
Abstract
The use of Electronic Nicotine Delivery Systems (ENDS), or vaping devices, has raised concerns about their potential impact on oral health, particularly periodontal disease. While traditional smoking is a well-established risk factor for periodontal disease, the biological and microbial effects of ENDS use are less well understood. Our study examined how vaping and vaping behaviors influence the subgingival plaque microbiome and the associated metabolic pathways that may contribute to oral disease. We enrolled 70 healthy adults aged 18–35, including 48 regular ENDS users and 22 non-vaping controls. ENDS users were categorized by puffing behavior into low, medium, and high flow groups using a validated topography device. All participants underwent periodontal screening and provided saliva and subgingival plaque samples. To evaluate exposure profiles, ENDS users also submitted their personal devices for emissions testing, and volatile organic compounds (VOCs) were collected and analyzed using gas chromatography-mass spectrometry and high-performance liquid chromatography. Compared to non-vapers, ENDS users demonstrated distinct shifts in their oral microbiome, with reductions in beneficial taxa and increased bacteria associated with inflammation and periodontal disease. These changes were more pronounced in high-puff volume users, who also exhibited lower microbial diversity. Functional profiling revealed vaping-associated enrichment of pathways related to lipid metabolism, inflammation, and xenobiotic degradation. Untargeted salivary metabolomics identified metabolic disruptions consistent with these functional shifts. Integrative network analyses incorporating VOC measurements demonstrated correlations between microbial composition, puff volume, and metabolic disruptions, particularly in lipid-regulated and inflammatory pathways. To our knowledge, this is one of the first studies to integrate vaping behavior, oral microbiome profiling, salivary metabolomics, and VOC emissions analysis in a human cohort. These findings suggest ENDS use, especially at higher intensities, may disrupt oral microbial and metabolic homeostasis through both biological and chemical pathways potentially enhancing periodontal disease risk. These patterns point to potential biological and chemical pathways of concern, warranting further investigation and informing public health priorities.
Similar content being viewed by others


Introduction
Electronic Nicotine Delivery Systems (ENDS) are battery-powered devices that heat and aerosolize liquids containing ingredients including propylene glycol, glycerol as humectants, various flavors, and often, nicotine1. Vaping refers to the act of inhaling aerosol generated by ENDS devices, commonly known as e-cigarettes or vape pens. Approximately 7.0% of adults in the United States vaped in 2024, with prevalence as high as 13.6% among young adults between the ages of 18 and 342. Vaping is not without risk, since it exposes the vaper to substances with known adverse health effects including nicotine, particulate matter, volatile organic compounds, and heavy metals3. The extent of exposure is likely influenced by vaping behavior, device, and the constituents of the e-liquid itself. Modifiable factors such as vaping behavior, coil temperature, and e-liquid formula may affect levels of exposure to nicotine, metals, and toxic carbonyls such as formaldehyde, aldehyde, and acrolein4,5.
In recent years, puffing topography assessments have been used to understand individual vaping behavior6. To quantify variations in vaping behavior and determine actual use, puffing topography devices have been utilized and validated for combustible cigarettes7 and ENDS8,9. Commercially available puffing topography devices such as the Clinical Research Support System (CReSS) analyze vaping behavior characteristics using a subject’s own ENDS10. Puffing topography evaluations revealed their utility in assessing real-world vaping behavior, providing important insights into determinants related to differential hazardous aerosol exposure11,12. Specifically, individual variations in puffing topography likely contribute to health effects due to differential puff volumes, puff frequency, and puff duration influencing exposure to toxic ENDS emission components and health related outcomes6.
Recent studies investigating the impact of ENDS use on oral health indicate a potential association between vaping and various oral symptoms and clinical outcomes, including oral mucosal lesions, dental caries, tooth sensitivity, and indicators of periodontal disease such as increased plaque accumulation, deeper probing depths, and greater bone loss13. Periodontal diseases, including gingivitis and periodontitis, are inflammatory conditions affecting the gums and supporting bone14. The initial stage of periodontal disease, known as gingivitis, is a mild, reversible condition characterized by irritation, swelling, redness, and bleeding14. In the advanced stage of the disease, periodontitis, inflammation spreads from the gingival epithelium to the underlying connective tissue, leading to the destruction of collagen fibers15. This breakdown of connective tissue results in clinical attachment loss, marking the transition from gingivitis to periodontitis16. As the disease progresses, osteoclast activation promotes alveolar bone resorption, which can ultimately lead to tooth mobility and loss17. Not only is periodontal disease a major threat to oral health14, research documents an association between periodontal disease and a range of systemic conditions including cardiovascular disease, gastrointestinal and colorectal cancer, diabetes, Alzheimer’s disease, as well as respiratory tract infections and adverse pregnancy outcomes18. While emerging evidence supports a link between ENDS use and periodontal disease, the biological mechanisms underlying this association remain unclear.
Because periodontal disease has an underlying polymicrobial etiology, the oral microbiome is an appropriate target of research to investigate the oral health outcomes of ENDS use. Emerging research suggests vaping may alter the oral microbiome. For example, studies have reported increased relative abundance of Pseudomonadota (formerly Proteobacteria) and shifts in taxa such as Rothia and Haemophilus among ENDS users19,20. While Rothia species are generally considered oral commensals, certain species such as R. mucilaginosa have been implicated as opportunistic pathogens21, and R. dentocariosa has been identified in association with periodontal disease22. These findings may not indicate direct pathogenicity but rather reflect microbial dysbiosis or ecological shifts that could contribute to disease risk23,24.
Importantly, prior investigations have not examined how vaping behavior (e.g., puff volume, duration) modulates the oral microbiome and host metabolic responses, despite evidence that topography-driven exposure differences may influence health outcomes6. Furthermore, while shifts in microbial taxa have been reported19,20, the functional implications of these changes—particularly their linkage to periodontal inflammation via metabolomic pathways—are poorly understood. The present study addresses these gaps by integrating puffing topography data with multi-omics (microbiome and metabolome) profiling to elucidate mechanistic connections between vaping behavior, microbial dysbiosis, and periodontal disease risk. The purpose of this study, therefore, was to identify microbial taxonomic and functional alterations associated with vaping, vaping behaviors, and periodontal disease.
Methods
Participant population and specimen collection
This study, approved by the Advarra Institutional Review Board (Protocol No. H22081), was conducted in full accordance with research guidelines/regulations established by the Declaration of Helsinki. Written informed consent was obtained from all 70 participants who were enrolled at the Chemical Insights Research Institute, UL Research Institutes (Marietta, GA, USA). Participants included 48 ENDS users and 22 non-vaping (and non-smoking) controls, aged 18–35 years, fluent in English, and with at least 16 natural teeth. ENDS users met the following criteria: exclusive ENDS use (no cigarettes or other tobacco use); vaping either daily or on at least 20 of the past 30 days; having used ENDS on at least 100 occasions; and having started ENDS use more than 90 days prior to enrollment. Excluded were individuals using a nicotine patch or currently using any other tobacco products, active marijuana use, pregnancy or lactation, diabetes, HIV infection, current use of immunosuppressant medications or antibiotics (as self-reported within the past 60 days), oral prophylactic procedures in the past 3 months or a household member already enrolled in the study. Participants underwent periodontal screening, saliva collection, puffing topography assessment, and subgingival plaque collection.
Periodontal screening examination
All participants received a screening conducted by a licensed dental hygienist using the Community Periodontal Index of Treatment Needs (CPITN)25,26. While not diagnostic, the CPITN is a widely used screening tool for estimating community periodontal prevention or treatment needs27,28,29,30. Per sextant, the deepest measured pocket and bleeding on probing are used to score the CPITN, with scores ranging from 0 (healthy gingiva) to 4 (pathological pockets ≥ 6 mm). Given the exploratory nature of this study and the need for a rapid, standardized assessment method, full clinical periodontal parameters (e.g., full-mouth probing depths, clinical attachment loss, plaque index) were not collected.
