
Abstract
This study investigated the prevalence and genomic epidemiology of SARS-CoV-2 variants as part of the TQT-COVID-19 Project, which aimed to expand COVID-19 testing, quarantine, and telemonitoring strategies. A total of 177 samples were analyzed between July 2022 and April 2023 from patients of 17 Primary Health Care (PHC) units in Salvador, Bahia, Brazil. Sequencing was done using Oxford Nanopore MinION technology and bioinformatics analysis for variant typing and phylodynamic spread assessment. Most participants were women (68.5%), under 39 years old (48.9%), used public transportation (66.4%), had no comorbidities (66.1%), presented mild COVID-19 symptoms (40.8%), and had received a second booster dose of a COVID-19 vaccine (88.2%). Nevertheless, 11.8% had never received a COVID-19 vaccine. The most prevalent Omicron subvariants were BA.5 (32.6%), BA.4 (21.7%), B (18.8%), and BQ.1 (26.3%), with other variants like XBB also identified. Vaccinated and unvaccinated individuals had different spike gene mutations. While multiple variant introductions occurred over time, SARS-CoV-2 geographic spread was mainly localized, with occasional rapid spread associated with inter-city travel or containment failures. These findings underscore the need to track viral spread and mutations to develop targeted strategies for breaking transmission chains.
Similar content being viewed by others

Introduction
By July 27, 2025, more than 778 million cases and 7 million deaths from COVID-19 had been recorded globally, with Brazil accounting for approximately 37.7 million cases and 703,005 deaths1. The country’s socioeconomic inequality significantly influenced the distribution of the disease, with disproportionate impacts in more vulnerable regions, where access to healthcare and surveillance was limited2,3. Salvador, the second most populous city in Northeastern Brazil, reported its first case on March 6, 20204, accumulating 470,079 cases and 8,835 deaths by July 20255. Despite its regional relevance, there is a scarcity of in-depth genomic studies on COVID-19 in the city, especially in areas with high social vulnerability.
About a year after the first COVID-19 cases, vaccines were introduced and successfully contributed to pandemic control, significantly reducing the risk of severe disease and deaths associated with COVID-19. However, the effectiveness of vaccines against SARS-CoV-2 and other therapeutic developments may be limited by the emergence of new variants, as SARS-CoV-2 continuously mutated during the pandemic, affecting transmissibility, immune escape, and clinical severity in different ways6. The identification and monitoring of these variants are possible through genomic sequencing, which enables their classification as variants of interest or concern (VOC)7. The introduction of a new variant, especially those classified as VOC, can affect the course of an epidemic or pandemic in various ways—impacting case and death numbers, reinfection rates, vaccine effectiveness, and potentially prolonging the pandemic’s duration8,9,10. Genomic surveillance strategies allow for the implementation of rapid public health responses, such as containment measures and vaccine updates11. The World Health Organization encouraged countries to intensify sequencing efforts and share data on public platforms, promoting the formation of international consortia to coordinate these actions. Various initiatives and consortia have been formed to coordinate sequencing operations in several countries12. In this context, the TQT COVID-19 study was implemented in healthcare units in vulnerable neighborhoods of Brazilian capitals, including Salvador, focusing on non-pharmaceutical strategies such as testing, quarantine, and telemonitoring. The current study site covers a specific area of the city of Salvador, comprised of neighborhoods in the Cabula-Beiru health district, a region characterized by high economic and social vulnerability, where the populations are disproportionately affected by COVID-19.
In addition to detecting variants, genetic sequencing and phylogenetic analysis allow for the identification of viral introduction events in different locations. Studies have shown multiple introductions and sustained transmission of SARS-CoV-2 in regions such as Scotland13Germany¹4and Peru15. In Brazil, phylogenetic and phylogeographic analyses revealed that by September 2021, the country had become an epicenter of the pandemic, with widespread circulation of diverse lineages in all states16. Despite advances in genomic surveillance, gaps remain in understanding the virus’s evolutionary and spatial dynamics at national and subnational levels, particularly in socially vulnerable urban contexts. Another important use for genomic data includes monitoring the emergence of mutations in the spike (S) protein of SARS-CoV-2 with the potential to increase viral transmissibility and interfere with vaccine efficacy. While some countries relied on one or two vaccine manufacturers only, Brazil employed vaccines from different laboratories, including AstraZeneca, Sinovac Biotech (CoronaVac), Pfizer, Janssen (Johnson & Johnson), and, to a lesser extent, Moderna17,18. In this multi-vaccine scenario, no studies monitor and model the impact of mutations in the S gene. Therefore, this study aimed to identify the prevalence of SARS-CoV-2 infection and use genomics to identify VOC, the viral geographic spread, and mutations in the S gene of SARS-CoV-2 from patients of primary healthcare (PHC) services located in socioeconomically vulnerable neighborhoods in Salvador, Brazil.
Results
Epidemiological profile
Out of the 1,025 participants from Salvador who had samples collected for RT-PCR, 174 participants were selected for this study (average Ct ≤ 15.4). Among these, the majority of participants were female, under the age of 39, self-reporting as Black ethnicity, and received both doses of the COVID-19 vaccine. Most reported cases were mild, although about a third had no classified symptoms. Transportation options such as taxis, Uber, trains, subways, and buses were the most commonly used among the participants. Additionally, most participants had at least one comorbidity. (Table 1).
Molecular typing and phylogenetic analyses of the SARS-CoV-2
We analyzed 174 patient samples, classifying the sequencing and lineage according to the Pango lineage. The resulting analysis included an ML phylogenetic tree illustrating the diversity and relationships among the variants (Fig. 1). The BA.5 variant and its sublineages (BA.5.1, BA.5.2.1, BA.5.3.1, BA.5.1.15) were the most frequent, found in 57 samples (32.6%). This was followed by the BQ.1 variant and its sublineages (BQ.1.1.36, BQ.1.1.60) with 46 samples (26.3%), BA.4 lineage and its sublineage BA.4.1 with 38 samples (21.7%), B with 33 samples (18.8%), and XBB.1 with 1 sample (0.6%).
In Fig. 1, the relationships among the different lineages are visualized, with each lineage illustrated in a distinct color for easy identification and comparison. The phylogeographic results indicate considerable variation in the speed and patterns of SARS-CoV-2 spread in Salvador. The high variation in the mean branch dispersal rate suggests multiple virus introduction events in the population, potentially related to travel and population movements. This finding is supported by the median of the mean branch dispersal rate (vbranch), 213.8691 km/year, with a 95% credible interval ranging from 62.08517 to 20406.07 km/year. These values indicate a high variation in dispersal speed among different virus lineages, suggesting the presence of multiple introduction events and heterogeneous dissemination patterns.
The lower variability observed in the weighted dispersal rate (vweighted) and the weighted diffusion coefficient suggests that slower-spreading lineages may establish more stably in the population. The median weighted dispersal rate was significantly lower, at 6.480699 km/year, with an HPD interval of 2.536906 to 410.7303 km/year. Compared to the mean branch dispersal rate, the lower variability in this parameter indicates that longer-duration lineages exhibit a smaller impact on the weighted dispersal. This suggests that while some lineages spread rapidly, others establish themselves and persist in the population for longer, contributing to the virus’s persistence.

Maximum likelihood phylogeny containing high-quality genome sequences. Phylogenetic Tree of SARS-CoV-2 Variants and Lineages Identified in Patients from Salvador Between July 2022 and November 2022.
