaçao, only five of the six species were detected in Bermuda: M. mirabilis, M. pharensis, M. carmabi, M. decactis and M. formosa. From these, only M. carmabi, M. mirabilis and M. decactis were previously unequivocally reported for Bermuda58,60,90,91. Cautious re-evaluation of dried M. pharensis and M. formosa specimens led us to maintain the original in situ identification and to assume the species are present in Bermuda. There are no obvious records of M. senaria presence in Bermuda. We note that due to the distinct morphological appearance of most M. decactis-like specimens collected from deep locations (> 30 m) in Bermuda (columnar in shape, as compared to the more knobby colonies collected from shallow locations in Bermuda and Curaçao) the notation M. cf. decactis has been assigned to these specimens.
Of 133 Madracis samples included in RADseq (n=85 and n=48 for Curaçao and Bermuda, respectively), only 115 passed the initial QC and filtering steps and only 95 had more than 95% of loci genotyped, leaving 282,328 out of possible 287,387 sites in the analysis.
Sequence-based analyses
Sequence-based ML phylogenies were roughly concordant with current taxa delimitation based on the six recognized Madracis morphospecies (Fig. 2 and Fig. S1). Each currently recognized Madracis morphospecies forms its own clade and is well supported, with one notable exception. Several specimens identified as M. pharensis did not cluster within the well-supported M. pharensis clade with Bermuda specimens, but rather fell either within distinct clades representing other species (e.g., within M. decactis, M. senaria or M. carmabi), or formed their own lineages as sister groups to some of the other clades, e.g. relative to the M. carmabi/M. formosa clade (specimens DC4585 from 60 m and DC4395 from 90 m), or relative to the M. decactis/M. mirabilis clade (specimen DC4945 from 15 m depth). All specimens identified as M. cf. decactis were collected in deep locations in Bermuda and all cluster within the M. decactis clade. Remarkably, the split between the Bermuda and Curaçao populations of M. mirabilis is robust (Fig. S1), suggesting that there is limited gene flow between the two regions. There is also strong differentiation within some of the other clades, namely M. carmabi, M. mirabilis, M. senaria and M. formosa, but not matching geographic origin. Phylogenetic reconstructions using different thresholds for the number of loci included show nearly identical topologies: as one constrains the amount of accepted missing data (trees with loci genotyped for at least 5%, 25% and 50% of the individuals; Fig. S2a-c), the only difference in the tree topologies is the position of the three unassigned M. pharensis lineages. In comparison to the ML tree in Fig. 2, the tree with only samples with >50% loci genotyped (Fig. S3a) is different only when it shows the M. pharensis group being non-supported. There is necessarily a low number of loci shared between the outgroup and the Madracis samples due to a large evolutionary distance between them. Bayesian inference on the same data (Fig. S3b) when only one specimen per species is included shows a different topology, albeit weakly supported, with M. senaria being the closest Madracis taxon to the root.

Maximum likelihood phylogeny (A), where the branch tip colors correspond to Madracis morphospecies (Mcar = M. carmabi, Mcfd = M. cf. decactis, Mdec = M. decactis, Mfor = M. formosa, Mmir = M. mirabilis, Mpha = M. pharensis, Msen = M. senaria, and Mspp = unidentified Madracis species). ML tree was re-ran to include all samples with >10% loci genotyped, and outgroup branch has been truncated. Colours in the phylogenetic tree (A) match those of Fig. 1. Probability of membership STRUCTURE (B) and genotype composition plots resulting from DAPC (C) are shown for k=5 through k=7, respectively. Results shown are not an average across permuted runs, but rather represent the most common iteration for each k. Further information in D: country locality are either BA (Bermuda) or CU (Curaçao), depth categories are <10, 11-20, 21-40, 41-60, or >60 meters, and symbiont type either B7 or B15 (note: B13 and B7 lineages are not separable by our methodology).
SNP-based analyses
Overall genetic structure based on NJ phylogeny (Fig. S4) corroborated the general topology obtained from the sequence-based analyses (Fig. 2). Here, the position of the three unassigned M. pharensis lineages aligned well with the ML trees where less missing data is allowed (loci genotyped for at least 50% of the individuals; Fig. S2c). Permuted STRUCTURE runs from k=2 up to k=8 (panels in Fig. S5) increasingly separated into clusters matching species clades represented in the ML/NJ trees, further supporting the structure predefined by the currently accepted Madracis species. As k increased in the STRUCTURE analyses, M. senaria was first to be recognized as a separate group (including two specimens of M. pharensis, see Fig S5), followed by M. mirabilis and M. decactis (including M. cf. decactis, and a few M. pharensis specimens), and then later by the main M. pharensis clade (all specimens originated from Bermuda). Next at k=6, two groups of M. mirabilis became apparent, mostly corresponding to geographic separation between Bermuda and Curaçao. Finally, at k=7 the separation of all currently recognized Madracis species is achieved (Fig. 2). Three M. pharensis individuals (basal or sister lineages in the ML/NJ trees) showed persistent signs of admixture in the permuted runs when k=3 or greater (Fig. S5a-b), and were thus flagged as unassigned (<0.8 cut-off value for mean ancestry assignment). These putatively admixed individuals included the shallow M. pharensis lineage (DC4945; which carried a strong signal from M. mirabilis and M. decactis), and the two deep M. pharensis lineages represented by DC4585 at 60 m depth (admixed between the main M. pharensis, M. mirabilis and M. formosa) and DC4395 at 90 m (admixed between M. formosa, the main M. pharensis and M. carmabi). These lineages tended to be assigned to their own cluster in some of the permuted runs while in others they exhibited a bipartinioned assignment typical of F1 hybrids. Overall, most of the permutations yielded a complex admixture signal. At low k values, partitioned STRUCTURE analyses per clade (Fig. S6a-c) corroborated the admixed nature of the three M. pharensis lineages mentioned above, while those at higher k values tended to assign a new cluster to each of these unassigned M. pharensis samples. In addition, the same pattern became evident for the two M. pharensis individuals (DC4576 and 2Mpha1) that clustered within M. senaria on the tree (Fig. 2).
DAPC analyses (Fig. 2) were less sensitive to detecting admixture signals but mostly corroborated the overall structure described above where clades represent the recognized Madracis morphospecies. Again, M. formosa and M. carmabi were the last clades to fall into separate clusters (at k=6 and k=7), and M. mirabilis again tended to separate by sampling origin. The three highly admixed M. pharensis individuals illustrate major discrepancies between the STRUCTURE and DAPC patterns and cluster into groups that do not match their relative positions in the ML tree (but always in the same DAPC as the M. formosa individuals). A M. pharensis individual (2Mpha1) that clustered with M. senaria in the STRUCTURE analysis, clustered with M. formosa in the DAPC. Further discrepancies between these two clustering approaches are found within the M. decactis and M. mirabilis clades, with several individuals (almost all originated in Curaçao) also clustering with the more distant M. formosa (at k=7), or with M. formosa and M. carmabi (at k=5).
Coalescent-based SNAPP analyses (Fig. 3A) based on unlinked biallelic SNPs with 0% missing data (3,548 SNPs included) subdivided the genus into the six commonly recognized species with high node support values (Bayesian posterior probabilities of 1; two individuals per species; Fig. S7a). Addition of the STRUCTURE-unassigned M. pharensis lineages into the analyses generated less supported topologies (Fig. S7), likely due to the rapid reduction in the number of SNPs covered by each SNAPP analysis. However, this analysis still showed high support (all Bayesian posterior probabilities >0.99; 2,791 SNPs included) for the M. pharensis lineage at 90 m (DC4395), suggesting the existence of M. pharensis lineages with sufficient differentiation to potentially be considered separated (cryptic) species. These will henceforth be referred to as “shallow”, “deep” and “very deep” M. pharensis lineages, according to their depth of origin (tentatively <20m, 40-60m and >80m, respectively).