Saliva collection via passive drool technique
All participants were asked to refrain from consuming any food or liquid, other than water, for at least 3 h prior to their visit. Briefly, at their in-person visit, participants were instructed to rinse out their mouth thoroughly with sterile filtered water and were provided Salimetrics® saliva collection aids (Part No. 5016.04). Saliva collection commenced 10–20 min after the mouth rinse. The collection aid was attached to a sterile 1.5 mL screw cap tube, and under guidance from a trained and licensed dental hygienist, participants allowed saliva to pool and then directed it into the collection aid and subsequently into the sterile screw cap tube. After collection, the saliva was spun for 10 min and the supernatants were aliquoted into sterile microcentrifuge tubes, stored in a Halt™ Protease and Phosphatase Inhibitor cocktail (1% v/v, ThermoScientific, Waltham, MA)31,32,33,34, then frozen at -80° until further analysis. Precautions were taken to avoid any sample contamination.
Puffing topography and vaping behavior assessments
Puff volume and other parameters (flow rate, peak flow, inter-puff interval, puff duration, and time of peak) were measured in a subset of participants and used to categorize them by flow rate: low flow vapers (n = 7), medium flow vapers (n = 5), and high flow vapers (n = 5). Puffing topography was assessed near the end of participant recruitment, increasing the sample size from 10 in a previous study35 to 17 in the current analysis. Participants used their personal e-cigarette device inserted into the Clinical Research Support System (CReSS; Pocket, Borgwaldt-KC, Richmond, VA, USA), a system validated to evaluate puffing topography35, using brand-specific adaptors as previously described11,36. After recording each participant’s puffing topography, their device was inserted into an automated vaping generation system programmed to simulate their individual vaping behavior based on their puffing topography profile37. The emissions generated were collected to analyze exposures associated with usage of their specific device. The primary goal of this puffing topography assessment was to classify participants into vaping flow rate categories (low, medium, high) based on puff volume, which served as a grouping variable for downstream microbiome and metabolic analyses as demonstrated in our recent studies where detailed puffing topography data were summarized and published12,38.
Volatile organic compound (VOC) emission analysis
ENDS emissions were collected on Tenax ® TA (300 mg) 60/80 mesh sorbent tube and DNPH cartridge for VOC and aldehyde analysis, respectively, as described previously12. Background ambient levels and conditions were also collected prior to experimentation and accounted for in ENDS VOC emissions analyses. Collected VOC and aldehyde samples were analyzed with thermal desorption gas chromatography/mass spectrometry (TD-GC/MS) and high-performance liquid chromatography (HPLC) at an ISO/IEC 17,025 accredited indoor air quality laboratory. The data collected from the TD-GC/MS were evaluated to ensure they met the guidelines for identification at the limit of quantification (LOQ).
Subgingival plaque collection
Subgingival plaque specimens were collected from all participants using protocols based on the Human Microbiome Project39. Briefly, a sterile curette was inserted into the gingival sulcus to collect subgingival plaque samples. To standardize and ensure broad special coverage of the mouth, one site from each sextant was randomly chosen for sampling, consistent with methods used in prior oral microbiome studies40. Plaque from each sampled site within a participant was pooled to generate a single composite sample per individual. Samples were pooled, preserved in TE buffer (Invitrogen, Carlsbad, CA), and stored at − 80 °C until microbial analysis.
DNA isolation and amplicon synthesis and sequencing
Plaque samples were sent to the Emory Integrated Genomics Core (Atlanta, GA, USA). DNA was isolated using the Qiagen DNeasy Powersoil Kit (cat# 12888, Qiagen, Hilden, Germany). Libraries were prepared with a modified Illumina 16 S Meta-genomic Sequencing Library Preparation workflow41, amplifying the third and fourth hypervariable region (v3/v4) of the 16 S rRNA gene. Final libraries, approximately 630 base pairs (bp) in length, were pooled in equal amounts based on fluorescence quantification, and quantitated via quantitative Polymerase Chain Reaction (qPCR) (cat# KK4824, Kapa Biosystems, Wilmington, MA, USA). The pooled library was sequenced on an Illumina MiSeq using the MiSeq reagent kit v3 600 cycle chemistry (cat# MS-102-3003, Illumina, Inc., San Diego, CA, USA) at a loading density of 6–8 pM with 20% PhiX, generating roughly 20 million, 300 bp paired-end reads.
Quality control and taxonomic annotation
All samples were collected using standard sterile technique. Consistent Powersoil PRO (QIAGEN) reagents were used throughout DNA extraction with a positive control (Escherichia coli bacterial pellet) and DNA extraction reagents alone (negative control) to confirm no contamination in the extraction kit reagent. Additional controls were used during the 16 S library prep process (positive: mock community with known microbiome diversity; negative: sterile water) to validate the library prep.
Amplicon sequence reads in compressed fastq.gz format were produced and checked for quality control with FastQC and MultiQC packages as performed previously42,43. Reads were analyzed with Quantitative Insights Into Microbial Ecology (QIIME2) 2020.234 using DADA2 for data denoising and dereplication44, resulting in an amplicon sequencing variant (ASV) feature table. DADA2 read trimming and truncation parameters of trim-left-f and trim-left-r 30 and trunc-len-f and trunc-len-r 240 were used. Taxonomic assignment used v3-v4 reads and GreenGenes (v13_8, 99% clustered OTUs)45, Silva v13246, and the Human Oral Microbiome Database (HOMD) v15.247 through QIIME2 taxonomy modules48. Phylum-level names have been updated to reflect current phylogenetic nomenclature49 while retaining HOMD designations for clinical traceability. Reference reads were pulled via QIIME2 feature-classifier extract-reads, trimming as above to match the raw reads using v3/v4 primers, then fitted to a naïve-Bayesian classifier using QIIME2 feature-classifier fit-classifier-naïve-bayes, and applied to the ASV feature table using QIIME2 feature-classifier sklearn48. The HOMD database best resolved our Zymo microbial mock community controls (ZymoBIOMICS Microbial Community Standard, Zymo Research, Catalog No. D6300; https://www.zymoresearch.com/products/zymobiomics-microbial-community-standard) and positive controls, which are an 8 bacterial taxa + 2 yeast taxa community control and an E. coli pellet respectively, thus HOMD taxonomic assignment was used and data were exported into .BIOM format for downstream analyses. For taxa where species-level classification was unresolved, the designation ‘NA’ indicates that the ASV could not be confidently assigned to a known species within the reference database (i.e. HOMD). These were retained at the genus level for ecological analyses but excluded from species-specific interpretations.
Microbiome analyses
We selected Shannon and Chao1 indices to assess alpha diversity because they capture complementary aspects of the oral microbiome. Higher Chao1 scores indicate more species present50, while the Shannon index reflects richness and evenness51. Communities dominated by one or a few species exhibit a low Shannon score, while communities with equally distributed abundance exhibit high evenness. Beta diversity, which measures similarity/dissimilarity between communities, was calculated using Bray-Curtis (abundance-weighted) distances. Communities were visualized on a principal coordinates analysis (PCoA) plot based on these distance matrices to assess any clustering by groups of interest. The significance of the cluster differences was assessed using the permutational multivariate analysis of variance (PERMANOVA).
The Linear Decomposition Model (LDM)52 was used to determine differences at the individual ASV level with permutation-based p-values in terms of relative abundance, controlling for a 5% false discovery rate (FDR). Significant p-values were reported as potential signals for future investigation, even if they did not persist after FDR correction. This allowed for the identification of potential biological relevance, as signals that do not survive stringent multiple testing correction might still indicate interesting patterns that warrant further exploration in future studies. Any taxa absent in at least 30% of samples were excluded to improve statistical power and focus the analysis on taxa with a minimum prevalence across the study population, reducing noise from rare or sparsely detected taxa. Taxa, by relative abundance, were tested for differences using LDM’s global test of the microbiome effect. To account for the difference in sample size, random sampling with replacement was implemented to match group sizes and the global test repeated for 1,000 permutations. Sugar intake (self-reported as the average number of teaspoons consumed per day) was included as a covariate, along with sex, age, and race, given their known influence on oral microbiome composition and function53.
Microbiome functional profiling and prediction
Raw 16 S rRNA sequencing read counts, along with DNA sequences and HOMD taxonomic assignments were further used for functional profiling using the Microbiomeanalyst 2.054 and Tax4Fun255. Raw read counts were filtered by removing features with low abundance (count < 4 and prevalence < 30%) and low variance (< 10%). Filtered data were normalized by total sum scaling. KEGG Orthologs (KOs) counts56,57,58 were then predicted and generated using Tax4Fun2 package55. Predicted KOs were used for functional profiling and pathway enrichment analysis, adjusted for sex, age, race, and sugar intake.
