
Abstract
Klebsiella pneumoniae carbapenemase (KPC) is a frequent and widespread carbapenemase, with over 260 variants identified. While KPC often evolves resistance to ceftazidime-avibactam, cefiderocol remains a key treatment option. Some variants, such as KPC-33 (D179Y), reduce cefiderocol susceptibility, but typically with only modest MIC increases. However, KPC’s genetic adaptability raises concern that further mutations could lead to high-level resistance, compromising cefiderocol’s efficacy. To anticipate this risk, we explored the mutational potential of blaKPC-2, blaKPC-3, and blaKPC-33 using random mutagenesis followed by 10-day selection under increasing cefiderocol pressure and whole genome sequencing. Libraries of 105, 104, and 105 mutants, respectively, yielded isolates with significantly elevated MICs, some exceeding 32 mg/L. All resistant clones shared a phenotype marked by cross-resistance to cefiderocol, ceftazidime, ceftazidime-avibactam, cefixime, and piperacillin, but restored susceptibility to carbapenems and most other β-lactams. Our findings highlight that no single mutation enables KPC to efficiently hydrolyze cefiderocol. Instead, high-level resistance requires a combination of enzymatic mutations and chromosomal alterations—such as disruptions in cirA and ybiX—suggesting a multifactorial and stepwise evolutionary pathway. Notably, ybiX has not previously been associated with cefiderocol resistance. These results underscore the importance of ongoing surveillance to detect emerging cefiderocol resistance in KPC-producing Enterobacterales.
Similar content being viewed by others
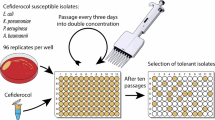


Klebsiella pneumoniae carbapenemase (KPC) stands as the most prevalent carbapenemase enzyme globally, with an endemic presence across North and South America, China, Israel, Greece, and Italy1,2. KPC exhibits a wide-ranging resistance profile, encompassing most beta-lactams including carbapenems and classical beta-lactamase inhibitors. New beta-lactamase inhibitors are effective against KPC beta-lactamase (avibactam, vaborbactam, relebactam …) and showed effectiveness against KPC-producing Enterobacterales3,4,5. However, blaKPC diversified rapidly with 260 clinical variants described to date (June 2025). The clinical success of KPC variants is attributed to intricate molecular mechanisms and a remarkable genetic adaptability, though our understanding of these processes remains incomplete6,7. While the combination of ceftazidime-avibactam (CZA) is recommended as the first-line treatment for systemic infections caused by KPC-producing Enterobacterales (KPE), many KPC variants have been reported to be resistant to CZA6,8,9. Recent studies also highlight the emergence of clinical variants that confer cross-resistance to newly introduced therapeutic agents such as meropenem-vaborbactam (MEV) or imipenem-relebactam (IMR)10,11,12. In this context, cefiderocol has become a therapeutic option since its introduction in 20209.
Cefiderocol is a siderophore cephalosporin sharing side chains similar to those of both cefepime and ceftazidime conferring stability against most beta-lactamases13,14,15. The chlorocatechol group binds covalently to extracellular Fe3 + , allowing its fast and active transport into the bacterial periplasm by iron TonB-dependent transporters (TBDT) such as FepA, Fiu or CirA13,14,16. However, recently cross-resistance to both CZA and cefiderocol has been described in some KPC variants and notably in KPC-33 and KPC-31 which are derived from the KPC-2 and KPC-3 β-lactamases by the addition of the D179Y substitution which is located within the omega loop, a mutational hotspot17,18,19,20,21. The D179Y mutation is observed in 26 out of 242 (11%) of KPC variants with KPC-33 and KPC-31, being prominent in global epidemiology6. The structural analogy between cefiderocol and ceftazidime could be one of the origins of this cross-resistance, corroborated by the identification of common molecular and enzymatic mechanisms18,22. While described mutants currently confer a modest increase in cefiderocol MICs—close to EUCAST breackpoints (2 mg/L), it is possible that the natural evolution of KPC-2 or one of its variant, along with the accumulation of mutational events with time, could further increase the level of cefiderocol resistance in the future preventing it’s potential use in therapeutics.
In this context, we aimed to explore the evolutionary potential of three prevalent KPC variants—blaKPC-2 and blaKPC-3, the most common globally, and blaKPC-33, which is associated with elevated cefiderocol MICs—to develop high-level cefiderocol resistance. To do so, we constructed mutant libraries via random mutagenesis to evaluate whether combinations of mutations could be selected that significantly increase cefiderocol resistance.