Analysis of SARS-CoV-2 dispersion dynamics
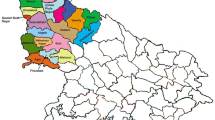
Figure 2A presents a map of Salvador, highlighting the SARS-CoV-2 dispersion routes. The phylogeographic analysis yielded a median diffusion coefficient of 12,540 km² yr⁻¹ (95% HPD: 03,210–1,180,00 km2 yr–1). When branch lengths were weighted by their temporal duration, the estimate decreased to 4,860 km2 yr–1 (HPD: 1,280–248,000 km2 yr–1), indicating that long, slow transmission chains contribute modestly to net geographic expansion. Branch-specific dispersal rates were highly heterogeneous: BA.5.1, BQ.1.1, and XBB.1 exhibited mean branch speeds of 250–18,000 km yr–1, whereas BA.4.1 remained confined mainly to the metropolitan area. The extremely broad HPD reflects two distinct diffusion regimes: most branches show local movement within Salvador, while a small set of early, poorly dated branches inflates the upper tail. Excluding those outliers narrows the HPD to 4,800–92,000 km2 yr–1, a range consistent with state-level rather than nationwide spread. Together, these findings reveal predominantly local transmission punctuated by occasional long-distance introductions that disproportionately shape overall diffusion metrics.
Figure 2B presents a phylogenetic cluster transition matrix. The green square in the upper left corner, marked with 0, coincides with the same area in Fig. 2A and indicates no documented transitions within this cluster. In contrast, the central purple square with the number 86, aligned with the area of highest concentration on the map in Fig. 2A, reveals a high frequency of transitions within this cluster, suggesting intense mutation or exchange activity within this group.

(A) Locator maps for Brazil/Bahia and the city of Salvador. Curved lines depict inferred diffusion paths between participants’ residential-address areas (neighbourhoods). Colour encodes path density (single-hue gradient) and does not represent lineage in this panel. Line width is proportional to the number of inferred transitions; circles mark neighbourhood centroids derived from residential addresses. (B) Transition matrix between residential-address areas. Each cell shows the number of inferred transitions from the source area (rows) to the destination area (columns) under a discrete phylogeographic Markov-jump model. Squares on the diagonal denote intra-area transitions; off-diagonal circles denote inter-area transitions. The numeral inside each symbol is the total count. Colours encode the source cluster and are panel-specific (they do not reuse the lineage palette from Fig. 1). Cells marked “0” indicate no transitions. Shape encoding and numeric counts provide redundancy for grayscale viewing.
Here, we have genomic and geospatial data focusing on variations in diffusion coefficients and isolation by distance (IBD). The original diffusion coefficient (Doriginal) had a median of 29,594.63 km²/year, with a 95% credible interval ranging from 5,737.911 to 8,537,430 km²/year. This coefficient reflects the spatial heterogeneity of the virus’s spread, suggesting that some lineages may spread rapidly in specific areas. In contrast, the weighted diffusion coefficient (Dweighted) had a median of 4,831.094 km²/year, with the highest probability density (HPD) interval ranging from 1,602.515 to 1,124,966 km²/year. Like the weighted speed, the weighted diffusion coefficient shows less variation, suggesting that long-duration events contribute less to overall diffusion.
Isolation-by-distance tests showed weak and non-significant correlations between patristic and geographic distances (Spearman rS median = − 0.029, HPD [− 0.043, 0.057]; Pearson rP#1 median = 0.042, HPD [− 0.031, 0.050]; log-transformed Pearson rP#2 median = − 0.001, HPD [− 0.045, 0.008]), indicating that geographic distance alone did not structure transmission during the study period. This pattern is consistent with prior work in Brazil and across Latin American cities, demonstrating that SARS-CoV-2 dissemination tracks mobility flows and urban connectivity more than Euclidean distance19,20.
The results in Fig. 3A show two main concentrations of values for the weighted diffusion coefficient, corresponding to two areas of higher concentration observed in the phylogeographic dispersion analysis described in Fig. 2A. The first concentration is located in the lower range of the weighted diffusion coefficient, with a relatively high mean branch variation, ranging between 5 and 15 units. The second concentration occurs in a higher range for the mean weighted diffusion coefficient, though with greater dispersion, reflected in the diffusion coefficient variation among branches, reaching values above 20 units. These two main concentrations are also observed in the kernel density analysis for weighted dispersion speed (Fig. S1).
Our findings suggest a heterogeneous dispersion of SARS-CoV-2 in Salvador, with certain lineages experiencing rapid expansion in specific areas. Phylogeographic analysis indicates that the virus’s spread is complex and influenced by multiple epidemiological and social factors.
Figure 3B illustrates the relationship between accumulated migration distance (in kilometers) and time, representing the spatial spread of SARS-CoV-2 over an observational period. The graph’s lines may represent observed dispersion trajectories for different lineages or viral spread events. The highlighted (red) line indicates the average or a specific trajectory of interest. At the beginning of the observational period, most lines remain close to the horizontal axis, indicating a low accumulated migration distance. This behavior suggests an initial phase of localized dispersion, with slight variation in migration distance among the different events. As time progresses, the trajectories diverge, gradually increasing the accumulated migration distance. This phase indicates a moderate geographic expansion of the virus, with some lineages spreading to more distant areas. After approximately 600 km, several lines show a sharp increase in the accumulated migration distance. This pattern suggests rapid and long-distance dispersion events, possibly associated with significant epidemiological outbreaks, intercity or international travel, or superspreader events. At the end of the observational period, the lines converge, indicating a common trend of increasing accumulated migration distance. This may reflect a stabilization phase after rapid and extensive dispersion, where different lineages reach similar geographic spread.

(A) Kernel Density Estimates of the Weighted Diffusion Coefficient Parameters. The figure displays a two-dimensional kernel density plot representing the estimated distribution of weighted diffusion coefficients from a phylogeographic dataset. The x-axis indicates the mean value of the weighted diffusion coefficient, and the y-axis reflects the variation of the diffusion coefficient among branches. The shaded areas represent the highest posterior density (HPD) intervals, with contours corresponding to the 50%, 75%, and 95% HPD intervals. The darkest region in the center of the plot delimits the 50% HPD interval, suggesting the highest probability for the actual values of the estimated parameters. Progressively lighter regions indicate the 75% and 95% HPD intervals, providing a hierarchical view of the uncertainty associated with the diffusion parameter estimates. (B) Temporal Dynamics of Migration Distance. This figure demonstrates the relationship between accumulated migration distance (in kilometers) and time, representing the spatial spread of a phenomenon over an observational period.
Analysis of mutations observed in the SARS-CoV-2 Spike protein in vaccinated and unvaccinated patients
Spike amino-acid substitutions documented in Salvador were interpreted in the context of the protein’s domain architecture and then organised by vaccination category as presented in Fig. 4 and Table S1. Because Table S1 lists the presence of specific substitutions by group, rather than full per-sample variant matrices or within-group frequencies, the description below is intentionally qualitative. The emphasis is on anatomical location within Spike and a plausible functional context. No inferential statistics were applied to test for associations between vaccination status and mutation patterns.
Across the entries in Table S1, substitutions map to several functional segments of Spike. These include the signal peptide and the N-terminal domain, the receptor-binding domain and its adjacent shoulder, the activation corridor around the S1/S2 junction and the early S2 fusion machinery, and the distal cytosolic tail. Many of the reported positions lie on solvent-exposed loops that influence antigenic presentation, whereas others occur in regions implicated in proteolytic priming, trimer stability, and membrane fusion. Considered together, the observations outline two broad regimes of variation. One regime is concentrated on the N-terminal surface, where subtle backbone and side-chain changes can reshape epitope geometry and alter antibody accessibility. A second regime is distributed across activation and fusion elements that control the efficiency and timing of entry. A smaller subset of substitutions sits within or adjacent to the receptor-binding domain shoulder, where local polarity and packing can affect the equilibrium between the “up” and “down” states of the RBD and, consequently, the exposure of the ACE2 interface.