Untargeted salivary high-resolution metabolomics
Salivary metabolites were extracted by adding 750 µL of ice-cold methanol into 300 µL of saliva. After centrifugation and removal of supernatants, samples were vacuum dried, reconstituted in 60 µL of HPLC diluent (95% water, 5% acetonitrile, 0.1% formic acid), sonicated, and centrifugated at 16,000 ×g for 8 min. Supernatants were then randomly loaded onto the HPLC autosampler and analyzed using an Agilent 6545 Q-TOF mass spectrometer (Agilent Technologies, Santa Clara, CA) equipped with ESI in positive ionization mode, utilizing a Waters™ ACQUITY UPLC HSS T3 column (100Å, 1.8 μm, 2.1 × 100 mm, Waters Corporation, Milford, MA). Mass data (m/z 70-1000) were obtained using Agilent MassHunter Acquisition software (v. B.06). Mass accuracy was improved by infusing Agilent Reference Mass Correction Solution (G1969-85001). MS/MS was performed in a data dependent acquisition mode. The calculated relative standard deviation for retention time was 0.27%; 8.4% for area, and 10.9% for height, all within the 30% accepted maximum tolerance for relative standard deviation for metabolomic studies using mass spectrometry59. A subset of data (10 ENDS users and 5 controls) was previously assessed in a separate companion study to evaluate ENDS-induced respiratory health effects. In our current study these metabolite alterations have been reanalyzed via a distinct statistical approach (described below) with larger sample sizes to examine microbiome-metabolome correlation and functional analysis.
Microbiome-metabolome functional analysis
Metabolomics has been widely adopted alongside microbiome analysis to decipher the functional connection between microbial communities and the host’s biological metabolic phenotype, in which metabolites play key roles in host-microbiome interactions60. In this study, paired microbiome-metabolome functional analysis was carried out using MicrobiomeAnalyst 2.054. Significant metabolic features were identified based on limma ANOVA, adjusting for race, sex, age, and sugar intake. Pathway enrichment analysis using mummichog61 was applied to the study-specific microbially originated compounds identified from the significant metabolic features to reveal significant pathways (FDR < 0.01, compound hits > 7). Significant microbial taxa at LDM p < 0.05 were then integrated with significant metabolic features for pathway enrichment analysis using mummichog61, focusing on the study-specific microbially originated compounds. Correlations between significant taxa and metabolites were visualized using a distance-based correlation method (correlation threshold > 0.2, p < 0.05)62. Procrustes analysis evaluated correlations between the microbiome and metabolome in a low-dimensional space63. Data Integration Analysis for Biomarker discovery using Latent cOmponents (DIABLO) was used for multi-omics biomarker exploration64.
Network integration analysis using xMWAS. To support the findings from microbiome-metabolome functional analysis, xMWAS65 was used to test the hypotheses in large-scale data sets with high-dimensional omics predictors. xMWAS is a platform-independent topological framework developed for high-dimensional network integration analysis and identification of topological modules of functionally related molecular and biological features, with proven understanding of disease pathophysiology and complex molecular interactions65. Significant taxa from each comparison were integrated with salivary metabolomics data using xMWAS based on the partial least-squares (PLS) regression method65. VOC-metabolome-microbiome network analysis was also performed to explore vaping behavior-exposure-health outcome interactions. A correlation threshold of |r| > 0.4 and p < 0.05 was set for community detection. The network was visualized using Cytoscape (version 3.9.1) with Prefuse Force OpenCL layout. Metabolic features identified from the network analysis were further used for pathway enrichment analysis using mummichog 2.6.061,66.
Statistical analysis
Statistical analyses were conducted using the GraphPad Prism software (version 10.0.2) or R version 4.1.3 (The R Foundation). The final study sample included 70 subgingival specimens. Because the analysis was conducted on an initial subset of 70 participants from a larger ongoing study to generate preliminary data, no formal power calculations were conducted, however, the sample size is consistent with similar exploratory oral microbiome studies31,38, and results will inform future adequately powered analyses. Descriptive statistics were used to illustrate demographic characteristics and ENDS usage patterns. Puff volume data were available for a limited subset of vapers; thus, stratified analyses by puff volume (low, medium, high) were conducted solely for exploratory purposes. Demographic comparisons were not conducted within puff volume groups due to small sample sizes. The subgingival microbiome was assessed through three comparisons: (1) ENDS usage, comparing all vapers to non-vapers; (2) Stratification of a subset of vapers based on puff volume, with groups defined as low (< 90 ml/puff; n = 7)), medium (90–150 ml/puff; n = 5), high (> 150 ml/puff; n = 5); and (3) Stratification of vapers and non-vapers according to CPITN status. T-test or one-way ANOVA with post hoc testing using Fisher’s LSD was performed to determine statistical significance. All multivariate analyses, including Linear Decomposition Model (LDM) testing and microbiome functional profiling, were adjusted for sex, age, race, and sugar intake to control for potential confounding effects of demographic and behavioral factors known to influence oral microbiome composition and metabolism. Values with p < 0.05 were considered significant.
Results
Participant Characteristics
Table 1 provides the basic characteristics of recruited study participants. Significant differences were observed in racial composition between vaper and non-vaper groups, however, there were no significant differences observed in terms of CPITN scores, sex, age, or sugar intake. Nicotine concentrations in participants’ e-liquids were self-reported and ranged from 2.5 to 5%. Reported flavors included fruit, dessert, and tobacco/menthol varieties. Exploratory analyses examining microbiome differences by periodontal status within vapers and non-vapers are presented in Supplementary Text 1, Table S1 and Fig. S1. Pathway enrichment findings among participants with higher CPITN scores are provided in Supplementary Text 2.
Vaping associated alterations in plaque microbiota
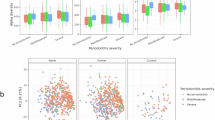
Bioinformatic processing of all samples yielded 3,158 taxa, which were reduced to 153 after filtering for low-abundance taxa. At the phylum level, Bacteroidota (formerly Bacteroidetes) was significantly decreased and Spirochaetota (formerly Spirochaetes) significantly increased in vapers versus non-vapers (p < 0.05). Spirochaetota and Synergistota (formerly Synergistetes) were significantly increased in low- and medium-puff volume vapers, respectively, compared to non-vapers (p < 0.05). Spirochaetota showed a 4.05-fold increase in high-puff volume vapers compared to non-vapers, though this did not reach statistical significance (p = 0.069). Figure 1A illustrates microbiome comparisons between non-vaper and vaper groups, as well as across puff volume groups at the genus level. Six taxa, including Abiotrophia defectiva, Leptotrichia , Veillonella , Stomatobaculum longum, Streptococcus, and Capnocytophaga, were positively correlated with puff volume (Fig. S2). See Supplementary Table 2 for significantly different genera and Fig. S3 for phylum-level differences.

Plaque microbiome 16s rRNA analysis. (A) Shows abundance at genus level (subset shown) for non-vapers vs. vapers, low puff volume, medium puff volume, and high puff volume group. (B) Shows a-diversity (Chao1 index) between non-vapers and puff volume groups. (C) Enriched KEGG pathways associated with significant KOs after covariant adjustment for sex, age, race, and sugar intake.
At the species level (Table 2), 11 taxa differentiated vapers from non-vapers after adjusting for sex, age, race, and sugar intake. Of these, seven were depleted and four enriched in vapers. Six taxa were positively correlated with puff volume (Figure S3). Notably, multiple ASVs could be assigned to a single taxon. For example, two Fusobacterium NAs were identified in the medium-puff volume vapers vs. non-vapers comparison. All six differentiating taxa in the medium-puff volume group were enriched relative to non-vapers. Alpha diversity, as measured by Chao1 index, significantly differed between high-puff volume vapers and both non-vapers and medium-puff volume vapers (Fig. 1B). Beta diversity differences were not identified in any of the comparisons (Fig.S4).