Results
Mutant libraries from random mutagenesis experiment
Mutant libraries were generated by random mutagenesis, yielding approximately 1.3 × 105, 3 × 104 and 1 × 105 colonies for blaKPC-2, blaKPC-3 and blaKPC-33 respectively, with each colony representing a distinct variant. Colonies were scraped off the agar into LB-glycerol (40%), mixed and stored at − 80 °C. The average mutagenesis efficiency, determined from 100 randomly selected colonies, was 3.6 substitution-type mutations per allele.
In vitro enrichment experiment and selection of mutants
Cefiderocol MICs of the ancestral variants blaKPC-2, blaKPC-3 and blaKPC-33 were 0.25, 0.12, and 2 mg/L respectively. The increase of MICs over the ten-day exposure period is shown in Fig. 1. By day 10, an increase in MICs was observed for all three exposed mutant libraries: > 32, 2 and 8 mg/L corresponding to an increase by > 128-fold, 17-fold, and fourfold respectively for the libraries derived from blaKPC-2, blaKPC-3 and blaKPC-33. Starting from the well that grew at the highest cefiderocol concentration, we re-isolated 10 µL of culture by plating on LB-tetracycline. Ten colonies from each of these enrichment experiments were randomly selected for characterization.

Results of the enrichment experiment of mutant libraries under increasing concentrations of cefiderocol (10 days of evolution).
Phenotypic characterization: Antibiotic susceptibility testing allowed the identification of a common resistance phenotypic profile for all tested mutants after the ten-day exposure to increasing concentrations of cefiderocol. Mutants were all resistant to ceftazidime and CZA, cefixime, and piperacillin but were susceptible to carbapenems (Table 1, Supplementary table 1 and Fig. 2). So, mutations enriched in our experiment (especially from blaKPC-2 and blaKPC-3) modified the resistance capacity of the KPC protein. Also, the cefiderocol MICs increased differently depending on the initial variant (Table 1).

Kiviat diagram showing the distribution of mean inhibition zone diameters (mm) for the principal antibiotic molecules tested on selected variants (blue—variants enriched from KPC-2 (D179Y-D209V), orange—variants from KPC-3 (L169P), grey variants from KPC-33 (R6H, F20L + /- G291S) as well as ancestral KPC-2 variant (yellow). AMX, Amoxicillin; AMX/CLAV, Amoxicillin/clavulanate; PIP, Piperacillin; PTZ, Piperacillin/tazobactam; AZT, Aztreonam; FEP, Cefepime; FIX, Cefixime; CTX, Cefotaxime; FOX, Cefoxitin; CAZ, Ceftazidime; CZA, Ceftazidime-avibactam; IMP, Imipenem; MER, Meropenem Diameters: millimeters; Blue points: EUCAST Breakpoints.
Genotypic characterization: Sanger sequencing was performed on the 10 isolated colonies from each enrichment experiment. From blaKPC-2mutants’ library, we identified only the combination of D179Y and D209V mutations on all tested mutants (n = 10) which had cefiderocol MIC > 32 mg/L (corresponding to a > 128-fold increase compared to wild-type blaKPC-2). From blaKPC-3 (H274Y), we identified only the L169P mutation (n = 10) with cefiderocol MIC of 2 mg/L (17-fold increase compared to wild-type blaKPC-3) and from blaKPC-33 (D179Y) mutants’ library, we identified either the F20L (n = 6), the F20L-G291S (n = 2) or the R6H-F20L (n = 2) mutations, all conferring cefiderocol MIC of 8 mg/L corresponding to a fourfold increase compared to blaKPC-33.
To assess the direct impact of these mutations in the blaKPC on cefiderocol and other beta-lactams resistance, mutated pBR322 plasmids were extracted from selected strains. Antibiotic susceptibility testing and cefiderocol MIC were then determined after re-electroporating the plasmids into new E. coli TOP10. A decrease in cefiderocol MICs was observed following re-electroporation showing that the KPC mutations alone do not fully explain the increase in cefiderocol MIC suggesting the contribution of other molecular mechanisms within the genomes to confer high levels of resistance to cefiderocol (Table 1).
Exploration of associated molecular mechanisms impacting cefiderocol resistance
To explore the presence of underlying molecular mechanisms, WGS was performed on one representative isolate of each mutant libraries (KPC-2n2 from KPC-2, KPC-3n4 from KPC-3 and KPC-33n6 from KPC-33) (Raw reads and fasta are available PRJNA1262690).