In the unvaccinated group (Fig. 4A), two substitutions were recorded, namely L5F and A701V. L5F falls within the signal peptide at the extreme amino terminus. This short hydrophobic segment directs co-translational targeting to the secretory pathway and contributes to the initial steps of folding. Replacing leucine with phenylalanine conserves hydrophobicity but introduces a bulkier aromatic ring. Such a change is more plausibly linked to local hydrophobic packing during biogenesis than to direct modulation of receptor engagement. The second substitution, A701V, is positioned just downstream of the S1/S2 junction in the activation corridor that coordinates furin priming and the conformational rearrangements required for fusion. Substituting alanine for valine slightly increases side-chain volume and hydrophobicity. This change may influence packing near the cleavage loop and, by extension, the kinetics of priming or the stability of the prefusion trimer. Taken together, these two positions flank the entry process in time, with one acting early during synthesis and trafficking and the other adjacent to the proteolytic checkpoint that licenses fusion.
Among recipients of two doses of AstraZeneca (Fig. 4B), two substitutions were observed, A27P and L822F. A27P is located in the N-terminal domain, within a flexible, solvent-exposed loop that forms part of a recognised antigenic surface. Introduction of proline tends to restrict backbone motion and can impose a defined bend in the local structure. In this context, A27P has the potential to reshape loop geometry and thereby adjust the presentation of nearby epitopes, while remaining physically distant from the ACE2 contact surface. L822F resides in early S2, near the fusion peptide and adjacent helices that refold during the transition from the prefusion to the postfusion state. Substitution of leucine with phenylalanine increases side-chain bulk and aromaticity, which can alter helix–helix packing within the trimeric core. Even modest changes in this region may shift the energy landscape of membrane fusion and thereby influence entry efficiency. The co-occurrence of an N-terminal domain surface edit and an early S2 structural edit within the same vaccination category illustrates how antigen display and fusogenic machinery can be tuned independently.
In the CoronaVac group (Fig. 4C), two substitutions were noted, F59S and K786N. F59S lies in the early S1 segment immediately N-terminal to the folded N-terminal domain. Exchange of a bulky hydrophobic phenylalanine for a polar serine alters local packing and hydrogen-bonding networks that help scaffold the N-terminal surface. K786N occurs in early S2, within a helical region that supports the prefusion architecture and sits between the S1/S2 and S2′ activation sites. Removing a positive charge at this position by replacing lysine with asparagine changes the local electrostatic environment. This can, in principle, influence trimer stability or the trajectory of the fusion transition. Considered together, the CoronaVac entries again link a surface-exposed adjustment near the N terminus with a modification in the core elements that execute fusion.
In the Pfizer group (Figure D), three substitutions were reported, P272S in the N-terminal domain and a neighbouring pair on the receptor-binding domain shoulder, namely F375S and A376T. P272S replaces a conformationally restrictive proline with a more flexible serine within an N-terminal loop. Increased backbone flexibility at this position can remodel epitope geometry and modulate accessibility for neutralising antibodies that target the N-terminal surface. F375S and A376T sit on the interior shoulder of the receptor-binding domain, in a microenvironment that influences the equilibrium between RBD “up” and RBD “down” conformations. Both substitutions increase local polarity, in one case by exchanging phenylalanine for serine and in the other by exchanging alanine for threonine. Additional polarity introduces new hydrogen-bonding options and can rewire short hydrogen-bond ladders and van der Waals contacts. Because these interactions propagate toward the receptor-binding ridge, even infrequent changes at 375 and 376 have plausible consequences for epitope exposure and the dynamics of ACE2 engagement.
In the Janssen group (Fig. 4E), one substitution was observed, H1271L, located near the extreme carboxyl terminus within the cytosolic tail. This region does not participate in receptor engagement or the large-scale conformational rearrangements on the virion exterior. It does, however, contribute to intracellular trafficking, retention signals, and incorporation of Spike into budding virions. Replacing histidine with leucine removes an ionisable side chain and increases hydrophobic character. Such a modification can influence interactions with the membrane or with host trafficking factors and, in turn, could affect the density of Spike at the virion surface, even though it acts at a stage that is downstream of entry.
For individuals with a combined CoronaVac/Pfizer schedule, a single N-terminal domain substitution, K202N, was recorded. Position 202 lies on an exposed loop that is outside the receptor-binding domain. The lysine-to-asparagine change removes a positive charge and introduces a neutral polar side chain. This shift is capable of altering local electrostatics and the organisation of solvent at the antigenic surface. Changes of this type are consistent with fine-scale shaping of antibody-facing loops rather than with direct effects on ACE2 binding.
When viewed against the linear and three-dimensional organisation of Spike, the substitutions in Table S1 display a consistent structural logic. Several vaccination categories include changes that face the exterior on the N-terminal domain, such as A27P in AstraZeneca recipients, F59S just upstream of the N-terminal domain in CoronaVac recipients, K202N in the mixed schedule, and P272S in Pfizer recipients. These positions sit on loops that constitute dominant antibody targets. Their locations make them well-suited to modulate antigenicity by altering loop rigidity, electrostatics, or solvent structuring, while leaving the receptor-binding interface untouched. A second cluster of changes lies within the activation and fusion corridor, including A701V in unvaccinated individuals as well as K786N and L822F in the CoronaVac and AstraZeneca categories. Side-chain chemistry in this corridor can affect the efficiency of proteolytic priming, the stability of the prefusion state, and the mechanics of trimer refolding. The pair F375S and A376T in Pfizer recipients occupies a third, mechanistically coherent niche that acts on conformational control of the receptor-binding domain, with potential effects on both antibody recognition and receptor accessibility. Finally, H1271L in Janssen recipients draws attention to a distal segment in the cytosolic tail, where variation can act on intracellular handling and incorporation of Spike rather than on external entry steps.
Interpretation of these patterns requires restraint. Table S1 enumerates which substitutions were observed within each vaccination category, but does not provide per-group frequencies, complete per-genome variant matrices, or covariates such as age or sampling week. Apparent clustering of specific substitutions by category should therefore be understood as a qualitative map of where changes occurred in the sequences represented, rather than evidence of vaccine-specific selection or enrichment. Within these limits, the structural distribution of the reported positions is informative. The affected segments align with domains known to shape antigen presentation, proteolytic processing, trimer stability, and the fusion transition. The joint presence of N-terminal loop edits, activation-corridor substitutions, receptor-binding domain shoulder polarity shifts, and a distal cytosolic-tail change suggests that, within this dataset, Spike variation touched multiple mechanistic levers that together determine how the protein is displayed, primed, and deployed during entry.
In summary, the Salvador entries in Table S1 outline a Spike mutational landscape with apparent internal coherence. Adjustments on the N-terminal surface, modifications within the activation corridor and early S2, polarity shifts on the receptor-binding domain shoulder, and a cytosolic-tail substitution together sketch a plausible spectrum of Spike tuning across antigenicity, processing, and fusion. Within the descriptive scope of the tabulated observations, the narrative above situates each reported substitution in its most relevant structural context and clarifies how the set of changes, considered as a whole, could influence the balance among immune recognition, receptor engagement, and the mechanics of membrane fusion, without making statistical claims about associations with vaccination status.

Spike Protein Mutations. Three-dimensional structure of the SARS-CoV-2 spike protein highlighting mutations in red, identified in the genomes of individuals: (A) - unvaccinated with the first and second doses; (B) - vaccinated with AstraZeneca for the first and second doses; (C) - vaccinated with Coronavac for the first and second doses; (D) - vaccinated with Pfizer for the first and second doses; E - vaccinated with Jansen for the first and second doses. The enlarged regions show specific mutations in the subunits of the spike protein.
Discussion
This study reflected the dynamics of SARS-CoV-2 variants spread in the Cabula-Beiru region of El Salvador, combining epidemiological data with sequencing data in a socioeconomically vulnerable area. The predominant profile is of female, Black individuals under 39 who use public transportation. This epidemiological data reveals essential characteristics of the selected population, consistent with the demographic census data 2022 from the Brazilian Institute of Geography and Statistics (IBGE - a governmental agency responsible for producing and disseminating statistical and geographic information about Brazil)21. COVID-19 vaccination appears to be present in a significant portion of the sample, higher than the state’s monthly average of 31.4%22, possibly indicating protection against more severe forms of the disease. However, the lack of detailed symptom information for some participants limits the analysis of case severity. At least one comorbidity in about one-third of participants is a relevant factor to consider in assessing the individual risk for complications from COVID-19 infection. This highlights the need for target containment and mitigation strategies to protect the most vulnerable, including older people and individuals with comorbidities.