Microbiome functional profiling
Adjusting for sex, age, race and sugar intake, functional profiling identified 11 KEGG pathways56,57,58 that were significantly enriched in vapers, including those related to amino acid metabolism, nucleotide metabolism, and xenobiotics biodegradation (Fig. 1C). Puff volume analyses revealed distinct pathway enrichment across groups (Fig. 1C). Drug metabolism pathways were consistently altered in the overall vaper group and across all puff volume groups compared to non-vapers. Other pathway differences included energy metabolism, lipid regulation, glycan, and benzoate pathways, and various amino acid metabolic processes.
Paired microbiome-metabolome functional analysis
Procrustes analysis showed strong concordance between microbiome and metabolome ordinations across all comparisons (Fig.S5, sum of squares ≥ 0.85, correlation ≥ 0.21, p = 0.001), indicating the functional correlation between these two omics spaces and the potential feedback between plaque microbial communities and the host salivary metabolome. DIABLO analysis identified taxa (e.g., Capnocytophaga NA1, Saccharibacteria TM7 spp. HMT347, Fusobacterium NA1, and Achromobacter xylosoxidans) and metabolites (e.g., carotenoids B-G and acetylornithine) as key features distinguishing vapers and puff volume groups (Fig.S6). The top 10 distinctive signatures for each omics layer are shown in Fig. S6.
As shown in Fig. 2A and Table S3, 30 study-specific microbially derived metabolites annotated with unique m/z and retention time differed significantly between vapers and non-vapers (p < 0.05). Among these, several nicotine-related metabolites—including nicotine and 2,6-dihydroxypseudooxynicotine—were positively correlated with Corynebacterium durum ,and A. xylosoxidans. Additional increases in microbial-origin nicotine metabolites are also presented in Fig. S7. Cobalt-sirohydrochlorin (a bacterial cobalt chelating product) and lipoic acid (a cofactor for TCA cycle enzymes) were highly correlated with C. durum, C. sp. HMT 326, Lachnoanaerobaculum umeaense, and A. xylosoxidans. Other significantly associated metabolites included amino acids (arginine, phenylalanine) and several antibiotics (streptomycin, neomycin A (neamine) and neomycin D (paromamine), and several kanamycin derivatives). Note, a significant decline in amino acids, antibiotics, and associated microbial-origin metabolites was observed (Figs. S8–S9). Decreases in energy and lipid-related metabolites were noted in Fig. S10, while vitamin and cofactor alterations are shown in Fig. S11.
Puff volume-stratified comparisons revealed additional microbiome-metabolome associations (Figs. 2B–D and Table S4–S6). In the low-puff volume vs. non-vaper comparison, 7 unique study-specific metabolites with putative annotation were significantly associated (p < 0.05), including para-aminobenzoic acid. This metabolite, known to regulate folate synthesis and enhance Porphyromonas gingivalis colonization and survival – thereby increasing periodontitis risk67 - was significantly correlated with Serratia marcescens. In the medium-puff volume group vs. non-vaper comparison, 16 unique study-specific metabolites were identified, including nicotine derivatives, xenobiotic metabolites (e.g., xylene/ethylbenzene, para-aminobenzoic acid, cresol/benzyl alcohol, etc.), and succinyl-CoA/methylmalonyl-CoA, benzylsuccinyl-CoA, and sulfoacetyl-CoA. These were associated with taxa including Fusobacterium, Actinomyces gerencseriae, Dialister invisus, and A. xylosoxidans (Table S5). In the high-puff volume group vs. non-vapers, 22 unique metabolites were annotated, including nicotine metabolites, several xenobiotics, and tryptophan and kynurenine metabolites, which were associated with taxa such as Granulicatella adiacens and L. umeaense.
Consistent with microbiome functional profiling results, 15 significantly enriched pathways were identified in the vaper vs. non-vaper comparison (FDR < 0.01, compound hits > 7), including those related to amino acid metabolism (D-amino acids, arginine, proline, histidine, lysine), secondary metabolite synthesis (monobactam, neomycin, kanamycin and gentamicin biosynthesis), polyketide production, O-antigen nucleotide sugar biosynthesis, nucleotide metabolism, and vitamin/cofactor biosynthesis (Fig. 2E). Four pathways (pyrimidine, purine, lysine, and monobactam) overlapped with those identified by microbiome functional profiling in the comparison between vapers and non-vapers (Fig. S12). Additional overlaps emerged in puff volume-stratified analysis: the low-puff volume group shared glyoxylate and dicarboxylate, pyrimidine, and sugar biosynthesis pathways; the medium group shared overlap with degradation pathways (pinene, camphor, geraniol, valine, leucine, isoleucine, and benzoate), and biosynthesis pathways (phenylalanine, tyrosine, and tryptophan); and the high-puff volume group shared enrichment in arginine and proline metabolism.

Paired microbiome-metabolome correlation and functional analysis. (A–D) show significant correlations (noted by * which was determined by distance correlation test) between microbial taxa and metabolic features for non-vapers vs. vapers (A), non-vapers vs. low puff volume group (B), non-vapers vs. medium puff volume group (C), non-vapers vs. high puff volume group (D). (E) Shows functional pathway enrichment associated with significant taxa and significant metabolic features for each comparison after covariant adjustment for sex, age, race, and sugar intake.
Integrative microbiome-metabolome network analysis
To explore host-microbiome metabolic interactions, we used xMWAS65 to integrate significant microbial taxa with metabolomic features. As shown in Fig. 3, vaping group comparisons revealed topological communities linking bacteria and functionally related metabolic pathways. In the vapers vs. non-vapers network (Fig. 3A), 17 significant pathways were enriched across three of seven modules. C. durum (centrality = 0.004) and L. umeaense (centrality = 0.005) were linked to 1133 metabolic features and eight pathways related to inflammation (leukotriene, arachidonic acid, prostaglandin formation, putative anti-inflammatory metabolites formation), bile acid biosynthesis, vitamin A, fatty acid activation, and de novo fatty acid biosynthesis. Porphyromonas sp. HMT 930 (centrality = 1) was associated with 608 metabolic features and nine lipid-centric pathways, including the TCA cycle. Streptococcus NA1 clustered with 315 metabolic features linked to tryptophan and purine metabolism.
In the low-puff volume group (Fig. 3B), 13 metabolic pathways emerged across four network clusters. Lachnoanaerobaculum NA (centrality = 0.72), C. sp. HMT 326 (centrality = 1), and Leptotrichia NA (centrality = 0.998) formed a large cluster (1236 metabolic features) mapping to nine metabolic pathways focused on lipid regulation. Streptococcus mutans (centrality = 0.042) and Actinomyces NA (centrality = 0.051) clustered with 828 metabolic features associated with prostaglandin formation and starch/sucrose metabolism. Campylobacter gracilis was associated with 119 metabolic features, mapped to the alkaloid biosynthesis II pathway, while A. xylosoxidans (centrality = 0.006) and S. marcescens (centrality = 0.006) clustered with 381 metabolic features, associated with the porphyrin pathway.
For the medium-puff volume comparison, 15 metabolic pathways were identified across three modules (Fig. 3C). A. gerencseriae (centrality = 0.044) was correlated with 179 metabolic features and pathways related to carbohydrate and lipid metabolism, xenobiotic degradation, vitamin A metabolism, porphyrin metabolism, pyrimidine metabolsim, and glycan biosynthesis. S. marcescens (centrality = 1), Fusobacterium NA1 (centrality = 0.872), Fusobacterium NA2 (centrality = 0.808), and D. invisus (centrality = 0.809) were linked to carnitine shuttle activity, tryptophan metabolism, and fatty acid β-oxidation.
For the high-puff volume group, 26 metabolic pathways were identified for five network communities (Fig. 3D). Streptococcus NA2 (centrality = 1) formed the largest cluster with 2676 metabolic features, and was associated with lipid-regulated pathways, including peroxisomal fatty acid oxidation, carnitine shuttle, and propanoate metabolism. L. umeaense and Selenomonas noxia were associated with 1232 metabolic features and 10 pathways, largely involving lipid metabolism and inflammation. G. adiacens was associated with 1,158 metabolic features and eight pathways, including lipid-regulated pathways, valine, leucine and isoleucine degradation, and glycosylphosphatidylinositol (GPI)-anchor biosynthesis which has been implicated in bacterial colonization and biofilm formation68. Actinomyces NA and D. invisus were associated with 125 metabolic features and four metabolic pathways: valine, leucine and isoleucine degradation, starch and sucrose, dimethyl-branched-chain fatty acid mitochondrial β-oxidation, and caffeine metabolism.