No newly acquired mutations were detected in genes encoding porins, iron TonB-dependent transporters (TBDT) such as fepA, fiu or cirA or penicillin-binding proteins (PBPs) (Supplementary table 2).
In comparison to the reference genome, all analyzed genomes exhibited at least one insertion in the cirA gene, resulting in a truncated protein. In details, insertion sequences (IS) identified with ISFinder as ISKpn72, ISKpn8 and IS1X2 were present in cirA gene of KPC-2n2, KPC-3n4 and KPC-33n6 isolates respectively. The integration occurred in the same region of the gene but at different sites (Supplementary Fig. 1).
In the KPC-2n2 we observed the insertion of another IS, ISKpn8 in the intergenic region upstream of ybiX that is part of an operon with fiu (Supplementary Fig. 1).
Deciphering the impact of each gene in cefiderocol MICs
To assess the contribution of cirA, ybiX and the combination of cirA/ybiX in the increase of cefiderocol resistance, MICs were assessed on Keio (E. coli K12) WT, and the same strain with ΔcirA, ΔybiX, or ∆cirA/∆ybiX knockout mutants (the latter has been constructed for this study) with and without the pBR322-KPC-2n2 plasmid containing blaKPC-2 (D179Y-D209V), and pBR322-KPC-33 (D179Y) (Table 2).
Cefiderocol MICs were initially very low (< 0.03 mg/L) in Keio WT and ΔybiX strains, but higher in ΔcirA (0.12 mg/L). Introducing blaKPC-33 (D179Y) or blaKPC-2 (with D179Y-D209V) increased MICs to 0.5 mg/L in Keio WT, 0.5–1 mg/L in ΔybiX, and 2–4 mg/L in ΔcirA. The ΔcirA/ΔybiX double mutant showed a dramatic MIC rise—from 0.5 mg/L without plasmid to > 32 mg/L with either KPC variant (Table 2).
Discussion
Using random mutagenesis to generate extensive diversity in three KPC alleles: blaKPC-2, blaKPC-3, and blaKPC-33 (the latter being already described with increased cefiderocol MICs18) we evaluated mutational possibilities to confer high-level resistance to cefiderocol. Our mutagenesis allowed the generation of approximately 104–105 mutants per allele (with mean 3.6-mutations/alleles), corresponding in total to about 3–4 × 105 mutations. The hypothesis driving this work is that the accumulation of mutations in blaKPC could enhance the cefiderocol resistance conferred by the KPC enzyme. A scenario similar to what was observed with TEM β-lactamases in response to ceftazidime a few decades earlier23.
Enrichment experiment under increasing concentrations of cefiderocol resulted in MIC increases, reaching > 32, 2, and 8 mg/L for libraries derived from blaKPC-2, blaKPC-3, and blaKPC-33, respectively. Mutants selected at the highest concentrations showed phenotypic convergence, notably a restored susceptibility to carbapenems, suggesting an evolutionary trade-off: mutations conferring cefiderocol resistance may also increase sensitivity to carbapenems and influence susceptibility to most other β-lactams. Overall, the mutants exhibited cross-resistance to ceftazidime and ceftazidime-avibactam, along with increased cefiderocol MICs. The cross-resistance to CZA and cefiderocol has recently been documented in the literature among clinical KPC variants, sometimes associated with other resistance mechanisms or beta-lactamases17,24,25. Although these variants can be selected during CZA treatment, our results raise the concerning possibility of rapid selection of CZA resistant-mutants also under cefiderocol treatment21,24. This likely stems from shared structural features between ceftazidime and cefiderocol and a similar hydrolytic mechanism by KPC variants for the two drugs22.
Among mutants derived from blaKPC-2 or blaKPC-3, only a single clone was enriched and subsequently dominated the population, as evidenced by identical sequences in all analysed mutants (10/10 clones). The L169P substitution selected from blaKPC-3 is associated with a moderate increase in cefiderocol MICs. This mutation has been previously reported in five clinical KPC variants and is often associated with other mutations (blaKPC-35, blaKPC-46, blaKPC-48, blaKPC-138, and blaKPC-155) exhibiting varying resistance phenotypes to beta-lactams6,26,27,28. Interestingly, the D179Y mutation, frequently observed in clinical KPC variants and known to increase cefiderocol resistance, was selected in blaKPC-2 alongside the D209V mutation. The catalytic efficiency of KPC-2 has been described to be approximately 20 times lower for cefiderocol compared to ceftazidime22. Nevertheless, the acquisition of the D179Y substitution significantly enhances the hydrolytic capacity of KPC-2 towards cefiderocol by increasing the enzyme’s affinity for this antibiotic substrate22. The D209V mutation is not located directly on the essential loops involved in the conformation of the active site of the enzyme and has not been previously reported. Mutant libraries derived from blaKPC-33 (D179Y) did not yield other mutations that further enhanced the resistance capabilities of the KPC enzyme underscoring the pivotal role of D179Y in cefiderocol resistance and the constrained evolutionary path toward high-level resistance in KPC enzymes.