By January 2022, the Omicron variant had become the predominant strain throughout Brazil. The predominance of the BA.4, BA.5, and BQ.1 lineages, including their sublineages, aligns with the global spread of these variants in recent periods, suggesting diversification and possible local evolution of these variants in Salvador. According to Rodrigues et al.23the rapid spread of the Omicron variant is due to multiple mutations in the spike protein, which may increase its affinity for the host cell receptor and allow it to evade immunity from previous vaccinations or infections. In addition to spike protein mutations, the rapid spread of this variant is also explained by the increase in global air travel, where locations with high case incidence and international connectivity can potentially contribute to variant exports if early outbreaks are not controlled24. The identification of multiple samples of the clade 22E-BQ. 1 (Fig. 1) underscores the importance of continuous surveillance and the need to monitor these variants due to their potential impact on vaccine efficacy and viral transmission25.
The COVID-19 pandemic has shown rapid evolution and global spread, necessitating continuous monitoring and detailed analyses of viral variants. Genomic surveillance is crucial for understanding the dispersion dynamics and evolution of the virus in different geographic regions. The study of SARS-CoV-2 dispersion data in Salvador, as shown in Fig. 2A, highlights significant transmission patterns with a high concentration of events in the city center. The distinction between different lineages and dispersion events, indicated by the various colors of points on the map, is critical to understanding the virus’s genetic diversity and the impact of emerging variants. Phylogenetic studies have demonstrated that SARS-CoV-2 variants can have different transmissibility and virulence profiles, which may influence propagation patterns and infection severity26. The emergence of highly transmissible SARS-CoV-2 variants and the importance of genomic epidemiology in tracking these variants27 has also been emphasized. The dispersion routes and the concentration of transmission events in central Salvador may be associated with highly transmissible variants, as evidenced by this study. Faria et al.28 observed similar intense dissemination patterns in central and high-density areas, correlating transmission event concentrations with viral activity in the city center. This observation for Salvador, where the high density of lines in the center indicates more intense virus spread, reflects high transmission in urban central areas and can inform public health actions to reduce transmission, infection, and disease severity. These results are consistent with the rapid evolution of SARS-CoV-2 and the high connectivity in densely populated urban areas, facilitating virus spread and the genetic diversity observed in circulating lineages 19. Multiple lineages and frequent transitions between clusters underscore the importance of continuous genomic surveillance, effective control measures to mitigate virus spread, and the emergence of new variants.
According to the phylogeographic results, the absence of a solid isolation by distance signal in El Salvador suggests that geographic distance does not significantly hinder the virus’s spread. This pattern is consistent with studies showing that higher urban mobility and population density facilitate the intra-city spread of SARS-CoV-2, particularly in densely populated central neighborhoods and areas with intensive bus usage20,29. This epidemiological behavior is critical for formulating control and mitigation strategies, as it highlights the need for measures that consider population mobility and local social dynamics.
The phylogenetic cluster transition matrix analysis offers a detailed view of SARS-CoV-2 variation and transmission dynamics. The contiguous green squares along the diagonal represent Cluster 1 (PCUs 3–5), in which no transitions were inferred, indicating genetic stability within that local transmission network. In contrast, the purple square annotated “86” marks the intensive bidirectional exchange between Cluster 2 and all other PCUs, highlighting an area of elevated viral activity. Faria et al.28 comprehensively analyze genetic diversity and transmission dynamics in São Paulo. The observation of high transition frequencies within clusters, as indicated in the purple square, is consistent with identifying local emerging and transmissible variants that characterized the scenario in Salvador. The stability observed in some clusters also reflects variability and transmission control in specific areas. Rambaut et al.30 discuss identifying and characterizing new SARS-CoV-2 lineages with specific mutations. The pattern observed in the transition matrix, mainly the high frequency of transitions within the purple cluster, may be related to the emergence and spread of new lineages with significant mutations.
The variability in weighted diffusion coefficients indicates different regimes of SARS-CoV-2 dispersion in the population studied in Salvador (Fig. 3A). The concentration of lower-weighted diffusion coefficient values suggests that, in many cases, the virus spread occurred relatively locally, with slight variation among branches. On the other hand, the concentration of higher values, though less frequent, indicates larger-scale dispersion events, possibly associated with broader epidemiological outbreaks or multiple introductions in different locations. Epidemiologically, these data are significant because they highlight the heterogeneity in SARS-CoV-2 spread dynamics. The observed variation in weighted diffusion coefficients reflects how human mobility, population density, and social behavior can influence virus spread. Identifying these concentrations can help develop more effective control strategies, directing efforts to areas at higher risk of widespread virus dispersion and implementing preventive measures in areas with more localized spread. Moreover, the variability in weighted diffusion suggests that public health interventions must be adaptive and specific to different local contexts, recognizing that virus spread may not be uniform throughout Salvador. Continuous genomic surveillance and rapid interventions could mitigate outbreaks and limit the spread of SARS-CoV-2 in complex urban settings31.
The Temporal Dynamics of Migration Distance graph clearly visualizes SARS-CoV-2’s spatial spread over time, highlighting distinct phases and identifying rapid dispersion events. The graph allows for specifying different phases of SARS-CoV-2 spread, from an initial localized phase to rapid long-distance dispersion events. Understanding these phases is crucial for developing intervention strategies at different epidemic stages32. The abrupt increase in migration distance can be attributed to superspreader events, where infected individuals quickly spread the virus to multiple distant locations. These events require special attention and swift action to contain the spread. The analysis of trajectories can guide the planning of containment measures, such as mobility restrictions, quarantines, and contact tracing in areas identified as hotspots for rapid dispersion. Specific interventions may be more effective if implemented during early slow-growth phases, preventing outbreak escalation. Information on the temporal dynamics of migration helps continuously adapt public health policies, ensuring that responses are proportional to the epidemic’s current phase and the observed spread pattern33. As the world faces an ongoing risk of infections and associated social and economic disruptions, increasing and coordinating efforts to safely monitor and contain the spread of various Omicron subvariants will help control the pandemic30.
The mutations observed in samples from vaccinated and unvaccinated patients may have several epidemiological and clinical implications. Mutations that alter ACE2 receptor binding, such as S: L5F, S:A27P, and S: F375S, can influence viral entry efficiency and antibody recognition34,35,36. Mutations that increase ACE2 affinity may enhance transmissibility, whereas others reshape the spike surface to evade neutralising antibodies derived from prior infection or vaccination. On the other hand, spike protein mutations may enable the virus to escape neutralization by antibodies generated from previous infections or vaccination37. Mutations such as S: A701V and S: L822F, observed in vaccinated patients, suggest viral adaptations in response to immune pressure, potentially reducing the effectiveness of existing vaccines and resulting in immune evasion. Conversely, mutations that affect spike trimer stability, such as S: L822F and S: H1271L, can impact viral fusion and entry into host cells, influencing the virus’s pathogenicity by altering spike protein stability. The different mutations observed in patients vaccinated with different vaccines suggest that the immune response may be driving the selection of specific viral variants. This highlights the importance of continuously monitoring SARS-CoV-2 mutations to adapt vaccines and therapies in response to emerging variants, impacting differentiated immune responses. The mutations observed in the Salvador population underscore the importance of monitoring and characterizing variants to ensure that COVID-19 control measures remain effective.
Better control of the COVID-19 pandemic will likely depend on high vaccine coverage and efficacy and continued adherence to effective and sustainable public health measures38. According to Velavan et al.39current data analysis would be essential to assess the impacts of the new generation of COVID-19 vaccines targeting Omicron subvariants, which would support the development of strategies to improve healthcare system preparedness against harmful health effects.