Paired microbiome-metabolome network analysis using xMWAS for non-vapers vs. vapers. (A) Non-vapers vs. Low-puff volume vapers. (B) Non-vapers vs. Medium-puff volume vapers. (C) Non-vapers vs. high-puff volume vapers (D). Pathway enrichment analysis was performed following network analysis using mummichog. Inset shows the network cluster associated with each comparison identified with xMWAS, where red lines indicate positive correlation and blue lines indicate negative correlation. Plots are color-coded to match the taxa and network cluster color scheme. FA, fatty acid; GPI, glycosylphosphatidylinositol.
VOC-metabolome-microbiome network analysis
To explore the vaping behavior-exposure-health outcome continuum, we integrated VOCs, microbiome, and metabolome data using xMWAS. Seven network clusters were identified (Fig. 4), with cluster 1 exhibiting strong puff volume dependency (Fig. 4A). Notable VOCs in this cluster included hexanal, nonanal, 1,2-propanediol, 1,2,3-butanetriol, toluene, butanoic acid, and total VOCs (Fig. 4B). Puff-volume dependent microbial taxa (Fig. 4C) and metabolic features (Fig. 4D) were also enriched in cluster 1, including Streptococcus NA2 and Actinomyces NA, particularly in the comparison between non-vapers and high-puff volume vapers. Pathway enrichment analysis of cluster 1 metabolic features revealed keratan sulfate degradation, selenoamino acid metabolism, and glycosphingolipid biosynthesis. UDP-N-acetyl-D-glucosamine was noted as the hub metabolite, implicated in bacterial surface glycoprotein synthesis and virulence69. As puff volume and VOC exposure increased, levels of folate, propenoyl-CoA, propionyl-carnitine decreased, whereas levels of adenylylselenate, leukotriene C4, taurolithocholate, and chenodeoxycholoyltaurine increased.

Integrative VOC-microbiome-metabolome network analysis using xMWAS for vapers with puff volume. (A) Network clusters associated with xMWAS, where red lines indicate positive correlation and blue lines indicate negative correlation. Heatmap of VOCs (B), microbial taxa (C), and metabolic features (D) associated with cluster 1. (E) Pathway enrichment analysis was performed following network analysis using mummichog using metabolic features from (D). UDP-GlcNAc, UDP-N-acetyl-D-glucosamine; CDCT, Chenodeoxycholoyltaurine; TLC, Taurolithocholate.
Discussion
The increased prevalence of ENDS use in the United States raises concerns about the health risks associated with vaping behavior, particularly regarding the impact of vaping on oral health. Emerging literature suggests ENDS use is associated with specific alterations in the oral microbiome, which may contribute to an increased risk of periodontal disease23,70,71. In this study, we observed that 87.5% of vapers had CPITN scores ≥ 3, compared to 68.2% of non-vapers. While the CPITN is primarily a screening tool27,28,29,30, our study observed a trend of higher scores among vapers compared to non-vapers, suggesting a potential need for periodontal treatment. As Fig. 5 illustrates, this study identified unique alterations in the subgingival plaque microbiome of vapers, with distinct microbial and metabolic signatures linked to puff volume, along with complex host-microbiome metabolic interactions.

High-level summary for overall vaping and puff-volume specific responses in the subgingival plaque microbiome and salivary metabolome.
The taxonomic alterations observed between vapers and non-vapers suggest a shift toward a periodontal disease-associated microbiome, characterized by a reduction in commensal and protective bacteria and an increase in potentially pathogenic taxa. At the phylum level, vapers had decreased Bacteroidota and increased Spirochaetota. At the genus level, commensals such as Capnocytophaga and Streptococcus (protective against periopathogens like P. gingivalis72 were depleted, while Fusobacterium and Saccharibacteria (formerly TM7), known to be associated with inflammatory diseases like periodontal disease73,74, were enriched. At the species level, the depletion of C. durum and increased abundance of the keystone periodontal pathogen, Treponema denticola75, suggests that vaping disrupts the balance of the subgingival microbiome, potentially contributing to periodontal disease. Stratification by CPITN scores confirms that disruptions in the subgingival ecosystem may be a driving force in the development of periodontal disease among vapers (Table S1). Supporting this, the microbial nicotine metabolite 2,6-dihydroxypseudooxynicotine—elevated in vapers—was significantly associated with the depletion of C. durum and enrichment of A. xylosoxidans, suggesting that nicotine metabolism may drive compositional shifts by selecting for stress-tolerant or opportunistic taxa, thus contributing to a disease-promoting microbiome.
Comparisons between non-vapers and vapers by puff volume revealed distinct microbial and metabolic shifts. In low-puff volume vapers, S. marcescens, known to be associated with periodontal disease76, and S. mutans, a known caries-causing pathogen were enriched, while several commensal bacteria were depleted. Paired microbiome-metabolome analyses further identified shifts in metabolites—including lipoate, phenol/benzenol, and UDP-N-acetyl-3-(1-carboxyvinyl)-D-glucosamine—linked to lipid metabolism and microbial glycan biosynthesis. These findings suggest that even low levels of vaping may disrupt host-microbe interactions and key physiologic pathways, potentially contributing to inflammation and oral health risks. Medium-puff volume vapers showed enrichment of taxa associated with periodontal disease and biofilm formation (A. xylosoxidans, S. marcescens, Fusobacterium NA, D. invisus, A. gerencseriae)77,78,79,80,81, as well as alterations in hormone-related pathways, such as androgen and estrogen biosynthesis. High-puff volume vapers exhibited enrichment of Streptococcus NA2, strongly associated with VOCs (e.g., toluene, hexanal, and butanoic acid) and glycan metabolism pathways, suggesting disruption of cellular communication and immune responses82. The depletion of commensals like L. umeaense and G. adiacens, along with vaping-related changes in salivary pH83, oral cavity temperature84, dry mouth85 may further alter the oral environment. Integrative network analyses supported the association between Streptococcus NA2 and glycan metabolism, reinforcing the potential for chronic, high-volume ENDS use to drive microbial and metabolic dysfunction.
Our integrative analyses also highlight unique effects of medium-puff volume vaping. Notably, drug metabolism and benzoate degradation pathways were enriched only in this group, suggesting that medium-level ENDS exposure may reach a threshold sufficient to induce microbial metabolic adaptation—particularly to xenobiotics like nicotine, xylene, and ethylbenzene commonly found in aerosols86. These shifts were accompanied by a moderate reduction in microbial diversity and may reflect the subgingival microbiome’s attempt to buffer toxic exposure through functional compensation. Moreover, our VOC-microbiome-metabolome network analysis identified glycan-related pathways—such as keratan sulfate degradation and glycosphingolipid biosynthesis—linked to microbial taxa and VOCs in a puff volume–dependent manner. These pathways, interconnected through UDP-N-acetyl-D-glucosamine, play critical roles in host-pathogen interactions and may contribute to immune modulation and tissue remodeling87.
While other studies reported higher alpha diversity in the oral microbiome of vapers compared to non-vapers19,71,88, our study did not, possibly due to the use of subgingival plaque compared to saliva or soft tissue swabs in other studies. Though overall alpha diversity did not differ significantly between non-vapers and vapers, high-puff volume vapers had significantly lower alpha diversity compared to non-vapers and medium-puff volume vapers, suggesting a stronger impact of higher puff volume vaping exposure on microbial diversity. This reduced microbial diversity among high-puff volume vapers was matched by decreased functional potential, as reflected by the predicted KOs, likely due to fewer species being available to perform key functions. Despite the lower KO functional potential, xMWAS analysis identified a significant increase in altered metabolic pathways in high-puff volume vapers, likely reflecting the different sensitivities of the two analytical methods and the compensatory activation of alternative metabolic pathways by surviving microbial species in response to the stress induced by high-puff volume vaping. Supporting this was the identification of 26 significantly altered metabolic pathways in high-puff volume vapers, nearly double the number found in low and medium-puff volume vapers, highlighting the significant impact of high-puff volume vaping on both the diversity and function of the subgingival microbiome. These shifts may also drive functional adaptations in the microbial community. Furthermore, increased particulate matter emissions with higher puff volume12, along with elevated levels of leukotriene C4, taurolithocholate, and chenodeoxycholoyltaurine, suggest that chronic exposure may intensify inflammatory responses and contribute to periodontal disease progression89,90,91,92,93.