Composite including A) species trees of Madracis inferred from coalescent-based SNAPP analyses, B) ABBA-BABA introgression tests run by fixing M. senaria as ancestral case, and C) macromorphological features (ID and CD distances, or inter-columellar distance and calice diameter, respectively) for the different recognized Madracis morphospecies and lineages resulting from RADseq analyses. Codes correspond to the following Madracis morphospecies (Mcar = M. carmabi, Mdec = M. decactis, Mfor = M. formosa, Mmir = M. mirabilis, Mpha = M. pharensis, Msen = M. senaria). Colours as in Fig. 1 and 2, with the exception of Madracis pharensis, now shown in different tones of green according to depth groups (“Mpha1_shallow”, “Mpha2_deep” and “Mpha3_very deep”). In A) the cloudograms for 14 samples and 2,791 SNPs illustrate the best species delimitation models from bi-allelic SNP data, and node labels show Bayesian posterior probabilities. In B) the three panels show, respectively from top to bottom, 1) the “BBAA”, or more stringent output, 2) the “Dmin” output, and 3) the “tree” output, based on the best ML phylogenetic tree. Heatmap color for each pair of cases (of Madracis species) gives the related admixture evidence based on the D-statistic. Entries with depth information (i.e., “Mpha15”, “Mpha90” and “Mpha60”) are based on one specimen only, and two “deep” lineages of M. pharensis are separated based on origin: BA (Bermuda) or CU (Curaçao). In C), all groups include a large number of specimens observed using light microscopy. Data for two reference taxa (M. pharensis (from Mediterranean) and M. kirby) are based on data published in Benzoni et al.67.
Hybridization tests
ABBA-BABA tests run by fixing different ancestral cases (M. senaria or M. pharensis to reflect the different potential evolutionary histories of Madracis) recurrently signaled the same lineages as being potentially involved in introgressive hybridization events (Fig. 3B). In tests utilizing a user-supplied tree, species typical of shallow versus deep habitats (i.e., M. mirabilis, M. decactis, M. senaria and “shallow” M. pharensis vs M. carmabi, M. formosa and “deep” and “very deep” M. pharensis) all appeared to exchange more genes (with any shallow vs deep counterpart) than might be expected with no hybridization. When M. senaria was set as the ancestral case, both the BBAA tests and the minD tests typically implicated MsenxMpha (M. pharensis-looking M. senaria genotypes) in hybridization with M. carmabi and with the unassigned M. pharensis lineages from 15 m and 60 m. When M. pharensis was set as ancestral case (Fig. S8), BBAA tests were suggestive of hybridization between M. senaria and the unassigned M. pharensis lineage from 15 m, between the unassigned M. pharensis lineage from 60 m and M. mirabilis, and between the two unassigned M. pharensis lineages (15 and 60 m). Dmin tests when M. pharensis was set as the ancestral case were again suggestive of hybridization between the two unassigned M. pharensis lineages (15 and 60 m). In all cases, there was statistical support for hybridization among the deeper unassigned M. pharensis lineage from 90 m (“very deep”) and its shallower counterparts. Overall, hybridization appears to be mostly mediated by these unassigned and highly admixed members of M. pharensis, which appear to be setting a bridge between the shallower and deeper clades of Madracis.
Symbiodiniaceae reads
Filtering of symbiont loci allowed the assignment of a Symbiodiniaceae genotype to most specimens sequenced by RADseq (Fig. 2). Trees generated from a set of random RAD loci matching the reference Symbiodiniaceae B1 nuclear genome typically showed two divergent branches matching the known separation of Madracis-associated Symbiodiniaceae into two broad lineages within the Breviolum genus: the Symbiodiniaceae B15 type and the nearly identical B13 and B7 types, according to known symbiont specificity by the host (i.e., M. senaria, M. decactis and M. mirabilis exclusively associated with B7/B13; and M. formosa, M. carmabi and M. pharensis with either one or both B7 and B15 lineages; see Frade et al.56). The Breviolum B13 and B7 lineages were not separable by our methodology. Our clustering approach assigned the most probable symbiont to each sample and did not allow for identification of more than one dominant symbiont lineage per host. Many Madracis species appear to associate with a single dominant Symbiodiniaceae type, and when the symbiont data is superimposed on the host tree, it becomes clear there is high specificity, particularly for M. cf. decactis, M. decactis, M. mirabilis, M. carmabi and M. senaria. The exceptions are M. formosa and M. pharensis, which are able to associate with more than one symbiont lineage.
Sequencing of mitochondrial ATP8 gene
Sequencing of the mitochondrial ATP8 gene was performed for just a fraction of the Madracis samples (the ones collected before 2015). In total, the ATP8 gene was sequenced in 114 specimens, with 102 yielding good quality sequences (of these, n=31 specimens were characterized by morphometrics, but only n=6 also yielded RADseq data). Maximum likelihood recovered a phylogeny with low robustness (Fig. S9), with most branches having low support and in which the Madracis morphospecies are not clearly separated into different clades. The exception is on the most divergent part of the tree. This topology clearly separates the Mediterranean Sea M. pharensis from all other Madracis (99.5% support). M. senaria specimens subsequently cluster together with some M. pharensis specimens from Curaçao, one of which was also genotyped using RADseq and showed signs of admixture in the STRUCTURE plots at high k levels (DC4576; k=8; Fig. S6b). This specimen also showed mixed morphological traits of M. senaria and M. pharensis, with corallites of both 6 and 10 septa being present. No M. pharensis from the Caribbean clustered with the Mediterranean Sea specimens. Moreover, the ATP8 tree roughly matches the RAD tree, but with reduced robustness. There is a “shallow cluster” including all other M. pharensis samples associating with the shallow-water Symbiodiniaceae B7 type, all M. decactis samples and M. mirabilis. There is then another clade that includes all M. pharensis associating with symbiont type B15, plus all M. formosa (except one), and where the “very-deep” M. pharensis samples also seem to form their own (non supported) branch. Surprisingly, some M. mirabilis specimens also cluster with this “deep cluster”. M. carmabi occupies the most basal position in the tree, but with no support. Most Madracis species are characterized by large indels (20-60 bp in length) that distinguish them from the other groups. This is also true for two distinct groups within M. mirabilis (one of which shares indels with the M. formosa group) and the different depth clusters within M. pharensis.
Morphometrics
Macromorphological features measured using light microscopy separated several Madracis species based on their inter columella distance (ID) and calice diameter (CD) combination (n=139 in total, see Fig. 3C). PERMANOVA applied on LDA scores indicated statistically significant differences in multivariate space of ID and CD (pseudo F (11,143) = 15.934, R2 = 0.55, p < 0.001, 999 permutations) with yet considerable overlap amongst species (see Fig. S10). Accuracy of the model as measured by “leave-one-out” cross validation (i.e., correct classification of observed species by the LDA model) was 36% overall, but reached very high accuracy for particular species or lineages (e.g., 72% for M. decactis, 50% for M. formosa, 93% for M. pharensis “very deep”, or 43% for M. senaria). Pairwise PERMANOVA comparisons on LDA scores identified several statistically cohesive groups, namely one formed by M. formosa alone, one group formed by M. mirabilis and M. senaria, and one formed by M. pharensis specimens originating from 80-90 m (Mpha3_very deep), which clustered in a unique position represented by a very large ID and small CD (Fig. 3C; p<0.05 for all pairwise comparisons; see Table S2). The remaining Madracis species formed a broad group (Pairwise PERMANOVA; p>0.05 for all comparisons): M. decactis, M. carmabi, M. pharensis from “deep” (Mpha2_deep) and “shallow” habitats (Mpha1_shallow), and the Indo-Pacific coral M. kirby, all characterized by intermediate CD and low ID. The exception here were “deep” M. pharensis from Bermuda, which grouped with the Mediterranean M. pharensis (Table S2). In fact, the Caribbean “deep” M. pharensis clearly segregated into two distinct clouds based on regional origin (Bermuda with larger corallites, Curaçao with smaller corallites). No further ecological axis appeared to relate to the measured morphological variation. For example, “shallow” M. pharensis specimens associating either with B7 or B15 Symbiodiniaceae, did not differ in the measured traits (p>0.05). The only M. pharensis-looking specimen measured that belonged to the M. senaria branch (DC4576), actually showed CD and ID values similar to the M. senaria specimens.