Thus, it is possible to test hypotheses about the mode and pace of virus dispersion during emerging epidemics40 by integrating genomic and environmental data. Implementing targeted interventions in areas with high variation in dispersion rates may be crucial to containing the virus’s spread and reducing the COVID-19 burden in the population. Mobility restrictions, contact tracing campaigns, and increased vaccine coverage in high-mobility areas could effectively control transmission. Our data highlights the importance of implementing a genomic surveillance program to monitor future VOCs, evaluate prevention strategies, and inform suitable interventions and public policies in vulnerable areas. Furthermore, information gained through sequencing and analysis of dispersions can help rapidly detect and respond to new threats, control local outbreaks with greater precision, and update vaccines and preventive strategies based on genomic evidence.
Our data reveal a dual transmission regime: routine within-city spread characterised by diffusion coefficients < 10,000 km² yr⁻¹, and sporadic long-distance introductions that punctuate local chains. These spikes coincide with weeks of elevated inter-district mobility and disproportionately affect districts where two-dose vaccination coverage remains below 50%. Such findings argue for a tiered surveillance strategy in which real-time genomic data from high-mobility, low-coverage districts trigger rapid contact-tracing and surge vaccination.
The five spike substitutions—L452R, F486V, S494P, T572I, and P681H—were 3- to 7-fold more common in breakthrough infections, paralleling experimental evidence that these residues modulate antibody binding and fusogenicity. Continuous monitoring of their prevalence can therefore serve as an early warning for immune-escape lineages. Integrating diffusion-coefficient estimates with mutation-frequency dashboards would allow health authorities to pre-emptively deploy booster campaigns in districts where escape variants achieve a critical foothold.
In sum, coupling high-resolution phylogeography with mutation profiling provides a practical blueprint for adaptive control, enabling municipalities to align vaccine allocation, mobility restrictions, and genomic surveillance with the evolving epidemiological landscape.
In conclusion, the results of this analysis emphasize the complexity of SARS-CoV-2 evolution in response to vaccination and population mobility dynamics. Identifying specific mutations associated with vaccination status and the detailed phylogeographic analysis of lineages present in Salvador is crucial for anticipating challenges to vaccine efficacy and developing more effective control strategies. Continuous genomic surveillance and adapting public health policies are essential to mitigate the virus’s spread and minimize the COVID-19 burden on the population. Despite these findings, some limitations should be noted. The higher average number of vaccinated individuals in this study may have affected sample selection. The lack of information about the symptoms of one-third of the study population limits the understanding of clinical presentations, and the geographic scope of the study does not cover the entire city of Salvador, which limits the generalizability of the conclusions. Future research should aim to address these constraints.
Methods
Study design, participants, and setting
This study was part of a testing expansion project aimed at implementing and expanding testing, isolation, quarantine, and telemonitoring strategies for COVID-19 prevention in primary healthcare services in socioeconomically vulnerable neighborhoods of Salvador41. The study area in Salvador occurred in the Cabula-Beirú District, which has a population of 392,542 inhabitants distributed over an area of 25.89 km². The district has 12 primary healthcare units and five specialized units, covering 244,007 inhabitants42. This study was conducted following the principles of the National Health Council Resolutions 466/12 and 510/16 and international research ethics guidelines. The study was approved by the WHO Ethics Research Committee (ERC) (nº CERC.0128 A and nº CERC.0128B), and the city’s local ERC approved the study protocol (Salvador, ISC/UFBA: nº 53844121.4.1001.5030). Written informed consent was obtained from participants aged ≥ 18 years. They signed those aged 12 to 17; written assent and parental or guardian consent were also obtained.
Clinical samples collection
From July 2022 to November 2022, individuals who visited the primary health care units for rapid antigen diagnostic testing (Ag-RDT) were invited to participate in the study if they met the following criteria: being between the third and seventh days of COVID-19 symptoms or between the fifth and seventh days after contact with a positive case, regardless of prior vaccination or infection status. Those who tested positive on the Ag-RDT were asked if they had been in contact with anyone since the onset of symptoms. If they answered affirmatively, consent was requested to contact those individuals. Subsequently, contact tracing was performed via telemonitoring, phone calls, or text messages. When contacts were reached, they were invited to visit the healthcare unit for testing. The individuals diagnosed with COVID-19 were monitored via telehealth, with follow-ups every 48 h for patients without comorbidities and every 24 h for patients with comorbidities until the end of isolation or quarantine.
Individuals who tested positive on the Ag-RDT were invited to participate in a new sample collection aimed at performing RT-PCR and sequencing. All the samples were labeled and transported in tubes containing viral transport and inactivation medium (Invitrogen RNAlater Stabilization Solution®) and stored at -80 °C. During the five-month data collection period in 2022, within one health district in Salvador, a total of 1,025 participants tested positive for SARS-CoV-2 by RT-PCR. From these, a target sample of 200 participants was calculated, considering a 20% prevalence of the Omicron sublineage BA.1.1, a 95% confidence interval, and a design effect of 1. Samples were considered for sequencing only if the Ct value was under 30 (Ct < 30). Of the 200 samples selected, 174 were used for viral typing, phylogenetic, and spread analyses. This loss occurred due to problems in the preparation of some libraries. Participant anonymity was ensured throughout each step of the study.
Demographic data collection
Individuals who agreed to participate in the study were also invited to complete a socio-behavioral questionnaire41. Individuals over 12 answered directly, while for those under 12, the questionnaire was answered by their guardians or companions. Clinical-epidemiological data were collected and analyzed, including characteristics such as age, sex, race/ethnicity, type of transportation used, date of first symptoms, date of sample collection, smoking status, vaccination, symptoms (shortness of breath, chest pain, fever, chills, headache, dry and productive cough, fatigue, sore throat, runny nose, nasal congestion, nausea/vomiting, diarrhea, joint pain, muscle pain, abdominal pain, loss of smell and taste, changes in sleep patterns, psychomotor agitation, mood changes, eye irritation), and comorbidities (obesity, diabetes, cardiovascular disease, respiratory disease, cancer, hematologic disease, renal disease, autoimmune disease, immunodeficiency). Participants who tested positive were classified according to their symptomatology during the monitoring as asymptomatic, mild, moderate, severe, or critical, following the Brazilian Ministry of Health43 criteria. The vaccination schedule was assessed according to the first and second doses. A descriptive analysis of the variables was performed using frequency comparisons and Pearson’s chi-square test, considering p ≤ 0.05 and a 95% confidence interval, with the database created in Microsoft Office Excel and analyzed using STATA.
Molecular typing of the SARS-CoV-2
Molecular typing was performed on 174 samples positive for SARS-CoV-2. The Invitrogen Purelink™ Genomic RNA Kit (Invitrogen, São Paulo, SP, Brazil) was used according to the manufacturer’s protocol to extract genomic RNA from previously collected nasal swab samples. Real-time PCR for SARS-CoV-2 detection was performed in duplicate based on the protocol described by CDC et al.44 After real-time PCR, the samples were inactivated and stored at UFBA-IMS-CAT in a -70 °C freezer for further analysis, posing no risk of contamination or pathogen dissemination. mRNA was extracted using the PureLink™ RNA Mini Kit, following the manufacturer’s protocol. cDNA synthesis was done by reverse transcription (RT) from mRNA using the SuperScript® IV Reverse Transcriptase kit (Invitrogen).