To disentangle the respective contributions of KPC mutations and chromosomal mechanisms to cefiderocol resistance, we assessed their individual impacts. For that, the impact of newly acquired KPC mutations on cefiderocol resistance was subsequently assessed by isolating the plasmid containing the mutated blaKPC from enriched mutant and reintroducing it into a naïve E. coli TOP10 strain, devoid of prior evolutionary changes. We found that the resulting cefiderocol MICs attributed to the mutated KPC proteins, were elevated compared to the ancestral variants, though the increases were more moderate (maximum an eightfold increase with the plasmid from the KPC-2n2 mutant) (Table 1). These findings suggest that additional molecular mechanisms are likely at play, and that chromosomal mutations can help achieving high-level cefiderocol resistance in these KPC variants. We focused on the most common variants (blaKPC-2, blaKPC-3, and blaKPC-33), which most likely to diversify. However, we cannot exclude the possibility that the evolutionary trajectory would have been different starting with other KPC backgrounds.
A comprehensive analysis of the mutants via WGS and results from transformation of knockout mutants with plasmids harboring KPC variants strongly suggest the involvement of the cirA gene in the acquisition of in vitro resistance in our mutants. Indeed, the introduction of the pBR322-KPC-2n2 plasmid into Keio WT cells raised the cefiderocol MIC from 0.03 to 0.5 mg/L corresponding to a 16-fold increase, showing a direct impact of the D179Y and D209V mutations on cefiderocol, though the MIC remained below the EUCAST resistance threshold29. Deletion of cirA alone raised the MIC fourfold (0.12 vs. < 0.03 mg/L), consistent with impaired cefiderocol uptake30. Combining cirA deletion with KPC-2n2 further increased the MIC to 2 mg/L—16-fold above ΔcirA and over 66-fold above WT—highlighting a synergistic effect between chromosomal and plasmid-mediated mechanisms. These results are consistent with existing data, as TBDT are the primary targets for in vitro and in vivo resistance against cefiderocol in Enterobacterales31. Recent studies have demonstrated the impact of loss of function of CirA in acquiring high levels of resistance, particularly in Enterobacterales producing New Delhi Metallo-beta-lactamase (NDM)16,32. Moreover, a Chinese multicentric study suggested that restoring normal CirA function can reverse resistance in NDM-producing E. coli32. However, few studies reported the selection of mutations targeting the cirA gene that confer significant cefiderocol resistance in KPC-producing E. coli. Our results suggest that the production of mutated KPC-2, KPC-3, and KPC-33 by E. coli strains may act synergistically in combination with mutations affecting the cirA gene to reach high cefiderocol MIC.
In parallel, WGS revealed the insertion of ISKpn8 upstream of the ybiX genes of KPC-2n2 which is described here for the first time as associated with cefiderocol resistance. This gene encodes a putative iron uptake factor and operates within an operon alongside fiu, which codes for the outer membrane iron-catecholate transporter, both regulated by a common transcriptional regulator: the Ferric Uptake Regulator (Fur)33. ybiX is homologous to piuC in Pseudomonas aeruginosa, a gene implicated in siderophore uptake34 which has been linked to in vitro resistance against siderophore-based antibiotics35. A similar mechanism is likely responsible for the observed resistance in the E. coli mutant, supporting the hypothesis that ybiX is implicated in the iron acquisition pathway. Recent literature indicates that the in vitro resistance level to cefiderocol is influenced by the contribution of each key TBDT involved in antibiotic substrate transport, and that the combined loss of Fiu and CirA function in E. coli significantly increase the MIC30. Here, the KPC-2n2 strain exhibited the highest cefiderocol MIC—32 mg/L and is the one that combined an insertion in both cirA and upstream of ybiX (distancing the promoter and thereby impacting expression) genes alongside with the expression of pBR322-KPC-2 with the D179Y and D209V mutations. The double inactivation likely causes a strong restriction in cefiderocol uptake, explaining the marked increase in MIC (Fig. 3).