Specimens of M. senaria, M. decactis, M. formosa, M. carmabi and M. mirabilis/auretenra showed corallite characters (Table S3) consistent with those described for their assigned species, thus confirming our macro-morphology based identifications. Small differences were observed: the M. senaria specimens had much better development of meta-septa than previously described42 and the M. mirabilis/auretenra specimens had close-packed corallites (Fig. S11b), whereas Locke et al.60 show them as spaced. Note that corallite spacing and septal development are often affected by environment and corallite position on the colony (e.g. Locke et al.60).
Conversely, specimens tentatively identified as M. pharensis f. luciphila on the basis of macro-morphology often showed corallite features (see Table S3) that were not consistent with those described for the species41. Several specimens lacked pali and secondary septa, while one (DC4576) had a septal arrangement similar to M. senaria. Within these specimens, there were three distinct corallite morphologies that corresponded to clades/groups delineated by our RADseq analyses (Fig. 2):
-
1.
Corallite spacing compact hexagonal to close, 10 primary septa with moderate to large spines, reduced secondary septa, pali absent, columella body small to moderate with a styliform/point projection, prominent ICR with a distinct groove (except DC5542). Specimens were from 15 m depth and included DC4940, DC4954 and DC5542 (Fig. S11a; M. pharensis lineage 1, or “shallow”).
-
2.
Corallite spacing close to compact, 10 primary septa with large spines, secondary septa absent, pali absent, columella body medium to large with a reduced styliform/point projection (DC4685 conical), slight ICR (except DC8809 and DC4685). Specimens were from 40-45 m depth and included DC6979, DC8803, DC8805 and DC8809 in the RADseq tree (Fig. 2), and DC4685 (Fig. S11a; M. pharensis lineage 2, or “deep”).
-
3.
Corallite spacing close to separated, 10 primary septa with moderate to large spines, developed secondary septa and pali, columella body small/diffuse with styliform or point projection, slight inter-calicular ridge (ICR). Specimens were from 80-90 m depth and included DC4395 in the RADseq tree (Fig. 2), DC3619, DC4381 and DC4382 (Fig. S11a; M. pharensis lineage 3, or “very-deep”).
The remaining M. pharensis specimens had a range of corallite morphologies that we could not resolve into distinct groups. All the Madracis SEM specimens showed an inter-callicular ridge in a cellular or hexagonal arrangement that was well separated from the corallites. In some specimens these structures were only barely visible as a row of spines (Fig. S11a; M. pharensis lineage 3, or “very-deep”), while others had strong development, often with a “groove” structure separating them from the corallite walls (Fig. S11a; M. pharensis lineage 1, or “shallow”). For specimens in which the coenosteum was porous, an underlying wall structure was apparent, indicating that the ICR is a much larger structure than a ridge of spines in the coenosteum. The ICR is unusual in that it is a wall formation completely outside and separate from the corallites’ walls. Since the ICR is often only apparent as a simple row of spines, it has previously not been recognised as a structure separate to the coenosteum, except for Wells et al.41 who described it for M. myriaster and appears to have first named it.
Discussion
Species delimitation is a fundamental step for guiding conservation actions, but can be challenging when different lines of evidence give incongruent results. The Caribbean pocilloporid genus, Madracis, illustrates many of the problems underlying phylogenetic inference in corals: few morphological traits available for species delimitation and conflicting genetic versus morphological patterns58. These conflicting patterns are usually attributed to high levels of phenotypic plasticity, introgressive hybridization between lineages, occurrence of cryptic species or incomplete lineage sorting10,11,14,15,16,40. Based on an integrated systematics approach that uses RADseq data for phylogenomics plus information on morphology, reproduction and ecology, we were able to greatly improve phylogenetic resolution in the Madracis genus resulting in an even more robust species tree for Madracis in the Western Atlantic. Whereas our work partially supports the morphology-based taxonomy, it also builds a framework for future formal taxonomic revision in the Madracis genus, based on the presence of undescribed cryptic species (Fig.4). Moreover, multiple lines of evidence point to the widespread role of hybridization in shaping Caribbean Madracis species diversity.

Tentative representation of Madracis species diversity in the Caribbean and West Atlantic, including phylogenetic relationships (topology based on coalescent-based SNAPP analyses), in situ images of colony shape, SEM-images of corallite details, plus several diagnostic life history and ecological traits. Figure includes recognized Madracis species (names in black and colourful boxes in full line stroke) and putative new species (names in white and dashed line stroke) emerging from the present analyses. The accepted Madracis pharensis from the Mediterranean Sea (MED SEA) is added as reference. Corallite data from Mediterranean Sea Madracis pharensis (MED SEA) obtained from Benzoni et al.67, and symbiont type and depth distribution from Goodbody-Gringley et al.92. Reproductive period and symbiont acquisition strategy for Caribbean Madracis are based on pers. obs. (P. R. Frade and P. Bongaerts) and refer only to Curaçao. Relative position of candidate new species within Madracis pharensis in the Madracis tree are based on the Neighbour-Joining tree. For the “deep” Madracis pharensis lineage 2, only the Bermuda form is shown; the Curaçao form has substantially different corallite dimensions (see Fig. 3). Underwater photos by Pedro R. Frade.
Unparalleled phylogenetic resolution in Madracis
Prior to this study, of the six commonly recognized Caribbean species within the genus, only one, M. senaria, had genetic validation based on traditional ribosomal and nuclear genetic monomarkers. The remaining Madracis were characterized by extensive sharing of genetic variation and formed unresolved species complexes44,58. In this study, we show that increasing the amount of sequence data used in phylogenetic analysis (by using a RADseq approach) led to unparalleled phylogenetic resolution in the genus, with at least three of the six morphologically recognized species presenting reciprocal monophyly in the SNP-based NJ tree: M. senaria, M. mirabilis and M. formosa. However, M. carmabi, M. decactis and M. pharensis still form unresolved complexes, mostly because of the polyphyletic nature of M. pharensis. Morphologically, these three species have the same number of septa, but in terms of gross morphology can be placed along a continuum (see Fig. 2 and morphometrics Fig. 3c), with the more round/bulky M. pharensis tending to be confounded with the knobby M. decactis, which in turn tends to be similar in shape to the branching M. carmabi at times. This morphological continuum (potentially also affected by environmental plasticity) means that misidentification is possible and/or that morphological similarity may reflect recent or ongoing introgression. These two hypotheses are not mutually exclusive, as a history of recent introgression would most likely and to some degree also favour misidentification based on morphology14. Yet, in this study, we purposely avoided non-typical morphologies (i.e., morphologies not matching the colony morphologies in the original descriptions41,42,43) and these peculiar “in-between” morphologies are being analysed by our research team in a follow up study using whole-genome approaches. Instead, we suggest that introgression is responsible for the non-monophylies we observed in the phylogenetic clusters formed by M. carmabi, M. decactis and M. pharensis. Upon closer inspection, some M. pharensis phenotypes clustered in the M. carmabi and M. decactis branches and three M. pharensis lineages represented by three individuals exhibit strong signals of admixture (Fig. 2 and Fig. S5a-b).