The preparation of genomic libraries for SARS-CoV-2 was conducted using Oxford Nanopore MinION technology. The resulting cDNA was subjected to multiplex PCR using Q5 High Fidelity Hot-Start DNA Polymerase (New England Biolabs) and a set of specific primers developed by the Artic Network for whole genome sequencing of SARS-CoV-2 (version 3 and 4)45. Amplicons were purified using 1x AMPure XP Beads (Beckman Coulter) and quantified using Qubit 3.0 (ThermoFisher Scientific) with the Qubit™ dsDNA HS Assay Kit (ThermoFisher Scientific). DNA library preparation was performed using the Sequencing Kit LSK109 (Oxford Nanopore Technologies) and the Native Barcoding Kit (NBD104 and NBD114, Oxford Nanopore Technologies). Libraries were loaded onto an R9.4 flow cell (Oxford Nanopore Technologies). Negative controls were used at each sequencing step to check for potential contamination. The translation of raw electronic signals from the sequencer into base pairs was performed using Guppy v3.4.5, and barcode demultiplexing was carried out using qcat. Consensus sequences were generated using Genome Detective - DIAMOND, employing the viral subset of the SwissProt UniRef protein database. NCBI blastn sequences were used as references, and alignment was conducted with AGA (Annotated Genome Aligner) and MAFFT46. To ensure the quality of the genome sequences generated in this study and achieve the highest possible phylogenetic accuracy, only genomes > 29,000 bp and < 1% ambiguities were considered. Across the 174 genomes, sequencing achieved a mean breadth of coverage of 98.9% (minimum 92.1%) relative to the Wuhan-Hu-1 reference (NC_045512.2). The mean per-site read depth was 867× (inter-quartile range 510–1,220×), exceeding the minimum 200× depth recommended for high-confidence variant calling in Oxford Nanopore SARS-CoV-2 protocols.
Phylogenetic analysis and lineage classification
Sequences were aligned using MAFFT and submitted to IQ-TREE2 for maximum likelihood (ML) phylogenetic analysis, employing the General Time Reversible (GTR) nucleotide substitution model and a proportion of invariable sites (+ I) as selected by ModelFinder47. The initial topology of the maximum likelihood (ML) tree was utilized to estimate the number of viral transmission events. TreeTime48 was employed to convert this ML tree topology into a time-scaled tree, applying a constant average rate of 8.0 × 10 − 4 nucleotide substitutions per site per year, with outlier sequences excluded from the analysis. A migration model was then applied to the time-scaled phylogenetic tree in TreeTime, assigning locations to both the tips and internal nodes. The dated phylogeny and discrete phylogeographic model were inferred solely from the 174 genomes sequenced in this study. No external sequences were included because the objective was to reconstruct within-city transmission dynamics. Each discrete location (“state”) in the diffusion model corresponds to one of the 12 primary-care units (PCUs) from which samples were obtained. We used dynamic lineage classification, as specified in the Phylogenetic Assignment of Named Global Outbreak LINeages (version 3.1.7) protocol (PANGOLIN)30.
Phylogenetic and spread analyses
Before phylogeographic inference, the temporal signal was assessed by root-to-tip regression in TempEst v1.5.349. Temporal structure was deemed acceptable when the correlation coefficient exceeded 0.2. For the Salvador dataset, the regression of genetic distance against sampling date indicated clocklike evolution, r² = 0.4626 and correlation coefficient = 0.4567, supporting the use of molecular-clock models for time calibration.
A Bayesian framework was then applied to obtain a time-scaled phylogeny and to reconstruct spatial diffusion among residential-address areas in Salvador. Multiple-sequence alignments in FASTA format were parameterised in BEAUti to generate BEAST XML inputs, with latitude and longitude assigned to each genome to enable discrete spatial inference. Model specification in BEAUti included the substitution process, molecular-clock configuration, and definition of the discrete location states. BEAST sampled phylogenies and model parameters from the posterior distribution by Markov chain Monte Carlo, producing time-calibrated trees and location transition histories used in subsequent analyses.
Time-scaled trees were inferred under a GTR + I + Γ4 substitution model and an uncorrelated log-normal relaxed molecular clock, using a mean clock prior of 8 × 10– 4bstitutions per site per year and a log-normal prior on among-branch rate variation. Demographic history was accommodated with a Bayesian Skyline prior with ten coalescent groups, allowing flexible trajectories of adequate population size through time. The posterior tree sample was summarised as a maximum-clade-credibility tree, providing median node ages and 95% highest posterior density intervals for divergence events, reported in calendar time and used for all downstream temporal interpretations.
Spatial diffusion within the city was modelled as a discrete continuous-time Markov chain, each state corresponding to a residential-address area rather than to health-care facilities. Bayesian stochastic search variable selection was applied to the instantaneous rate matrix to identify well-supported routes, yielding a sparse set of links with high posterior support. Posterior Markov-jump counts were aggregated between pairs of areas to construct the transition matrix shown in the figure, and posterior-expected movements were rendered as curved paths on the map. In this formulation, the counts displayed in the matrix summarise the posterior number of state changes along branches of the time-scaled tree, and the mapped trajectories represent movements supported by the posterior, linking the temporal phylogeny to the observed spatial structure.
MCMC performance and reliability were evaluated rigorously. Two independent chains of 100,000,000 iterations were run, with samples recorded every 10,000 steps. The first 10% of each chain was discarded as burn-in. Convergence and adequate sampling were assessed in Tracer v1.7.1; effective sample sizes exceeded 250 for all key parameters, and independent runs yielded concordant posterior summaries. TreeAnnotator was used to summarise the posterior into a clade-credibility tree. Analyses were conducted with BEAST v1.10.x, specifically v1.10.4 for configuration and v1.10.5 for execution, with no material differences in posterior behaviour under the specified priors and models.
Taken together, these procedures provide the time-calibrated phylogeny used throughout the Results, including node-age estimates and associated uncertainty, and deliver the discrete phylogeographic reconstruction from which transition counts and spatial trajectories are derived. Figures and numerical summaries presented in the Results therefore represent posterior summaries from the Bayesian model, rather than point estimates from a single tree, ensuring that temporal and spatial inferences incorporate parameter uncertainty.
Analysis of patterns in the SARS-CoV-2 Spike protein associated with vaccination status
Sequencing reads were aligned to the SARS-CoV-2 reference genome (NCBI NC_045512.2) with the BWA-MEM (Burrows-Wheeler Aligner)50 algorithm, and BAM files were coordinate-sorted and indexed before variant discovery. Variant calling used a complementary, two-caller approach to balance sensitivity and specificity: Genome Analysis Toolkit (GATK)51 was applied for baseline quality score recalibration, variant discovery, and genotyping using standard hard-filter criteria to remove sites with low base or mapping quality, strand bias, or insufficient support; in parallel, LoFreq52 was run to detect low-frequency variants using per-base quality scores and error modelling. Callsets were merged and deduplicated, and positions failing caller-specific filters were excluded. Downstream interpretation was restricted to substitutions and indels in the Spike coding sequence. LoFreq allele-frequency estimates were retained in VCFs for quality control, but reporting in Table S1 is limited to the presence of specific substitutions by vaccination category rather than quantitative frequencies.
Retained Spike variants were functionally annotated and mapped onto the canonical domain architecture, including the signal peptide, N-terminal domain (NTD), receptor-binding domain (RBD), the S1/S2 junction and S2 regions, and the cytosolic tail. To provide structural context, affected residues were located on the trimeric Spike glycoprotein (PDB 6VSB) in PyMOL53 to distinguish substitutions on solvent-exposed antigenic surfaces from those within activation and fusion elements. Table S1 lists substitutions observed at least once within each vaccination category and is intended to document presence by group; accordingly, the accompanying text provides qualitative interpretation based on structural location and plausible functional context.