Interplay between blaKPC mutations and siderophore transport deficiencies driving high cefiderocol MICs.
Conclusion: In this study, we explored how libraries of mutants of blaKPC-2, blaKPC-3, and blaKPC-33 could adapt over a ten-day cefiderocol treatment. The resulting enriched mutants displayed similar phenotypes, including cross-resistance to both ceftazidime-avibactam and cefiderocol but with increased susceptibility to carbapenems and most other beta-lactams. While KPC mutations alone do not lead to high-level cefiderocol resistance in our experiment, we could hypothesize that they facilitate bacterial survival under cefiderocol treatment, creating opportunities for alternative resistance mechanisms, such as chromosomal mutations, to emerge. This intricate interplay between blaKPC mutations and siderophore transport deficiencies (Fig. 3) underscores the importance of integrated therapeutic and surveillance approaches to mitigate resistance emergence and maintain effective treatment options. These findings provide critical insights into the resistance mechanisms of cefiderocol in KPC-producing E. coli. They suggest that the evolutionary pathway to high-level cefiderocol resistance is not straightforward, relying on both enzymatic mutations and chromosomal adaptations.
Methods
KPC variants and mutagenesis experiments
blaKPC-2, blaKPC-3 (H274Y mutation) and blaKPC-33 (D179Y mutation) were cloned into the same pBR322 backbone (a low-copy-number plasmid ~ 15–20 copies/cell) using the native bla promoter and ribosome binding site, ensuring consistent transcriptional and translational context across constructs. They were sourced from our existing collection36 and used as templates for mutagenesis experiments. To explore KPC overall evolvability and diversity through random point mutations across the blaKPC gene, we employed a PCR protocol using error-prone DNA polymerase (Mutazyme II DNA polymerase, GeneMorph II Random Mutagenesis Kit, Agilent Technologies). The protocol was performed as specified by the manufacturer and optimized to achieve mutation frequency of around 5 nucleotides changes per KPC genes (with a size close to one kb). Primers KPC-PCRmuta-F (5′-TAACCCTGATAAATGCTTCAATAATATTGAAAAAGGAAGAGTATG-3′) and KPC-PCRmuta-R (5′-TAAATCAATCTAAAGTATATGAGTAAACTTGGTCTGACAGTTA-3′) were used to amplify the blaKPC gene. The mutated genes were then cloned using Gibson assembly (NEBuilderⓇ HiFi DNA Assembly, New England Biolabs) at a 5:1 ratio into the pBR322 plasmid, which had been pre-amplified with pBR322-pr-Gibson-F (5′-CATACTCTTCCTTTTTCAATATTATTGAAGCATTTATCAGGGTTA-3′) and pBR322-pr-Gibson-R (5′-TAACTGTCAGACCAAGTTTACTCATATACTTTAGATTGATTTA-3′) primers and purified using the QIAquick Gel Extraction Kit (Qiagen). After assembly, mutated plasmids were transformed by electroporation and expressed in One Shot™ TOP10 Electrocomp™ E. coli (Invitrogen) strains. Selection was performed on LB-tetracycline medium to generate the mutant libraries.
In vitro enrichment experiment
The enrichment experiment was designed to select for mutants with high-level resistance to cefiderocol. A 107 CFU/mL initial inoculum of the libraries of mutants were exposed to progressively increasing cefiderocol concentrations (ranging from 0.03 and up to 32 mg/L) in an iron-depleted Mueller–Hinton (MH) medium in microdilution plates (with 200 µl total volume). Each well was subcultured daily, allowing for a gradual escalation of cefiderocol concentration up to 32 mg/L. At the end of the ten-day period, viable resistant isolates that grew at the highest cefiderocol concentration were plated on LB-tetracycline medium, from which 10 colonies were randomly selected from each enrichment experiment and isolated for phenotypic and genotypic characterization.
Phenotypic characterization : The beta-lactams antibiotic susceptibility profile of the mutants were assessed both by the disk diffusion method on MH-agar medium and by broth microdilution plates using sensititre plates (ThermoFisher Scientific) in accordance with the last European Committee on Antimicrobial Susceptibility Testing guidelines (EUCAST)29. Additionally, the cefiderocol MIC was determined using the Bruker’s UMIC® kit and CZA MIC was determined using the E-test® method (BioMérieux, Marcy l’Etoile, France) on MH-agar medium.