Periodontal disease is a chronic inflammatory condition associated with subgingival microbial dysbiosis. Consistently, our integrative microbiome-metabolome network analysis revealed that typically beneficial taxa such as C. durum and L. umeaense94,95, were associated with major inflammatory response pathways in vapers, suggesting that ENDS exposure may trigger pathobiont activity from commensals like C. durum and L. umeaense94,95,96. Although their presence does not necessarily imply pathogenicity, xenobiotic exposure from ENDS may modulate the immunological function of these organisms, activating strong immunostimulatory properties that could be integral to periodontal disease development97. Studies have also reported elevated oral inflammation among vapers98, including our own study which demonstrated that inflammatory cytokines (IL-1β, IL-6, TNF-α) were elevated in the saliva samples of vapers compared to non-vapers38.
Collectively, the significant correlation between the subgingival plaque microbiome and the salivary metabolome of vapers overall and across puff-volumes suggests potential feedback mechanisms between microbial communities in the subgingival biofilm and host metabolomes in the saliva. Puff volume-specific microbial and metabolic signatures, including consistent identification of acetylornithine and salivary nicotine, underscore the dose-dependent effect of ENDS use on oral health. Together, these findings suggest that vaping reshapes the oral environment in ways that may promote inflammation and increase the risk of periodontal disease.
Limitations
Several limitations must be acknowledged. First, the sample size, particularly in the stratified puff-volume groups, was small, which may limit the generalizability of our findings. Nonetheless, this exploratory analysis provides preliminary insights and effect size estimates to inform future analyses. Larger studies are needed to confirm our results and to explore the full spectrum of microbial alterations associated with varying levels of vaping and poly tobacco exposure. Second, the cross-sectional design limits the ability to infer causality. Longitudinal studies are necessary to establish temporal relationships between vaping, microbial alterations, and periodontal health outcomes. Our findings highlight specific microbial targets for future longitudinal investigation. Third, periodontal status was assessed using the CPITN, a population-level screening tool that, while validated for estimating treatment needs, does not capture attachment loss or site-specific disease severity. As such, early or localized periodontitis may have been underestimated. Future studies could strengthen periodontal assessments by incorporating full mouth examinations per American Academy of Periodontology guidelines. Fourth, although we adjusted for several potential confounders (e.g., age, sex, race, and sugar intake), unmeasured factors such as oral hygiene practices, broader dietary patterns, and socioeconomic status may also influence the oral microbiome and periodontal outcomes. Fifth, although detailed puffing behavior parameters (e.g., puff duration, number of puffs, inter-puff interval) were recorded using the CReSS system, they were not analyzed or reported due to the small subsample size and the primary analytic focus on flow rate categories derived from puff volume. Future studies with larger samples could more comprehensively evaluate the role of specific vaping behaviors in shaping exposure profiles and biological outcomes. Next, the use of 16 S rRNA sequencing provides limited taxonomic resolution at the species level making it challenging to draw definitive conclusions from the differential abundance testing. Future work will incorporate targeted metagenomics or metatranscriptomic approaches to more precisely characterize these microbial shifts. Finally, while efforts were made to exclude participants who had recently used antibiotics, our screening was based on a 60-day self-report window, which may not fully capture the period required for oral microbiome recovery. This may represent a limitation in interpreting microbiome stability among participants with prior antibiotic use.
Conclusion
Our study highlights significant associations between vaping behavior and alterations in the subgingival plaque microbiome and salivary metabolome, particularly in relation to inflammatory pathways. Vapers exhibited microbial shifts that were closely linked to puff volume, with higher vaping intensity contributing to a depletion of beneficial bacteria and an enrichment of potentially pathogenic taxa. These alterations, along with disruptions in metabolic pathways involving lipids, amino acids, and xenobiotics, provide mechanistic evidence that ENDS use may increase the risk of periodontal disease through complex microbial and metabolic interactions. The integration of microbiome and metabolomic data in this study underscores the need for further research to elucidate the mechanisms driving these changes and to inform future public health interventions aimed at mitigating the oral health risks associated with ENDS use.
Data availability
The datasets analyzed in this article are available in the Sequence Read Archive, [Bioproject # PRJNA1173730 in https://www.ncbi.nlm.nih.gov/sra].
References
McRobbie, H. et al. Effects of switching to electronic cigarettes with and without concurrent smoking on exposure to nicotine, carbon monoxide, and acrolein. Cancer Prev. Res. 8, 873–878 (2015).
National Center for Health Statistics. Percentage of current electronic cigarette use for adults aged 18 and over, United States, 2019–2024. National Health Interview Survey. https://wwwn.cdc.gov/NHISDataQueryTool/SHS_adult/index.html (accessed 9 Aug 2025).
Esteban-Lopez, M. et al. Health effects and known pathology associated with the use of E-cigarettes. Toxicol. Rep. 9, 1357–1368 (2022).
Li, Y. et al. Impact of e-Liquid composition, coil temperature, and puff topography on the aerosol chemistry of electronic cigarettes. Chem. Res. Toxicol. 34, 1640–1654 (2021).
Zhao, D. et al. Effects of e-liquid flavor, nicotine content, and puff duration on metal emissions from electronic cigarettes. Environ. Res. 204, 112270 (2022).
Wadkin, R., Allen, C. & Fearon, I. M. E-cigarette puffing topography: the importance of assessing user behaviour to inform emissions testing. Drug Test. Anal. 15, 1222–1232 (2023).
Ross, K. C. & Juliano, L. M. Smoking through a topography device diminishes some of the acute rewarding effects of smoking. Nicotine Tob. Res. 18, 564–571 (2016).
Cunningham, A. et al. Development, validation and application of a device to measure e-cigarette users’ puffing topography. Sci. Rep. 6, 35071 (2016).
Adams, A. T., Mandel, I., Shats, A., Robin, A. & Choudhury, T. PuffPacket: A platform for unobtrusively tracking the fine-grained consumption patterns of E-cigarette users. In CHI Conference on Human Factors in Computing Systems; Honolulu HI USA, 1–12 (Association for Computing Machinery (ACM), 2020).
De Jesus, S. Smoking Topography. In Encyclopedia of Behavioral Medicine. (ed. Turner, M. G. J. R.) 1–2 (Springer, 2017).
Mikheev, V. B. et al. The application of commercially available mobile cigarette topography devices for E-cigarette vaping behavior measurements. Nicotine Tob. Res. 22, 681–688 (2020).
Jeon, J. et al. The role of puff volume in vaping emissions, inhalation risks, and metabolic perturbations: a pilot study. Sci. Rep. 14, 18949 (2024).
Yang, I., Sandeep, S. & Rodriguez, J. The oral health impact of electronic cigarette use: A systematic review. Nicotine Tob. Res. 50, 97–127 (2020).
About Periodontal. (Gum) Disease https://www.cdc.gov/oral-health/about/gum-periodontal-disease.html
Zhou, M. & Graves, D. T. Impact of the host response and osteoblast lineage cells on periodontal disease. Front. Immunol. 13, 998244 (2022).
Nanci, A. & Bosshardt, D. D. Structure of periodontal tissues in health and disease. Periodontol 2000. 40, 11–28 (2006).
Hienz, S. A., Paliwal, S. & Ivanovski, S. Mechanisms of bone resorption in periodontitis. J. Immunol. Res. 2015. (2015).
Bui, F. Q. et al. Association between periodontal pathogens and systemic disease. Biomed. J. 42, 27–35 (2019).
Chopyk, J. et al. Crotty Alexander LE: compositional differences in the oral microbiome of E-cigarette users. Front. Microbiol. 12, 599664 (2021).
Kumar, P., Clark, P., Brinkman, M. & Saxena, D. Novel nicotine delivery systems. Adv. Dent. Res. 30, 11–15 (2019).