Data availability
The DNA sequences generated and/or analysed during the current study are available in the GISAID EpiCoV repository (https://www.gisaid.org/) , Accession: EPI_ISL_13838063, EPI_ISL_13838064, EPI_ISL_13838065, EPI_ISL_13838066, EPI_ISL_13838067, EPI_ISL_13838024, EPI_ISL_13838068, EPI_ISL_13838025, EPI_ISL_13838069, EPI_ISL_13838026, EPI_ISL_13838027, EPI_ISL_13838028, EPI_ISL_13838029, EPI_ISL_13838060, EPI_ISL_13838061, EPI_ISL_13838062, EPI_ISL_13838052, EPI_ISL_13838053, EPI_ISL_13838054, EPI_ISL_13838055, EPI_ISL_13838056, EPI_ISL_13838057, EPI_ISL_13838058, EPI_ISL_13838059, EPI_ISL_13838090, EPI_ISL_13838091, EPI_ISL_13838092, EPI_ISL_13838093, EPI_ISL_13838050, EPI_ISL_13838094, EPI_ISL_13838051, EPI_ISL_13838041, EPI_ISL_13838085, EPI_ISL_13838042, EPI_ISL_13838086, EPI_ISL_13838043, EPI_ISL_13838087, EPI_ISL_13838044, EPI_ISL_13838088, EPI_ISL_13838045, EPI_ISL_13838089, EPI_ISL_13838046, EPI_ISL_13838047, EPI_ISL_13838048, EPI_ISL_13838049, EPI_ISL_13838080, EPI_ISL_13838081, EPI_ISL_13838082, EPI_ISL_13838083, EPI_ISL_13838040, EPI_ISL_13838084, EPI_ISL_13838030, EPI_ISL_13838074, EPI_ISL_13838031, EPI_ISL_13838075, EPI_ISL_13838032, EPI_ISL_13838076, EPI_ISL_13838033, EPI_ISL_13838077, EPI_ISL_13838034, EPI_ISL_13838078, EPI_ISL_13838035, EPI_ISL_13838079, EPI_ISL_13838036, EPI_ISL_13838037, EPI_ISL_13838038, EPI_ISL_13838039, EPI_ISL_13838070, EPI_ISL_13838071, EPI_ISL_13838072 and EPI_ISL_13838073). The datasets used and/or analyzed during the current study are available from the corresponding author upon reasonable request.
References
World Health Organization, WHO COVID-19 dashboard. Available at: https://data.who.int/dashboards/covid19/deaths?m49=001&n=c accessed on 08/14/2025.
Sott, M. K., Bender, M. S. & da Silva Baum, K. Covid-19 outbreak in brazil: health, social, political, and economic implications. Int. J. Health Serv. 52 (4), 442–454. https://doi.org/10.1177/00207314221122658 (2022).
Tomasiello, D. B., Vieira, J. P. B., Parga, J. P. F. A., Servo, L. M. S. & Pereira, R. H. M. Racial and income inequalities in access to healthcare in Brazilian cities. J. Transp. Health. 34 (101722). https://doi.org/10.1016/j.jth.2023.101722 (2024).
BAHIA. Secretaria da Saúde.COVID-19: Boletim Epidemiológico, nº 01, março. (2020). Available at: https://www.saude.ba.gov.br/temasdesaude/coronavirus/boletins-epidemiologicos-covid-19-2020/ accessed on 11/10/2024.
SALVADOR. Centro de Informações Estratégicas de Vigilância em Saúde (CIEVS) do município de Salvador. Boletim Epidemiológico, nº 07, july, 2025. Available at: http://www.cievs.saude.salvador.ba.gov.br/boletins-epidemiologicos/ accessed on 07/25/2025.
Peck, K. M. & Lauring, A. S. Complexities of viral mutation rates. J. Virol. 92 (14). https://doi.org/10.1128/jvi.01031-17 (2018).
Center for Disease, Control and Prevetion. What is Genomic Surveillance? Dec. 2. (2022). Available at: https://archive.cdc.gov/www_cdc_gov/coronavirus/2019-ncov/variants/genomic-surveillance.html (2024).
Choi, J. Y. & Smith, D. M. SARS-CoV-2 variants of concern. Yonsei Med. J. Nov. 62 (11), 961–968. https://doi.org/10.3349/ymj.2021.62.11.961 (2021).
Naveca, F. G. et al. COVID-19 in amazonas, brazil, was driven by the persistence of endemic lineages and P.1 emergence. Nat. Med. 27 (7), 1230–1238. https://doi.org/10.1038/s41591-021-01378-7 (2021).
Chen, J. et al. Emerging dominant SARS-CoV-2 variants. J. Chem. Inf. Model. 63 (1), 335–342. https://doi.org/10.1021/acs.jcim.2c01352 (2023).
Robishaw, J. D. et al. Genomic surveillance to combat COVID-19: challenges and opportunities. Lancet Microbe. 2 (9), e481–e484. https://doi.org/10.1016/S2666-5247(21)00121-X (2021).
Saravanan, K. A. et al. Role of genomics in combating COVID-19 pandemic. Gene 823, 146387. https://doi.org/10.1016/j.gene.2022.146387 (2022).
da Silva Filipe, A. et al. Genomic epidemiology reveals multiple introductions of SARS-CoV-2 from Mainland Europe into Scotland. Nat. Microbiol. 6 (1), 112–122. https://doi.org/10.1038/s41564-020-00838-z (2021).
Walker, A. et al. Genetic structure of SARS-CoV-2 reflects clonal Superspreading and multiple independent introduction events, North-Rhine westphalia, germany, February and March 2020. Euro. Surveillance: Bull. Europeen Sur Les Maladies transmissibles = Eur. Commun. Disease Bull. 25 (22), 2000746. https://doi.org/10.2807/1560-7917.ES.2020.25.22.2000746 (2020).
Juscamayta-López, E. et al. Phylogenomics reveals multiple introductions and early spread of SARS-CoV-2 into Peru. J. Med. Virol. 93 (10), 5961–5968. https://doi.org/10.1002/jmv.27167 (2021).
Giovanetti, M. et al. Genomic epidemiology of the SARS-CoV-2 epidemic in Brazil. Nat. Microbiol. 7, 1490–1500. https://doi.org/10.1038/s41564-022-01191-z (2022).
Hadj Hassine, I. Covid-19 vaccines and variants of concern: A review. Rev. Med. Virol. 32 (4). https://doi.org/10.1002/rmv.2313 (2022).
Zhou, Z., Zhu, Y. & Chu, M. Role of COVID-19 vaccines in SARS-CoV-2 variants. Front. Immunol. 13 https://doi.org/10.3389/fimmu.2022.898192 (2022).
Candido, D. S. et al. Evolution and epidemic spread of SARS-CoV-2 in Brazil. Science (New York, N.Y.). 369(6508), 1255–1260, https://doi.org/10.1126/science.abd2161 (2020).
Kephart, J. L. et al. The effect of population mobility on COVID-19 incidence in 314 Latin American cities: a longitudinal ecological study with mobile phone location data. Lancet Digit. Health. 3 (11), e716–e722. https://doi.org/10.1016/S2589-7500(21)00174-6 (2021).
Instituto Brasileiro De Geografia E Estatística (IBGE). Censo Brasileiro De (IBGE, 2022).
BAHIA. Superintendência de Estudos Econômicos e Sociais da Bahia, SEPLAN.COVID-19 na Bahia, 12 de março. https://infovis.sei.ba.gov.br/covid19/, accessed on 05/06/2024. (2024).
Rodrigues, E. S. et al. Epidemiology of the SARS-CoV-2 Omicron variant emergence in the Southeast Brazilian population. Microorganisms 12 (3), 449. https://doi.org/10.3390/microorganisms12030449 (2024).
Tegally, H. et al. Dispersal patterns and influence of air travel during the global expansion of SARS-CoV-2 variants of concern. Cell 186 (15), 3277–3290. https://doi.org/10.1016/j.cell.2023.06.001 (2023).
Ao, D., He, X., Hong, W. & Wei, X. The rapid rise of SARS-CoV-2 Omicron subvariants with immune evasion properties: XBB.1.5 and BQ.1.1 subvariants. MedComm. 4(2), e239, https://doi.org/10.1002/mco2.239 (2023).
Mahilkar, S. et al. SARS-CoV-2 variants: impact on biological and clinical outcome. Front. Med. 9 https://doi.org/10.3389/fmed.2022.995960 (2022).
Alpert, T. et al. Early introductions and transmission of SARS-CoV-2 variant B.1.1.7 in the united States. Cell 184 (10), 2595–2604. https://doi.org/10.1016/j.cell.2021.03.061 (2021).