Genotypic characterization of selected mutants
Sanger sequencing: for the selected mutants, the blaKPC gene carried by pBR322 plasmid was amplified using standard PCR protocol and submitted for Sanger sequencing (GenewizⓇ Europe, Azenta Life Sciences) to identify mutations in the blaKPC gene.
Whole genome sequencing: On ancestral strains as well as on one selected strain per enrichment experiment, genomic DNA extraction was performed using the Genomic DNA|gDNA Isolation Kits (Qiagen) for whole-genome sequencing (WGS). Libraries were prepared using the Illumina DNA Flex kit and sequenced on a NextSeq platform, using a NextSeq 500/550 Mid Output Kit v2.5 (300 cycles) (Illumina).
Bioinformatic analysis: Raw reads were trimmed using Trim Galore v0.4.4_dev and Trimmomatic v0.38 with a Phred score ≥ 20 and a minimum length of 50 bp. The quality of the trimmed reads was verified using FastQC v 0.11.8 and MultiQC v1.7. First, variant detection was performed based on the K12_DH5_alpha reference genome (GCF_002899475.1 NCBI accession). Alignment was carried out using BWA v 0.7.17-r1188, and variants were called using FreeBayes v 1.3.1–16. A custom python script was used to merge and generate a matrix of presence and absence of single nucleotide polymorphisms (SNPs). The pangenome was computed by constructing genomes of the samples with SPAdes v3.15.4. The presence and absence matrix was generated by Panaroo v1.5.0 based on Prokka v1.14 annotations. Both the SNPs and gene matrices were analyzed using an in-house R script to compare variations between cefiderocol resistant evolved strains and ancestral ones. Some insertions escaped detection by FreeBayes, particularly those with minimal overlap. To detect them, PanISa was used to specifically identify such cases and an R script was used to visualize the results.
Determination of the role of mutational events in Cefiderocol resistance
To differentiate between resistance conferred by the selected KPC variants and potential other chromosomal mechanisms, we extracted mutated pBR322-KPC plasmids from the evolved strains, re-electroporated them into new One Shot™ TOP10 Electrocomp™ E. coli, and reassessed the cefiderocol MICs and antibiotic susceptibility testing.
Thanks to genomic analysis, we identified genomic modifications in some genes of the TBDT family (cirA gene and ybiX). To assess the contribution of these genes in Cefiderocol resistance phenotypes, cefiderocol MICs were assessed on strains from the Keio collection as well as on newly constructed deleted mutant37: Keio WT, ΔcirA, ΔybiX, and ∆cirA/∆ybiX knockout mutants before and after transformation with pBR322 containing the mutated blaKPC. For the construction of double-gene deletion mutant, the DNA fragments ∆ybiX::kanamycin cassette (kmfrt) was amplified from E. coli BW25113∆ybiX::Kmfrt Keio strain. After removing the kanamycin cassette from E. coli BW25113∆cirA::Kmfrt by transforming with the recombinase plasmid pCP20, the PCR products of ∆ybiX::kmfrt were introduced on the native chromosomal location under the native promoter in BW25113∆cirA::frt using the λ-red linear recombinase plasmid pKOBEG to generate ∆cirA/∆ybiX construct38,39. The integrity of all cloned fragments and mutations was verified by PCR with specific primers and DNA sequencing (Supplementary table 3).
Data availability
Sequence data that support the findings of this study have been deposited in the European Nucleotide Archive with the primary accession code at PRJNA1262690.
References
Logan, L. K. & Weinstein, R. A. The epidemiology of carbapenem-resistant enterobacteriaceae: The impact and evolution of a global menace. J. Infect. Dis. 215, S28–S36 (2017).
Cui, X., Zhang, H. & Du, H. Carbapenemases in enterobacteriaceae: detection and antimicrobial therapy. Front. Microbiol. 10, 1823 (2019).
Karampatakis, T., Tsergouli, K. & Lowrie, K. Efficacy and safety of ceftazidime-avibactam compared to other antimicrobials for the treatment of infections caused by carbapenem-resistant Klebsiella pneumoniae strains, a systematic review and meta-analysis. Microb. Pathog. 179, 106090 (2023).
Zhanel, G. G. et al. Imipenem-relebactam and meropenem-vaborbactam: two novel carbapenem-β-lactamase inhibitor combinations. Drugs 78, 65–98 (2018).