Different species delimitation methods based on RADseq data provided similar results. The SNAPP species-delimitation model (Fig. 3 and Fig. S7), including only samples with no missing data, confirmed the general clades identified in the sequence-based phylogeny, which resolves the genus into the six currently recognized morphospecies. When the admixed M. pharensis individuals are included, the SNAPP analyses lose support, providing further evidence of the admixed nature of these specimens. Thus, our study was able to delineate Madracis species more robustly than earlier studies, with complex topologies/non-monophylies likely being the result of introgression44,58, an interpretation not previously possible due to the use of single markers as compared to our wide genomic coverage.
Genome-wide versus single marker analyses
Topological conflict between RAD and mitochondrial ATP8 phylogenies muddles the nature of species relationships. The most striking difference between datasets is that in the ATP8 phylogeny M. decactis, M. mirabilis and “shallow” M. pharensis form a single clade within which they even share one single haplotype. Such an unresolved clade resembles the result obtained with the nad5 mitochondrial marker58 and suggests either recent speciation in this group of species (although there is recognized slow evolution of the mitochondrial genome in corals, see e.g., Huang et al.21), the occurrence of introgressive hybridization, or that genes or fractions within the ATP8 marker are under strong selection. The recent confirmation that mitochondrial genomes are under strong purifying selection in both Hexacorallia and Octocorallia93 impairs their use in analyses that rely on assumptions of neutrality. Based on the mitochondrial phylogeny alone, taxa like M. decactis, the shallow M. pharensis and M. mirabilis do not appear to represent reproductively isolated species (sensu Mayr94). Mitochondrial markers reflect the history of maternal lineages, often incongruent with the species evolutionary history93,95,96. Although mitochondrial phylogenies may be a good first step to start assessing species relationships in corals, our study is concurrent with other findings demonstrating that an integrative approach that uses a higher number of more variable markers and ecological traits decisively improves our capacity to infer the evolutionary history of coral species, particularly for closely related coral taxa37,38.
Cryptic diversity within Madracis pharensis
Our RADseq approach facilitated an improved understanding of the hidden genetic diversity within Caribbean M. pharensis and the previously hypothesized depth divergence for this species66. The taxon, in the Caribbean, appears to include at least three distinct genetic lineages: 1) the “shallow” lineage (Mpha_1) that associates with Symbiodiniaceae type B7 and exhibits relatively low columellar distances (represented by a single specimen in the RAD dataset, but fairly represented in the ATP8 tree; see Fig. S9 and Fig. S11), 2) the “deep” lineage (Mpha_2) that associates with Symbiodiniaceae type B15 (and which could comprise two distinct genetic and morphological entities separated by origin: Curaçao vs Bermuda), and 3) the “very deep” lineage (Mpha_3; found here at 80-90 m, and for which only one specimen yielded sufficient RAD data, but fairly represented in the ATP8 dataset) that also associates with Symbiodiniaceae type B15, exhibits quite unique morphological traits (including at the SEM level) and is genetically closer to the deep-water branching species M. carmabi and M. formosa. Strikingly, the shallowest and deepest M. pharensis collected (all from Curaçao) are represented by highly admixed individuals, which suggests that these lineages may be the result of hybridization processes.
The taxonomic status of these M. pharensis-like admixed lineages remains uncertain, but they could hypothetically represent hybrid species where a fraction of the population is introgressing with the parental lineages, or they could constitute hybrid species that are already isolated from their parental lineages. While we would need to have a much larger number of such individuals in our analyses, the relative admixed nature of these individuals seems to point to recent hybridization and ongoing gene flow, in a kind of “two-island model”97. This is also supported by the introgression tests we ran, where hybridization appears to be mediated by these admixed members of M. pharensis, which act as an evolutionary bridge between the shallower and deeper clades of Madracis. An alternative is that represented by a “pulse model” of hybridization97, where an admixed lineage arose from two diverged parental lineages in the past, and hybridization has since stopped. Our introgression tests have not been developed to resolve between recent and ancient species contact and thus this remains an open question to be addressed in the future.
Formal taxonomic actions needed for Caribbean Madracis
Although we were able to attain a reasonably robust phylogeny, some incongruences still remain. Species delimitation in the Caribbean genus Madracis is still an unfinished exercise, as it becomes clear that there is cryptic diversity within what is identified as Madracis pharensis. Thus, one has to question the taxonomic position of the studied M. pharensis in relation to the Mediterranean Madracis pharensis (Heller, 1868) (from where the species is originally described) and to the Caribbean form named M. pharensis f. luciphila Wells, 1973 by some41,42,69,98. Although the level of genetic and morphological divergence within Caribbean M. pharensis that we observed is enough to consider it likely that cryptic species exist within the Caribbean M. pharensis, a formal taxonomic resolution is beyond the scope of this manuscript. Yet, it is therefore clear that a taxonomic revision is needed to formally separate the different species contained within M. pharensis and determine the position of M. pharensis f. luciphila within the defined lineages. However, we propose the following candidate taxa (see Fig. 4) to be included in a future study to ascertain their status as cryptic species of M. pharensis f. luciphila Wells, 1973:
-
1)
The “shallow” Madracis pharensis lineage 1 has a spongey coenosteum and non-conical columella; Specimens exhibit thick septa, mostly absent secondary septa or septal spines, mostly absent pali. This corresponds to shallow to mid-water (15-40 m) “M. pharensis” that associates with B7 symbiont and is only confirmed for Curaçao. None of the RAD-sequenced specimens (DC4945) was examined under SEM (but see specimens DC4940, DC4954 and DC5542 in the ATP8 tree Fig. S9 and Fig. S11).
-
2)
The “deep” Madracis pharensis lineage 2, originating from 40-60 m, represented by specimen DC6979 originating from Bermuda (plus specimens DC4685, and maybe DC5541 and, DC5558 in the ATP8 tree Fig. S9 and Fig. S11). Specimens exhibit thick septa, mostly absent secondary septa or septal spines, mostly absent pali. It is distinguishable from the shallow lineage as it presents a large conical columella and plain to pitted coenosteum, and absent second cycle of septa. This taxon associates with Symbiodiniaceae type B15. The Bermuda and Curaçao forms are clearly different based on corallite morphometrics, and may represent separate species. Interestingly, the Bermuda phenotype is not statistically different (in measured CD and ID) from the Mediterranean M. pharensis.
-
3)
The “very deep” Madracis pharensis lineage 3, originating from 80-90 m, including specimen DC4395 in the RADseq tree (plus specimens DC3619, DC4381 and DC4382 in ATP8 tree Fig. S9 and Fig. S11). Specimens exhibit thin septa, a small columella shaped as a blade, the prevalence of a second cycle of septa, abundant and conspicuous septal spines, prevalent pali, and a pitted to plain coenosteum. This taxon associates with Symbiodiniaceae type B15, and is genetically closer to the deep water branching species M. carmabi and M. formosa. Its distribution is hitherto only known for Curaçao.
Madracis phylogenomics corroborated by life histories
Besides the case of deep and shallow M. pharensis lineages differing in morphology and symbiont associations, the topology obtained from our reduced-representation genome sequencing is corroborated by many phenotypic traits observed in Madracis. The most striking case is that of M. senaria, a species characterized by having a well-defined temporal spawning window, in contrast to the all-year-round reproductive activity of all other species in the Madracis genus50. During this period M. senaria releases undeveloped egg-shaped larvae lacking the conspicuous zooxanthellae ring seen for other species (pers. obs. P. R. Frade and P. Bongaerts). This difference in reproductive period may constitute the single needed factor contributing to the reproductive isolation of M. senaria and its clearly divergent position within the Madracis tree. Although we cannot here conjecture on potential mechanisms leading to this phenotypic difference, similar examples of recent genetic divergence concomitant with functional differences in reproductive traits also exist in other coral genera such as Orbicella99, Fungia100 or Pachyseris40. Moreover, M. senaria is characterized by unique corallite morphology within the genus: six proto septa exerted from the corallite, in contrast with the ten uniform septa usually seen in most other Madracis (or eight in M. formosa).