Maraki, S. & Papadakis, I. S. Rothia mucilaginosa pneumonia: a literature review. Infect. Dis. (Lond). 47, 125–129 (2015).
Kataoka, H. et al. Rothia dentocariosa induces TNF-alpha production in a TLR2-dependent manner. Pathog. Dis. 71, 65–68 (2014).
Thomas, S. C. et al. Electronic cigarette use promotes a unique periodontal microbiome. mBio 13, e0007522 (2022).
Park, B. et al. The mediating roles of the oral microbiome in saliva and subgingival sites between e-cigarette smoking and gingival inflammation. BMC Microbiol. 23, 35 (2023).
Ainamo, J. et al. Development of the world health organization (WHO) community periodontal index of treatment needs (CPITN). Int. Dent. J. 32, 281–291 (1982).
Christensen, L. B., Petersen, P. E., Krustrup, U. & Kjøller, M. Self-reported oral hygiene practices among adults in Denmark. Community Dent. Health. 20, 229–235 (2003).
Nazir, M. et al. Global prevalence of periodontal disease and lack of its surveillance. ScientificWorldJournal. 2020, 2146160 (2020).
Tanik, A. Evaluation of the relationship of CPITN and DMFT index of adult patients in Turkey with their demographic characteristics: an epidemiological study. Biotechnol. Biotechnol. Equip. 33, 1626–1634 (2019).
Nijland, N. et al. External validation of a rapid, non-invasive tool for periodontitis screening in a medical care setting. Clin. Oral Investig. 25, 6661–6669 (2021).
Wereszczyński, M., Śmigiel, A., Tomaszewska, I. & Niedźwieńska, A. Investigating the relationship between periodontitis and specific memory processes in the search for cognitive markers of alzheimer’s disease risk. Sci. Rep. 13, 11555 (2023).
Alqahtani, S., Cooper, B., Spears, C. A., Wright, C. & Shannahan, J. Electronic nicotine delivery system-induced alterations in oral health via saliva assessment. Exp. Biol. Med. (Maywood). 245, 1319–1325 (2020).
Adamson, S. X., Wang, R., Wu, W., Cooper, B. & Shannahan, J. Metabolomic insights of macrophage responses to graphene nanoplatelets: role of scavenger receptor CD36. PLoS One. 13, e0207042 (2018).
Xia, L. et al. Pulmonary and neurological health effects associated with exposure to representative composite manufacturing emissions and corresponding alterations in Circulating metabolite profiles. Toxicol. Sci. 193, 62–79 (2023).
Setyabrata, D. et al. Proteomics and metabolomics profiling of meat exudate to determine the impact of postmortem aging on oxidative stability of beef muscles. Food Chem. X. 18, 100660 (2023).
Behar, R. Z., Hua, M. & Talbot, P. Puffing topography and nicotine intake of electronic cigarette users. PLoS One. 10, e0117222 (2015).
Perkins, K. A. & Karelitz, J. L. A procedure to standardize puff topography during evaluations of acute tobacco or electronic cigarette exposure. Nicotine Tob. Res. 22, 689–698 (2020).
Zhang, Q. et al. Characterization of an electronic nicotine delivery system (ENDS) aerosol generation platform to determine exposure risks. Toxics. 11. (2023).
He, X. et al. Multi-Omics assessment of puff Volume-Mediated salivary biomarkers of metal exposure and oxidative injury associated with electronic nicotine delivery systems. Environ. Health Perspect. 133, 17005 (2025).
McInnes, P. & Cutting, M. Manual of procedures for Human Microbiome Project: Core microbiome sampling Protocol A, HMP Protocol# 07 -001, version number: 12.0. Available: hmpdacc org/doc/sops_2/manual_of_procedures_v11 pdf (29 March -last update, 2012, October) 2010. 2010. (2010).
Takeshita, T. et al. Distinct composition of the oral Indigenous microbiota in South Korean and Japanese adults. Sci. Rep. 4, 6990 (2014).
16S Metagenomic Sequencing Library Preparation. https://support.illumina.com/documents/documentation/chemistry_documentation/16s/16s-metagenomic-library-prep-guide-15044223-b.pdf
Yang, I. et al. The oral Microbiome and inflammation in mild cognitive impairment. Exp. Gerontol. 111273 (2021).
Yang, I. et al. Subgingival microbiome in pregnancy and a potential relationship to early term birth. Front. Cell. Infect. Microbiol. 12, 873683 (2022).
Callahan, B. J. et al. DADA2: High-resolution sample inference from illumina amplicon data. Nat. Methods. 13, 581 (2016).
DeSantis, T. Z. et al. Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl. Environ. Microbiol. 72, 5069–5072 (2006).
Quast, C. et al. Glöckner fojnar: the SILVA ribosomal RNA gene database project: improved data processing and web-based tools. 41:D590–D596 (2012).
Chen, T. et al. The Human Oral Microbiome Database: a web accessible resource for investigating oral microbe taxonomic and genomic information. Database. 2010. (2010).
Bolyen, E. et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 37, 852–857 (2019).
Oren, A. & Garrity, G. M. Valid publication of the names of forty-two phyla of prokaryotes. Int. J. Syst. Evol. Microbiol. 71. (2021).
Lemos, L. N., Fulthorpe, R. R., Triplett, E. W. & Roesch, L. F. Rethinking microbial diversity analysis in the high throughput sequencing era. J. Microbiol. Methods. 86, 42–51 (2011).
Magurran, A. E. Measuring Biological Diversity (Wiley-Blackwell, 2013).
Hu, Y-J. & Satten, G. A. Testing hypotheses about the Microbiome using the linear decomposition model (LDM). Bioinformatics (2020).
Angarita-Díaz, M. D. P., Fong, C., Bedoya-Correa, C. M. & Cabrera-Arango, C. L. Does high sugar intake really alter the oral microbiota? A systematic review. Clin. Exp. Dent. Res. 8, 1376–1390 (2022).
Lu, Y. et al. MicrobiomeAnalyst 2.0: comprehensive statistical, functional and integrative analysis of Microbiome data. Nucleic Acids Res. 51, W310–W318 (2023).
Wemheuer, F. et al. Tax4Fun2: prediction of habitat-specific functional profiles and functional redundancy based on 16S rRNA gene sequences. Environ. Microbiome. 15, 11 (2020).
Kanehisa, M. Goto sjnar: KEGG: Kyoto encyclopedia of genes and genomes. 28, 27–30. (2000).
Kanehisa, M., Furumichi, M., Sato, Y., Matsuura, Y. & Ishiguro-Watanabe, M. KEGG: biological systems database as a model of the real world. Nucleic Acids Res. 53, D672–D677 (2025).
Kanehisa, M. Toward Understanding the origin and evolution of cellular organisms. Protein Sci. 28, 1947–1951 (2019).
Koek, M. M., Jellema, R. H., van der Greef, J., Tas, A. C. & Hankemeier, T. Quantitative metabolomics based on gas chromatography mass spectrometry: status and perspectives. Metabolomics. 7, 307–328 (2011).
Bauermeister, A., Mannochio-Russo, H., Costa-Lotufo, L. V., Jarmusch, A. K. & Dorrestein, P. C. Mass spectrometry-based metabolomics in microbiome investigations. Nat. Rev. Microbiol. 20, 143–160 (2022).
Li, S. et al. Predicting network activity from high throughput metabolomics. PLoS Comput. Biol. 9, e1003123 (2013).
Magnúsdóttir, S. et al. Generation of genome-scale metabolic reconstructions for 773 members of the human gut microbiota. Nat. Biotechnol. 35, 81–89 (2017).
Peres-Neto, P. R. & Jackson, D. A. How well do multivariate data sets match? The advantages of a procrustean superimposition approach over the mantel test. Oecologia 129, 169–178 (2001).
Singh, A. et al. Lê Cao KA: DIABLO: an integrative approach for identifying key molecular drivers from multi-omics assays. Bioinformatics 35, 3055–3062 (2019).
Uppal, K., Ma, C., Go, Y. M., Jones, D. P. & Wren, J. xMWAS: a data-driven integration and differential network analysis tool. Bioinformatics. 34, 701–702 (2018).