Zhanel, G. G. et al. Ceftazidime-avibactam: a novel cephalosporin/β-lactamase inhibitor combination. Drugs 73, 159–177 (2013).
Hobson, C. A. et al. Klebsiella pneumoniae carbapenemase variants resistant to ceftazidime-avibactam: An evolutionary overview. Antimicrob. Agents Chemother. 66, e0044722 (2022).
Ding, L. et al. Klebsiella pneumoniae carbapenemase variants: the new threat to global public health. Clin. Microbiol. Rev. 36, e0000823 (2023).
Paul, M. et al. European society of clinical microbiology and infectious diseases (ESCMID) guidelines for the treatment of infections caused by multidrug-resistant Gram-negative bacilli (endorsed by European society of intensive care medicine). Clin. Microbiol. Infect. 28, 521–547 (2022).
Tamma, P. D. et al. Infectious diseases society of America 2022 guidance on the treatment of extended-spectrum β-lactamase producing enterobacterales (ESBL-E), carbapenem-resistant enterobacterales (CRE), and pseudomonas aeruginosa with difficult-to-treat resistance (DTR-P. aeruginosa). Clin. Infect. Dis. 75, 187–212 (2022).
Gaibani, P., Amadesi, S., Lazzarotto, T. & Ambretti, S. Genome characterization of a Klebsiella pneumoniae co-producing OXA-181 and KPC-121 resistant to ceftazidime/avibactam, meropenem/vaborbactam, imipenem/relebactam and cefiderocol isolated from a critically ill patient. J. Glob. Antimicrob. Resist. 30, 262–264 (2022).
Gato, E. et al. In vitro development of imipenem/relebactam resistance in KPC-producing Klebsiella pneumoniae involves multiple mutations including OmpK36 disruption and KPC modification. Int. J. Antimicrob. Agents 62, 106935 (2023).
Lombardo, D., Ambretti, S., Lazzarotto, T. & Gaibani, P. In vitro activity of imipenem-relebactam against KPC-producing Klebsiella pneumoniae resistant to ceftazidime-avibactam and/or meropenem-vaborbactam. Clin. Microbiol. Infect. 28, 749–751 (2022).
El-Lababidi, R. M. & Rizk, J. G. Cefiderocol: A siderophore cephalosporin. Ann. Pharmacother. 54, 1215–1231 (2020).
Sato, T. & Yamawaki, K. Cefiderocol: Discovery, chemistry, and in vivo profiles of a novel siderophore cephalosporin. Clin. Infect. Dis. 69, S538–S543 (2019).
Aoki, T. et al. Cefiderocol (S-649266), A new siderophore cephalosporin exhibiting potent activities against Pseudomonas aeruginosa and other gram-negative pathogens including multi-drug resistant bacteria: Structure activity relationship. Eur. J. Med. Chem. 155, 847–868 (2018).
Jousset, A. B. et al. Rapid selection of a cefiderocol-resistant Escherichia coli producing NDM-5 associated with a single amino acid substitution in the CirA siderophore receptor. J. Antimicrob. Chemother. 78, 1125–1127 (2023).
Poirel, L., Sadek, M., Kusaksizoglu, A. & Nordmann, P. Co-resistance to ceftazidime-avibactam and cefiderocol in clinical isolates producing KPC variants. Eur. J. Clin. Microbiol. Infect. Dis. 41, 677–680 (2022).
Hobson, C. A. et al. Cross-resistance to cefiderocol and ceftazidime-avibactam in KPC β-lactamase mutants and the inoculum effect. Clin. Microbiol. Infect. 27(1172), e7-1172.e10 (2021).
Castillo-Polo, J. A. et al. Outbreak by KPC-62-producing ST307 Klebsiella pneumoniae isolates resistant to ceftazidime/avibactam and cefiderocol in a university hospital in Madrid Spain. J. Antimicrob. Chemother. 78, 1259–1264 (2023).
Amadesi, S. et al. Complete Genome sequence of a klebsiella pneumoniae strain carrying novel variant blaKPC-203, Cross-resistant to ceftazidime/avibactam and cefiderocol, but susceptible to carbapenems, isolated in Italy, 2023. Pathogens 13, 507 (2024).
Giufrè, M. et al. Detection of KPC-216, a novel KPC-3 variant, in a clinical isolate of Klebsiella pneumoniae ST101 Co-resistant to ceftazidime-avibactam and cefiderocol. Antibiotics 13, 507 (2024).