Symbiont diversity and its role in the depth differentiation of Madracis
Our study further resolves the relationship between the Madracis hosts and their symbionts. Each Madracis morphospecies associates with one unique dominant symbiont genotype, the exceptions being M. formosa and M. pharensis, where both B7 and B15 Symbiodiniaceae (genus Breviolum Parkinson and Lajeunesse, 2018) can be dominant in the same host population. This occurrence pattern is strongly influenced by the methodology used, which did not enable us to resolve between symbionts B7 and B13, or to identify cases of multiple dominant symbionts in the same sample (shown to exist by our fingerprinting technique). This is particularly relevant for Madracis species previously known to be able to establish associations with multiple types, namely M. carmabi, M. formosa and M. pharensis. Notably, the depth-related genetic divergence in the M. pharensis host is linked to that of the dominant endosymbiont lineages harbored by this morphospecies, with Symbiodiniaceae type B7 typically associating with the “shallow” M. pharensis lineage and type B15 associating with the “deep” and “very deep” lineages, thus corroborating and building upon previous findings58. This link between depth distribution of host and symbiont has been interpreted as being the result of putative adaptive divergence and ecological speciation56, a possibility discussed for other coral genera with similar depth-zonation patterns, such as Agaricia86 or Seriatopora101. Based on the available symbiont phylogenetic studies102, B15 appears as the ancestral symbiont in Madracis and B7 a more diverged lineage. Association with B15, a deep-water specialist56, was only observed in the “deeper cluster” of Madracis involving M. pharensis and M. formosa, but excluding M. carmabi (see Fig. 2). This raises the question whether the absence of B15 in M. carmabi is genetically determined (e.g., host-symbiont incompatibility), or whether it is simply a result of selection imposed by the abiotic environment (i.e., environmental filtering) and of the (narrower) depth-distribution of the M. carmabi host (25-35 m depth in Curaçao, as compared to the deeper M. formosa, 30-60 m, and to the wider M. pharensis, 5-90 m).
An additional hypothesis that could explain the prevalence of two dominant symbionts in the same Madracis species is cross-fertilization between two lineages associating with distinct symbionts. In this case, the expected symbiont vertical transmission typical of brooders (like Madracis) would lead to a particular morphospecies exhibiting a “foreign” symbiont only when an individual colony develops the phenotype passed on from the male parental colony, as it is expected that symbionts would have mostly been passed on from the maternal colony103. Another interesting point is that none of the M. pharensis genetic lineages in our study associated with more than one symbiont (e.g., either B7 or B15, but never B7 and B15 within the same cluster), which suggests that host-symbiont associations are based on selective forces where the symbiont plays a selective role (depth-light gradients, for example). This supports a subjacent model of selection leading to disruption, or ecological selection, instead of prevalent hybrid zones (or depth zones). The correlation between depth distribution and symbiont type among M. pharensis clades has been interpreted as evidence of ongoing disruptive selection and adaptive divergence within M. pharensis58. In this case, harbouring either a symbiont genetic type adapted to shallow or deeper lifestyle (e.g., light adaptation, see Frade et al.55) may create the peculiar conditions promoting assortative mating within each of these lineages, a phenomenon that could lead to ecological speciation even in the presence of gene flow104. For example, temperature-mediated selection has been suggested as a driver of marine ecological speciation in coastal fish species105. In deep sea Callogorgia corals, recent results have shown that adaptive divergence with depth is possible, therefore providing support to the depth-differentiation hypothesis106.
Conclusion
Our high resolution RADseq approach coupled with morphological and ecological traits resulted in great advances in resolving species relationships in the Caribbean coral genus Madracis and decisively supported morphological descriptions for most of the recognized species. This is remarkable given the large degree of shared genetic variation shown by previous studies and highlights the great potential of high throughput sequencing for generating thousands of loci that are able to assist in resolving phylogenetic problems relating to species boundaries in scleractinian corals.
Data availability
Sequencing and variant call datasets are accessible through https://doi.org/10.5281/zenodo.15120504 and microscopy images are available via MorphoBank (http://morphobank.org/permalink/?P5806). For further clarification contact corresponding author.
Change history
25 November 2025
A Correction to this paper has been published: https://doi.org/10.1038/s41598-025-28788-8
References
Hughes, T. P. et al. Coral reefs in the anthropocene. Nature 546, 82–90. https://doi.org/10.1038/nature22901 (2017).
Hughes, T. P. et al. Global warming and recurrent mass bleaching of corals. Nature 543, 373–377. https://doi.org/10.1038/nature21707 (2017).
Bongaerts, P. et al. Deep reefs are not universal refuges: Reseeding potential varies among coral species. Sci. Adv. https://doi.org/10.1126/sciadv.1602373 (2017).
Bridge, T. C. L. et al. A tenuis relationship: Traditional taxonomy obscures systematics and biogeography of the ‘Acropora tenuis’ (Scleractinia: Acroporidae) species complex. Zool. J. Linn. Soc. https://doi.org/10.1093/zoolinnean/zlad062 (2023).
Romano, S. L. & Palumbi, S. R. Evolution of scleractinian corals inferred from molecular systematics. Science 271, 640–642. https://doi.org/10.1126/science.271.5249.640 (1996).
Fukami, H. et al. Conventional taxonomy obscures deep divergence between Pacific and Atlantic corals. Nature 427, 832–835 (2004).
Fukami, H. et al. Mitochondrial and nuclear genes suggest that stony corals are monophyletic but most families of stony corals are not (Order Scleractinia, Class Anthozoa, Phylum Cnidaria). PLoS ONE 3, e3222 (2008).
Kitahara, M. V., Fukami, H., Benzoni, F. & Huang, D. in The Cnidaria, Past, Present and Future: The world of Medusa and her sisters (eds Stefano Goffredo & Zvy Dubinsky) 41-59 (Springer International Publishing, 2016).
Huang, D. W., Licuanan, W. Y., Baird, A. H. & Fukami, H. Cleaning up the “Bigmessidae”: Molecular phylogeny of scleractinian corals from faviidae, merulinidae pectiniidae and trachyphylliidae. BMC Evol. Biol. 11, 37. https://doi.org/10.1186/1471-2148-11-37 (2011).
Todd, P. A. Morphological plasticity in scleractinian corals. Biol. Rev. 83, 315–337 (2008).
Paz-García, D. A., Hellberg, M. E., García-de-León, F. J. & Balart, E. F. Switch between morphospecies of pocillopora corals. Am. Nat. 186, 434–440. https://doi.org/10.1086/682363 (2015).
Willis, B. L., van Oppen, M. J. H., Miller, D. J., Vollmer, S. V. & Ayre, D. J. The role of hybridization in the evolution of reef corals. Annu. Rev. Ecol. Evol. Syst. 37, 489–517 (2006).
Combosch, D. J. & Vollmer, S. V. Trans-Pacific RAD-Seq population genomics confirms introgressive hybridization in Eastern Pacific Pocillopora corals. Mol. Phylogen. Evol. 88, 154–162. https://doi.org/10.1016/j.ympev.2015.03.022 (2015).
Forsman, Z. H. et al. Coral hybridization or phenotypic variation? Genomic data reveal gene flow between Porites lobata and P. compressa. Mol. Phylogen. Evol. 111, 132–148. https://doi.org/10.1016/j.ympev.2017.03.023 (2017).