He, X. et al. Metabolomics of V. Toxicol. Appl. Pharmacol. 459, 116327 (2023).
Kuboniwa, M. et al. Metabolic crosstalk regulates Porphyromonas gingivalis colonization and virulence during oral polymicrobial infection. Nat. Microbiol. 2, 1493–1499 (2017).
Ikeda, E. et al. Japanese subgingival microbiota in health vs disease and their roles in predicted functions associated with periodontitis. Odontology 108, 280–291 (2020).
Naseem, S. & Konopka, J. B. N-acetylglucosamine regulates virulence properties in microbial pathogens. PLoS Pathog. 11, e1004947 (2015).
Charde, P., Ali, K. & Hamdan, N. Effects of e-cigarette smoking on periodontal health: A scoping review. PLoS Glob. Public. Health. 4, e0002311 (2024).
Ganesan, S. M. et al. Adverse effects of electronic cigarettes on the disease-naive oral Microbiome. Sci. Adv. 6, eaaz0108 (2020).
Abranches, J. et al. Biology of oral Streptococci. Microbiol Spectr. 6. (2018).
Bor, B., Bedree, J. K., Shi, W., McLean, J. S. & He, X. Saccharibacteria (TM7) in the human oral microbiome. J. Dent. Res. 98, 500–509 (2019).
Thurnheer, T., Karygianni, L., Flury, M. & Belibasakis, G. N. Species and subspecies differentially affect the composition and architecture of supra- and subgingival biofilms models. Front. Microbiol. 10, 1716 (2019).
Socransky, S. S. & Haffajee, A. D. Periodontal microbial ecology. Periodontology 2000 38, 135–187 (2005).
Vieira Colombo, A. P., Magalhães, C. B., Hartenbach, F. A. & Martins do Souto, R. Maciel Da Silva-Boghossian C: Periodontal-disease-associated biofilm: A reservoir for pathogens of medical importance. Microb. Pathog. 94, 27–34 (2016).
Vishnu, R. A., Alamelu, S., Arun, K. V., Sujitha, P. & Ganesh, P. R. Comparative evaluation of subgingival microbiome in healthy periodontium and gingivitis using next-generation sequencing technology: A case-control study. J. Indian Soc. Periodontol. 26, 224–229 (2022).
Colombo, A. P. et al. Comparisons of subgingival microbial profiles of refractory periodontitis, severe periodontitis, and periodontal health using the human oral microbe identification microarray. J. Periodontol. 80, 1421–1432 (2009).
Ribeiro, A. C., Matarazzo, F., Faveri, M., Zezell, D. M. & Mayer, M. P. Exploring bacterial diversity of endodontic microbiota by cloning and sequencing 16S rRNA. J. Endod. 37, 922–926 (2011).
Vielkind, P., Jentsch, H., Eschrich, K., Rodloff, A. C. & Stingu, C. S. Prevalence of actinomyces spp. In patients with chronic periodontitis. Int. J. Med. Microbiol. 305, 682–688 (2015).
Valour, F. et al. Actinomycosis: etiology, clinical features, diagnosis, treatment, and management. Infect. Drug Resist. 7, 183–197 (2014).
Baum, L. G. & Cobb, B. A. The direct and indirect effects of glycans on immune function. Glycobiology 27, 619–624 (2017).
Cichońska, D., Kusiak, A., Kochańska, B., Ochocińska, J. & Świetlik, D. Influence of electronic cigarettes on selected antibacterial properties of saliva. Int. J. Environ. Res. Public Health. 16, 4433 (2019).
McClelland, M. L., Sesoko, C. S., MacDonald, D. A., Davis, L. M. & McClelland, S. C. The immediate physiological effects of E-Cigarette use and exposure to secondhand E-Cigarette vapor. Respir. Care. 66, 943–950 (2021).
Guo, X., Hou, L., Peng, X. & Tang, F. The prevalence of Xerostomia among e-cigarette or combustible tobacco users: A systematic review and meta-analysis. Tob. Induc. Dis. 21, 22 (2023).
National Academies of Sciences E & Medicine Public Health Consequences of e-cigarettes (National Academies, 2018).
Jan, M. & Arora, S. Glycans in the host–pathogen interaction. In The Glycol. Apple Academic, 157–197 (2021).
Pushalkar, S. et al. Electronic cigarette aerosol modulates the oral microbiome and increases risk of infection. Iscience. 100884. (2020).
Kwon, J. W., Park, H. W., Kim, W. J., Kim, M. G. & Lee, S. J. Exposure to volatile organic compounds and airway inflammation. Environ. Health. 17, 65 (2018).
van der Toorn, M. et al. Critical role of aldehydes in cigarette smoke-induced acute airway inflammation. Respir. Res. 14, 45 (2013).
Dalton, K. R. et al. Microbiome alterations from volatile organic compounds (VOC) exposures among workers in salons primarily serving women of color. Environ. Res. 214, 114125 (2022).
Li, W. et al. Long-term exposure to ambient fine particulate matter and periodontitis: an observational study using nationally representative survey data. J. Clin. Periodontol. 51, 596–609 (2024).
Yang, T. H. et al. Personal exposure to particulate matter and inflammation among patients with periodontal disease. Sci. Total Environ. 502, 585–589 (2015).
Kamer, A. R. et al. Periodontal dysbiosis associates with reduced CSF Aβ42 in cognitively normal elderly. Alzheimers Dement. (Amst). 13, e12172 (2021).
Hedberg, M. E. et al. Lachnoanaerobaculum gen. Nov., a new genus in the lachnospiraceae: characterization of lachnoanaerobaculum Umeaense gen. Nov., sp. Nov., isolated from the human small intestine, and lachnoanaerobaculum orale sp. Nov., isolated from saliva, and reclassification of Eubacterium Saburreum (Prevot 1966) Holdeman and Moore 1970 as lachnoanaerobaculum Saburreum comb. Nov. Int. J. Syst. Evol. Microbiol. 62, 2685–2690 (2012).
Sugihara, K. & Kamada, N. Metabolic network of the gut microbiota in inflammatory bowel disease. Inflamm. Regen. 44, 11 (2024).
Jiao, Y., Hasegawa, M. & Inohara, N. The role of oral pathobionts in dysbiosis during periodontitis development. J. Dent. Res. 93, 539–546 (2014).
Maki, K. A. et al. The role of the oral Microbiome in smoking-related cardiovascular risk: a review of the literature exploring mechanisms and pathways. J. Transl. Med. 20, 584 (2022).
Acknowledgements
This study was supported in part by the Emory Integrated Genomics Core (EIGC) (RRID: SCR_023529), which is subsidized by the Emory University School of Medicine and is one of the Emory Integrated Core Facilities. Additional support was provided by the Georgia Clinical & Translational Science Alliance of the National Institutes of Health under Award Number UL1TR002378. The content is solely the responsibility of the authors and does not necessarily reflect the official views of the National Institutes of Health.This work was also supported by the Emory University Emory Integrated Computational Core Facility (RRID: SCR_023525).
Funding
This study was funded by the NIH-NIDCR 1R56DE031814-01.
Author information
Authors and Affiliations
Contributions
Study design was conceptualized by I.Y., X.H., C.W., and J.S. J.J. and P.C. acquired samples and subject data. R.A. processed raw sequence data through the bioinformatics pipeline, including quality control, taxonomic classification, and diversity analysis. H.C. performed data analysis, including differential abundance testing. X.H. conducted functional and integrative analyses, created figures, and visualized data. Data interpretation was carried out by I.Y., X.H., C.W., and J.S. Initial manuscript preparation and supplementary materials were handled by I.Y. and X.H. J.J., H.C., R.A., P.C., S.W., R.L., and M.B. critically reviewed the manuscript. Revisions and editing were done by I.Y., X.H., C.W., and J.S. All authors read and approved the final and revised manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval and consent to participate
This study was approved by Advarra Institutional Review Board (Protocol No. H22081) and all participants provided informed consent prior to participation.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Yang, I., He, X., Jeon, J. et al. The impact of vaping behavior on functional changes within the subgingival microbiome. Sci Rep 15, 34374 (2025). https://doi.org/10.1038/s41598-025-17121-y
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-17121-y