Birgy, A., Nnabuife, C. & Palzkill, T. The mechanism of ceftazidime and cefiderocol hydrolysis by D179Y variants of KPC carbapenemases is similar and involves the formation of a long-lived covalent intermediate. Antimicrob. Agents Chemother. 68, e0110823 (2024).
Salverda, M. L. M., De Visser, J. A. G. M. & Barlow, M. Natural evolution of TEM-1 β-lactamase: Experimental reconstruction and clinical relevance. FEMS Microbiol. Rev. 34, 1015–1036 (2010).
Fröhlich, C., Sørum, V., Tokuriki, N., Johnsen, P. J. & Samuelsen, Ø. Evolution of β-lactamase-mediated cefiderocol resistance. J. Antimicrob. Chemother. 77, 2429–2436 (2022).
Gaibani, P., Ambretti, S., Campoli, C., Viale, P. & Re, M. C. Genomic characterization of a Klebsiella pneumoniae ST1519 resistant to ceftazidime/avibactam carrying a novel KPC variant (KPC-36). Int. J. Antimicrob. Agents 55, 105816 (2020).
Hemarajata, P. & Humphries, R. M. Ceftazidime/avibactam resistance associated with L169P mutation in the omega loop of KPC-2. J. Antimicrob. Chemother. 74, 1241–1243 (2019).
Cano, Á. et al. Use of carbapenems in the combined treatment of emerging ceftazidime/avibactam-resistant and carbapenem-susceptible KPC-producing Klebsiella pneumoniae infections: Report of a case and review of the literature. J. Glob. Antimicrob. Resist. 22, 9–12 (2020).
Naas, T. et al. Beta-lactamase database (BLDB)—structure and function. J Enzyme Inhib Med Chem 32, 917–919 (2017).
European Committee on Antimicrobial Susceptibility Testing. Data from the EUCAST MIC distribution website, last accessed 26 Sept 2024. https://www.eucast.org (2024).
Ito, A. et al. In vitro antibacterial properties of cefiderocol, a novel siderophore cephalosporin, against gram-negative bacteria. Antimicrob. Agents Chemother. 62, e01454-e1517 (2018).
Kriz, R. et al. In vitro resistance development gives insights into molecular resistance mechanisms against cefiderocol. J. Antibiot. https://doi.org/10.1038/s41429-024-00762-y (2024).
Wang, Q. et al. Occurrence of high levels of cefiderocol resistance in carbapenem-resistant escherichia coli before its approval in China: A report from China CRE-network. Microbiol. Spectr. 10, e02670-e2721 (2022).
Seo, S. W. et al. Deciphering Fur transcriptional regulatory network highlights its complex role beyond iron metabolism in Escherichia coli. Nat. Commun. 5, 4910 (2014).
McHugh, J. P. et al. Global iron-dependent gene regulation in Escherichia coli. A new mechanism for iron homeostasis. J. Biol. Chem. 278, 29478–29486 (2003).
McPherson, C. J. et al. Clinically relevant Gram-negative resistance mechanisms have no effect on the efficacy of MC-1, a novel siderophore-conjugated monocarbam. Antimicrob. Agents Chemother. 56, 6334–6342 (2012).
Hobson, C. A. et al. Impact of anticancer chemotherapy on the extension of beta-lactamase spectrum: an example with KPC-type carbapenemase activity towards ceftazidime-avibactam. Sci. Rep. 10, 589 (2020).
Baba, T. et al. Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: The Keio collection. Mol. Syst. Biol. 2, 2006–0008 (2006).
Cherepanov, P. P. & Wackernagel, W. Gene disruption in Escherichia coli: TcR and KmR cassettes with the option of Flp-catalyzed excision of the antibiotic-resistance determinant. Gene 158, 9–14 (1995).
Chaveroche, M. K., Ghigo, J. M. & d’Enfert, C. A rapid method for efficient gene replacement in the filamentous fungus Aspergillus nidulans. Nucleic Acids Res. 28, E97 (2000).
Funding
This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
Author information
Authors and Affiliations
Contributions
Conceptualization: SH, SB, AB; Investigation: SH, YY; Formal analysis: SH, KL, MGM, IEM, YBB; Data curation: KL, MGM, IEM, YBB; Writing—original draft: SH; Writing—review & editing: SB, AB; Supervision: AB All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Hanna, S., La, K., Yoshii, Y. et al. Exploring mutational possibilities of KPC variants to reach high level resistance to cefiderocol. Sci Rep 15, 31312 (2025). https://doi.org/10.1038/s41598-025-17044-8
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-17044-8