Forsman, Z. H., Barshis, D. J., Hunter, C. L. & Toonen, R. J. Shape-shifting corals: Molecular markers show morphology is evolutionarily plastic in Porites. BMC Evol. Biol. 9, 45 (2009).
Boulay, J. N., Hellberg, M. E., Cortes, J. & Baums, I. B. Unrecognized coral species diversity masks differences in functional ecology. Proc. of the Royal Society B-Biological Sciences 281 ARTN 20131580 https://doi.org/10.1098/rspb.2013.1580 (2014).
Bongaerts, P. et al. Morphological stasis masks ecologically divergent coral species on tropical reefs. Curr. Biol. 31(2286), 2298. https://doi.org/10.1016/j.cub.2021.03.028 (2021).
Schmidt-Roach, S., Miller, K. J., Lundgren, P. & Andreakis, N. With eyes wide open: a revision of species within and closely related to the Pocillopora damicornis species complex (Scleractinia; Pocilloporidae) using morphology and genetics. Zool. J. Linn. Soc. 170, 1–33. https://doi.org/10.1111/zoj.12092 (2014).
Shearer, T. L., Van Oppen, M. J. H., Romano, S. L. & Worheide, G. Slow mitochondrial DNA sequence evolution in the Anthozoa (Cnidaria). Mol. Ecol. 11, 2475–2487 (2002).
Hellberg, M. E. No variation and low synonymous substitution rates in coral mtDNA despite high nuclear variation. BMC Evol. Biol. 6, 24. https://doi.org/10.1186/1471-2148-6-24 (2006).
Huang, D., Meier, R., Todd, P. A. & Chou, L. M. Slow mitochondrial COI sequence evolution at the base of the metazoan tree and its implications for DNA barcoding. J. Mol. Evol. 66, 167–174 (2008).
Chen, I. P. et al. Comparative analyses of coding and noncoding DNA regions indicate that acropora (anthozoa: scleractina) possesses a similar evolutionary tempo of nuclear vs. mitochondrial genomes as in plants. Mar. Biotechnol. 11, 141–152. https://doi.org/10.1007/s10126-008-9129-2 (2009).
Andrews, K. R., Good, J. M., Miller, M. R., Luikart, G. & Hohenlohe, P. A. Harnessing the power of RADseq for ecological and evolutionary genomics. Nat. Rev. Genet 17, 81–92. https://doi.org/10.1038/nrg.2015.28 (2016).
Baird, N. A. et al. Rapid SNP discovery and genetic mapping using sequenced RAD markers. PLoS ONE 3, e3376. https://doi.org/10.1371/journal.pone.0003376 (2008).
Miller, M. R., Dunham, J. P., Amores, A., Cresko, W. A. & Jonhson, E. A. Rapid and cost-effective polymorphism identification and genotyping using restriction site associated DNA (RAD) markers. Genome Res. 17, 240–248 (2007).
Rubin, B. E. R., Ree, R. H. & Moreau, C. S. Inferring Phylogenies from RAD Sequence Data. PLoS ONE 7, e33394. https://doi.org/10.1371/journal.pone.0033394 (2012).
Eaton, D. A. R. & Ree, R. H. Inferring phylogeny and introgression using RADseq data: An example from flowering plants (Pedicularis: Orobanchaceae). Syst. Biol. 62, 689–706. https://doi.org/10.1093/sysbio/syt032 (2013).
Bragg, J. G., Potter, S., Bi, K. & Moritz, C. Exon capture phylogenomics: efficacy across scales of divergence. Mol. Ecol. Resour. 16, 1059–1068. https://doi.org/10.1111/1755-0998.12449 (2016).
Wagner, C. E. et al. Genome-wide RAD sequence data provide unprecedented resolution of species boundaries and relationships in the Lake Victoria cichlid adaptive radiation. Mol. Ecol. 22, 787–798. https://doi.org/10.1111/mec.12023 (2013).
Pante, E. et al. Use of RAD sequencing for delimiting species. Heredity 114, 450–459. https://doi.org/10.1038/hdy.2014.105 (2015).
Quattrini, A. M. et al. Phylogeography of paramuricea: The role of depth and water mass in the evolution and distribution of deep-sea corals. Front. Marine Sci. https://doi.org/10.3389/fmars.2022.849402 (2022).
Herrera, S. & Shank, T. M. RAD sequencing enables unprecedented phylogenetic resolution and objective species delimitation in recalcitrant divergent taxa. Mol. Phylogen. Evol. 100, 70–79. https://doi.org/10.1016/j.ympev.2016.03.010 (2016).
Iguchi, A. et al. RADseq population genomics confirms divergence across closely related species in blue coral (Heliopora coerulea). BMC Evol. Biol. 19, 187. https://doi.org/10.1186/s12862-019-1522-0 (2019).
Quattrini, A. M. et al. A next generation approach to species delimitation reveals the role of hybridization in a cryptic species complex of corals. bioRxiv, 523936, https://doi.org/10.1101/523936 (2019).
Quek, Z. B. R. & Huang, D. Application of phylogenomic tools to unravel anthozoan evolution. Coral Reefs 41, 475–495. https://doi.org/10.1007/s00338-021-02072-3 (2022).
Johnston, E. C. et al. A genomic glance through the fog of plasticity and diversification in Pocillopora. Sci. Rep. 7, 5991. https://doi.org/10.1038/s41598-017-06085-3 (2017).
Rosser, N. L. et al. Phylogenomics provides new insight into evolutionary relationships and genealogical discordance in the reef-building coral genus Acropora. Proc. of the Royal Society B-Biological Sciences 284 ARTN 20162182 https://doi.org/10.1098/rspb.2016.2182 (2017)
van Oppen, M. J. H., McDonald, B. J., Willis, B. & Miller, D. J. The evolutionary history of the coral genus Acropora (Scleractinia, Cnidaria) based on a mitochondrial and a nuclear marker: Reticulation, incomplete lineage sorting, or morphological convergence?. Mol. Biol. Evol. 18, 1315–1329 (2001).
Dimond, J. L., Gamblewood, S. K. & Roberts, S. B. Genetic and epigenetic insight into morphospecies in a reef coral. Mol. Ecol. 26, 5031–5042. https://doi.org/10.1111/mec.14252 (2017).
Bongaerts, P. et al. Morphological stasis masks ecologically divergent coral species on tropical reefs. Curr. Biol. 31, 2286-2298.e2288. https://doi.org/10.1016/j.cub.2021.03.028 (2021).
Wells, J. W. New and old scleractinian Corals from Jamaica. Bull. Mar. Sci. 23, 16–55 (1973).
Wells, J. W. Two new hermatypic scleractinian corals from the West Indies. Bull. Mar. Sci. 23, 925–932 (1973).
Vermeij, M. J. A., Diekmann, O. E. & Bak, R. P. M. New species of scleractinian coral (Cnidaria, anthozoa), Madracis carmabi n. sp from the Caribbean. Bull. Mar. Sci. 73, 679–684 (2003).
Diekmann, O. E., Bak, R. P. M., Stam, W. T. & Olsen, J. L. Molecular genetic evidence for probable reticulate speciation in the coral genus Madracis from a Caribbean fringing reef slope. Mar. Biol. 139, 221–233 (2001).
Vermeij, M. J. A. & Bak, R. P. M. How are coral populations structured by light? Marine light regimes and the distribution of Madracis. Marine Ecology-Progress Series 233, 105–116 (2002).
Vermeij, M. J. A. & Bak, R. P. M. Corals on the move: Rambling of Madracis pharensis polyps early after settlement. Coral Reefs 21, 262–263 (2002).
Vermeij, M. J. A. & Bak, R. P. M. Species-specific population structure of closely related coral morphospecies along a depth gradient (5–60 m) over a Caribbean reef slope. Bull. Mar. Sci. 73, 725–744 (2003).
Vermeij, M. J. A., Delvoye, L., Nieuwland, G. & Bak, R. P. M. Patterns in fluorescence over a Caribbean reef slope: The coral genus Madracis. Photosynthetica 40, 423–429 (2002).
Vermeij, M. J. A., Sampayo, E., Broker, K. & Bak, R. P. M. The reproductive biology of closely related coral species: gametogenesis in Madracis from the southern Caribbean. Coral Reefs 23, 206–214 (2004).
Vermeij, M. J. A., Sampayo, E., Broker, K. & Bak, R. P. M. Variation in planulae release of closely related coral species. Marine Ecol.-Progress Series 247, 75–84 (2003).
Vermeij, M. J. A., Sandin, S. A. & Samhouri, J. F. Local habitat distribution determines the relative frequency and interbreeding potential for two Caribbean coral morphospecies. Evol. Ecol. 21, 27–47 (2007).
Kaandorp, J. A. et al. Simulation and analysis of flow patterns around the scleractinian coral Madracis mirabilis (Duchassaing and Michelotti). Philos. Trans. Royal Soc. London Series B-Biol.l Sci. 358, 1551–1557 (2003).
Kaandorp, J. A. et al. Morphogenesis of the branching reef coral Madracis mirabilis. Proc. of the Royal Society B-Biological Sciences 272, 127-133 (2005).
Filatov, M. V., Frade, P. R., Bak, R. P., Vermeij, M. J. & Kaandorp, J. A. Comparison between colony morphology and molecular phylogeny in the caribbean scleractinian coral genus madracis. PLoS ONE 8, e71287. https://doi.org/10.1371/journal.pone.0071287 (2013).
Frade, P. R., Bongaerts, P., Winkelhagen, A. J. S., Tonk, L. & Bak, R. P. M. In situ photobiology of corals over large depth ranges: A multivariate analysis on the roles of environment, host, and algal symbiont. Limnol. Oceanogr. 53, 2711–2723 (2008).
Frade, P. R., De Jongh, F., Vermeulen, F., Van Bleijswijk, J. & Bak, R. P. M. Variation in symbiont distribution between closely related coral species over large depth ranges. Mol. Ecol. 17, 691–703. https://doi.org/10.1111/J.1365-294x.2007.03612.X (2008).
Frade, P. R., Englebert, N., Faria, J., Visser, P. M. & Bak, R. P. M. Distribution and photobiology of Symbiodinium types in different light environments for three colour morphs of the coral Madracis pharensis: Is there more to it than total irradiance?. Coral Reefs 27, 913–925 (2008).
Frade, P. R. et al. Semi-permeable species boundaries in the coral genus Madracis: Introgression in a brooding coral system. Mol. Phylogen. Evol. 57, 1072–1090 (2010).
Fenner, D. P. Species distinctions among several Caribbean stony corals. Bull. Mar. Sci. 53, 1099–1116 (1993).
Locke, J. M., Weil, E. & Coates, K. A. A newly documented species of Madracis (Scleractinia : Pocilloporidae) from the Caribbean. Proc. Biol. Soc. Wash. 120 214-226 (2007).
Flot, J. F. et al. Incongruence between morphotypes and genetically delimited species in the coral genus Stylophora: phenotypic plasticity, morphological convergence, morphological stasis or interspecific hybridization?. BMC Ecol. 11, 22. https://doi.org/10.1186/1472-6785-11-22 (2011).
Stefani, F. et al. Comparison of morphological and genetic analyses reveals cryptic divergence and morphological plasticity in Stylophora (Cnidaria, Scleractinia). Coral Reefs 30, 1033–1049. https://doi.org/10.1007/s00338-011-0797-4 (2011).
Flot, J. F. et al. Mitochondrial sequences of Seriatopora corals show little agreement with morphology and reveal the duplication of a tRNA gene near the control region. Coral Reefs 27, 789–794. https://doi.org/10.1007/s00338-008-0407-2 (2008).
Pinzón, J. H. et al. Blind to morphology: Genetics identifies several widespread ecologically common species and few endemics among Indo-Pacific cauliflower corals (Pocillopora, Scleractinia). J. Biogeogr. 40, 1595–1608. https://doi.org/10.1111/jbi.12110 (2013).
Schmidt-Roach, S. et al. Assessing hidden species diversity in the coral Pocillopora damicornis from Eastern Australia. Coral Reefs 32, 161–172. https://doi.org/10.1007/s00338-012-0959-z (2013).
Bongaerts, P. et al. Deep down on a Caribbean reef: lower mesophotic depths harbor a specialized coral-endosymbiont community. Sci. Rep. 5, 7652. https://doi.org/10.1038/srep07652 (2015).
Benzoni, F. et al. Morphological and genetic divergence between Mediterranean and Caribbean populations of Madracis pharensis (Heller 1868) (Scleractinia, Pocilloporidae): too much for one species?. Zootaxa 4471, 473–492 (2018).
Zibrowius, H. Les scléractiniaires de la Méditerranée et de l’Atlantique nord-oriental. Mémoires de l’Institut Océanographique, Monaco 11, 1–55 (1980).
Cairns, S. D. A revision of the shallow-water azooxanthellate Scleractinia of the western Atlantic. Stud. Nat. History Caribbean Region 75, 1–231 (2000).
Russello, M. A., Waterhouse, M. D., Etter, P. D. & Johnson, E. A. From promise to practice: Pairing non-invasive sampling with genomics in conservation. Peerj 3, e1106. https://doi.org/10.7717/peerj.1106 (2015).
Reitzel, A. M., Herrera, S., Layden, M. J., Martindale, M. Q. & Shank, T. M. Going where traditional markers have not gone before: Utility of and promise for RAD sequencing in marine invertebrate phylogeography and population genomics. Mol. Ecol. 22, 2953–2970. https://doi.org/10.1111/mec.12228 (2013).
Eaton, D. A. R. & Overcast, I. ipyrad: Interactive assembly and analysis of RADseq datasets. Bioinformatics 36, 2592–2594. https://doi.org/10.1093/bioinformatics/btz966 (2020).
Shoguchi, E. et al. Draft assembly of the symbiodinium minutum nuclear genome reveals dinoflagellate gene structure. Curr. Biol. 23, 1399–1408. https://doi.org/10.1016/j.cub.2013.05.062 (2013).
Shoguchi, E., Shinzato, C., Hisata, K., Satoh, N. & Mungpakdee, S. The large mitochondrial genome of symbiodinium minutum reveals conserved noncoding sequences between dinoflagellates and apicomplexans. Genome Biol. Evol. 7, 2237–2244. https://doi.org/10.1093/gbe/evv137 (2015).
Danecek, P. et al. The variant call format and VCFtools. Bioinformatics 27, 2156–2158. https://doi.org/10.1093/bioinformatics/btr330 (2011).
Yu, G., Smith, D. K., Zhu, H., Guan, Y. & Lam, T.T.-Y. ggtree: An r package for visualization and annotation of phylogenetic trees with their covariates and other associated data. Methods Ecol. Evol. 8, 28–36. https://doi.org/10.1111/2041-210X.12628 (2017).
Jakobsson, M. & Rosenberg, N. A. CLUMPP: A cluster matching and permutation program for dealing with label switching and multimodality in analysis of population structure. Bioinformatics 23, 1801–1806. https://doi.org/10.1093/bioinformatics/btm233 (2007).
Jombart, T. adegenet: A R package for the multivariate analysis of genetic markers. Bioinformatics 24, 1403–1405. https://doi.org/10.1093/bioinformatics/btn129 (2008).
Bouckaert, R. et al. BEAST 2.5: advanced software platform for Bayesian evolutionary analysis. PLoS Comp Biol. 15, e1006650. https://doi.org/10.1371/journal.pcbi.1006650 (2019).
Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312–1313. https://doi.org/10.1093/bioinformatics/btu033 (2014).
Miller, M. A., Pfeiffer, W. & Schwartz, T. In 2010 Gateway computing environments workshop (GCE). 1-8.
Shinzato, C. et al. Using the Acropora digitifera genome to understand coral responses to environmental change. Nature 476, 320–323. https://doi.org/10.1038/nature10249 (2011).
Green, R. E. et al. A draft sequence of the Neandertal genome. Science 328, 710–722. https://doi.org/10.1126/science.1188021 (2010).
Malinsky, M., Matschiner, M. & Svardal, H. Dsuite - Fast D-statistics and related admixture evidence from VCF files. Mol. Ecol. Res. 21, 584–595. https://doi.org/10.1111/1755-0998.13265 (2021).
Li, H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. ArXiv 1303 (2013).
Bongaerts, P. et al. Sharing the slope: Depth partitioning of agariciid corals and associated Symbiodinium across shallow and mesophotic habitats (2–60 m) on a Caribbean reef. BMC Evol. Biol. https://doi.org/10.1186/1471-2148-13-205 (2013).
Flot, J. F., Magalon, H., Cruaud, C., Couloux, A. & Tillier, S. Patterns of genetic structure among Hawaiian corals of the genus Pocillopora yield clusters of individuals that are compatible with morphology. C. R. Biol. 331, 239–247 (2008).
Venables, W. N. & Ripley, B. D. Modern Applied Statistics with S. (Springer, 2002).
O'Leary, M. A. & Kaufman, S. G. MorphoBank 3.0: Web application for morphological phylogenetics and taxonomy. (2012).
Venn, A. A., Weber, F. K., Loram, J. E. & Jones, R. J. Deep zooxanthellate corals at the high latitude Bermuda Seamount. Coral Reefs 28, 135–135 (2009).
Fricke, H. W. & Meischner, D. Depth limits of Bermudan scleractinian corals: a submersible survey. Mar. Biol. 88, 175–187 (1985).
Goodbody-Gringley, G. et al. Irradiance driven trophic plasticity in the coral Madracis pharensis from the Eastern Mediterranean. Sci. Rep. 14, 3646. https://doi.org/10.1038/s41598-024-54217-3 (2024).
Quattrini, A. M. et al. Mito-nuclear discordance within Anthozoa, with notes on unique properties of their mitochondrial genomes. Sci. Rep. 13, 7443. https://doi.org/10.1038/s41598-023-34059-1 (2023).
Mayr, E. Animal Species and Evolution (Harvard University Press, 1963).
Currat, M., Ruedi, M., Petit, R. J. & Excoffier, L. The hidden side of invasions: Massive introgression by local genes. Evolution 62, 1908–1920. https://doi.org/10.1111/j.1558-5646.2008.00413.x (2008).
Gijsbers, J. C. et al. Global phylogenomic assessment of Leptoseris and Agaricia reveals substantial undescribed diversity at mesophotic depths. BMC Biol. 21, 147. https://doi.org/10.1186/s12915-023-01630-1 (2023).
Moran, B. M. et al. The genomic consequences of hybridization. Elife https://doi.org/10.7554/eLife.69016 (2021).
Cairns, S. D., Jaap, W. C. & Lang, J. C. in Gulf of Mexico Origins, Waters, and Biota. Biodiversity (eds D. L. Felder & D. K. Camp) 333-347 (Texas A and M Press, College Station Texas, 2009).
Knowlton, N., Mate, J. L., Guzman, H. M., Rowan, R. & Jara, J. Direct evidence for reproductive isolation among the three species of the Montastraea annularis complex in Central America (Panama and Honduras). Mar. Biol. 127, 705–711 (1997).
Grinblat, M. et al. Biogeography, reproductive biology and phylogenetic divergence within the Fungiidae (mushroom corals). Mol. Phylogen. Evol. 164, 107265. https://doi.org/10.1016/j.ympev.2021.107265 (2021).
Bongaerts, P. et al. Adaptive divergence in a scleractinian coral: physiological adaptation of Seriatopora hystrix to shallow and deep reef habitats BMC. Evol. Biol. 11, 303. https://doi.org/10.1186/1471-2148-11-303 (2011).
Diekmann, O. E., Olsen, J. L., Stam, W. T. & Bak, R. P. M. Genetic variation within Symbiodinium clade B from the coral genus Madracis in the Caribbean (Netherlands Antilles). Coral Reefs 22, 29–33 (2003).
Harrison, P. L. in Coral Reefs: An Ecosystem in Transition (eds Zvy Dubinsky & Noga Stambler) 59-85 (Springer Netherlands, 2011).
Prada, C. & Hellberg, M. E. Speciation-by-depth on coral reefs: Sympatric divergence with gene flow or cryptic transient isolation?. J. Evol. Biol. 34, 128–137. https://doi.org/10.1111/jeb.13731 (2021).
Teske, P. et al. Thermal selection as a driver of marine ecological speciation. Proc. of the Royal Society B 286 20182023, https://doi.org/10.1098/rspb.2018.2023 (2019).
Quattrini, A. M., Baums, I. B., Shank, T. M., Morrison, C. L. & Cordes, E. E. Testing the depth-differentiation hypothesis in a deepwater octocoral. Proc. of the Royal Society B-Biological Sciences 282 UNSP 20150008 https://doi.org/10.1098/rspb.2015.0008 (2015).
Acknowledgements
We would like to thank Mark Vermeij, Norbert Englebert, Kyra Hay and Carlos Winterdaal for logistical support during fieldwork, and Francesca Benzoni and Roberto Arrigoni for gently sharing ATP8 data from Madracis. We thank Adriaan ‘‘Dutch’’ Schrier, Laureen Schenk and the “Curasub” submersible team at Substation Curaçao: Barbara van Bebber, Bruce Brandt, Tico Christiaan, Rob Loendersloot, Barry Brown, Manuel Jove and Joe Oliver. The study is an ‘‘Ocean Heritage Foundation/Curaçao Sea Aquarium/Substation Curaçao’’ (OHF/CSA/SC) contribution. Submersible dives were funded by ‘‘The Explorers Club - Eddie Bauer Grant for Expeditions’’. The study was conducted as part of the “Catlin Seaview Survey” funded by Catlin Group Limited and the Global Change Institute. JAH was supported by a National Science Foundation (NSF) Ocean Sciences (OCE) Postdoctoral Research Fellowship (PRF) under grant no. OCE-2126069 and a generous private donation from Embi Tec. The authors thank two anonymous reviewers for their comments and suggestions.
Funding
National Science Foundation,OCE-2126069.
Author information
Authors and Affiliations
Contributions
PRF and PB conceived or designed the study; GGG acquired samples from Bermuda. PB, PRF and KRWL acquired the samples and prepared the ddRAD libraries; PB and PRF performed the bioinformatics; PB, PRF, JH, HS and PM performed genomic or morphological analyses; PRF, PB and JH wrote the manuscript; all authors edited the manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional Information
The original online version of this Article was revised: The original version of this Article contained errors in Figure 2. Full information regarding the corrections made can be found in the correction for this Article.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Frade, P.R., Hoey, J.A., Goodbody-Gringley, G. et al. Hybridization as driving force for cryptic species diversity in the Caribbean coral genus Madracis. Sci Rep 15, 33359 (2025). https://doi.org/10.1038/s41598-025-17038-6
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-17038-6