Faria, N. R. et al. Genomics and epidemiology of the P.1 SARS-CoV-2 lineage in Manaus, Brazil. Science (New York, N.Y.). 372(6544), 815–821, https://doi.org/10.1126/science.abh2644 (2021).
Araújo, J. L. B., Oliveira, E. A., Neto, L., Andrade, A. S., Furtado, V. Jr & J. S., & Unveiling the paths of COVID-19 in a large City based on public transportation data. Sci. Rep. 13 (1), 5761. https://doi.org/10.1038/s41598-023-32786-z (2023).
Rambaut, A. et al. A dynamic nomenclature proposal for SARS-CoV-2 lineages to assist genomic epidemiology. Nat. Microbiol. 5 (11), 1403–1407. https://doi.org/10.1038/s41564-020-0770-5 (2020).
Du, Z. et al. Systematic review and meta-analyses of Superspreading of SARS-CoV-2 infections. Transbound. Emerg. Dis. 69 (5), e3007–e3014. https://doi.org/10.1111/tbed.14655 (2022).
Lucas, P. C. C. et al. Institutional outbreaks of influenza-like illnesses in the state of São paulo: an analysis of the epidemiological profile during the COVID-19 pandemic. Public. Health. 221, 142–149. https://doi.org/10.1016/j.puhe.2023.06.018 (2023).
Assato, P. A. et al. Retrospective insights of the COVID-19 epidemic in the major Latin American city, São paulo, southeastern Brazil. Viruses 15 (2), 327. https://doi.org/10.3390/v15020327 (2023).
Dadonaite, B. et al. Spike deep mutational scanning helps predict success of SARS-CoV-2 clades. Nature 631 (8021), 617–626. https://doi.org/10.1038/s41586-024-07636-1 (2024).
Zhao, Z. et al. Omicron SARS-CoV-2 mutations stabilize Spike up-RBD conformation and lead to a non-RBM-binding monoclonal antibody escape. Nat. Commun. 13 (1), 4958. https://doi.org/10.1038/s41467-022-32665-7 (2022).
Kaleta, T. et al. Antibody escape and global spread of SARS-CoV-2 lineage A.27. Nat. Commun. 13 (1). https://doi.org/10.1038/s41467-022-28766-y (2022).
Candido, K. L. et al. Spike protein of SARS-CoV-2 variants: a brief review and practical implications. Brazilian J. Microbiol.. 53 (3), 1133–1157. https://doi.org/10.1007/s42770-022-00743-z (2022).
Talic, S. et al. Effectiveness of public health measures in reducing the incidence of COVID-19, SARS-CoV-2 transmission, and COVID-19 mortality: systematic review and meta-analysis. BMJ (Clinical Res. ed.). 375, e068302. https://doi.org/10.1136/bmj-2021-068302 (2021).
Velavan, T. P., Ntoumi, F., Kremsner, P. G., Lee, S. S. & Meyer, C. G. Emergence and geographic dominance of Omicron subvariants XBB/XBB.1.5 and BF.7 - the public health challenges. Int. J. Infect. Diseases: IJID : Official Publication Int. Soc. Infect. Dis. 128, 307–309. https://doi.org/10.1016/j.ijid.2023.01.024 (2023).
Dellicour, S., Rose, R. & Pybus, O. G. Explaining the geographic spread of emerging epidemics: a framework for comparing viral phylogenies and environmental landscape data. BMC Bioinform. 17, 82. https://doi.org/10.1186/s12859-016-0924-x (2016).
Magno, L. et al. Expansion of testing, isolation, quarantine, e-health and telemonitoring strategies in socioeconomically vulnerable neighbourhoods at primary healthcare in the fight against COVID-19 in brazil: a study protocol of a multisite testing intervention using a mixed method approach. BMJ Open. 13 (6), e068016. https://doi.org/10.1136/bmjopen-2022-068016 (2023).
BRASIL. Secretaria Municipal de Saúde. Distritos Sanitários: Distrito Sanitário Cabula/Beiru. Prefeitura de Salvador. Salvador [Internet]. 2024 [cited 2024 Jul 30]. Available from: http://www.saude.salvador.ba.gov.br/distritos-sanitarios/#1463062811469-2991b9bd-61ff (2024).
BRASIL & Guia de Vigilância Epidemiológica Covid-19: Emergência de Saúde Pública de Importância Nacional pela Doença pelo Coronavírus 2019 — Ministério da Saúde [Internet]. 2019 [cited 2024 Jul 24]. Available from: https://www.gov.br/saude/pt-br/assuntos/covid-19/publicacoes-tecnicas/guias-e-planos/guia-de-vigilancia-epidemiologica-covid-19/view (2019).
Centers for Disease Control and Prevention. CDC 2019-Novel Coronavirus. -nCoV) Real-Time RT-PCR Diagnostic Panel; Centers for Disease Control and Prevention: Atlanta, GA, USA, 2019. (2019).
Quick, J. nCoV-2019 Sequencing Protocol. Protocol; Berkeley, CA, USA: (2020).
Vilsker, M. et al. Genome detective: an automated system for virus identification from high-throughput sequencing data. Bioinf. (Oxford England). 35 (5), 871–873. https://doi.org/10.1093/bioinformatics/bty695 (2019).
Minh, B. Q. et al. IQ-TREE 2: new models and efficient methods for phylogenetic inference in the genomic era. Mol. Biol. Evol. 37 (5), 1530–1534. https://doi.org/10.1093/molbev/msaa015 (2020).
Sagulenko, P., Puller, V., Neher, R. A. & TreeTime Maximum-likelihood phylodynamic analysis. Virus Evol. 4 (1). https://doi.org/10.1093/ve/vex042 (2018).
Rambaut, A., Lam, T. T., Carvalho, M. & Pybus, O. G. L., Exploring the temporal structure of heterochronous sequences using TempEst (formerly Path-O-Gen). Virus evolution. 2(1), vew007, https://doi.org/10.1093/ve/vew007 (2016).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics (Oxford England). 25 (14), 1754–1760. https://doi.org/10.1093/bioinformatics/btp324 (2009).
McKenna, A. et al. The genome analysis toolkit: a mapreduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20 (9), 1297–1303. https://doi.org/10.1101/gr.107524.110 (2010).
Wilm, A. et al. LoFreq: a sequence-quality aware, ultra-sensitive variant caller for Uncovering cell-population heterogeneity from high-throughput sequencing datasets. Nucleic Acids Res. 40 (22), 11189–11201. https://doi.org/10.1093/nar/gks918 (2012).
Schrodinger, L. L. C. The PyMOL molecular graphics system. Version, v. 1, p. 8, (2015).
Acknowledgements
The authors would like to thank the patients who participated in this study.
Funding
PrEP1519 is funded by UNITAID (Process 2017–15-FIOTECPrEP) and the Ministry of Health of Brazil. This study was also supported by Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (Code 001). The funding agencies played no role in the design or conclusions of the study.
Author information
Authors and Affiliations
Contributions
ACSD, HBMO, TA, LM, FS, ID, TST, VGV, OCFJ, LAS, FKB, GBC, and LMM conceived and designed the study; LM, FS, ID, and TSST recruited the participants and collected the data and biological samples; ACSD, HBMO, AFL, JMFM, and LSS conducted the PCR and sequencing; ACSD, HBMO, and DSM performed the epidemiological analyses; ACSD, HBMO, TTSP, LM, and LMF performed bioinformatic analyses; ACSD, HBMO, and TTSP wrote the first draft of the manuscript; ACSD, HBMO, TTSP, LM, TST, FS, ID, LAS, AMSG, GBC and LM conducted subsequent reviews. All the authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics declarations and approval for human experiments
This study was conducted following the principles of the National Health Council Resolutions 466/12 and 510/16 and international research ethics guidelines. The study was approved by the WHO Ethics Research Committee (ERC) (nº CERC.0128 A and nº CERC.0128B) and each city’s local ERC approved the study protocol (Salvador, ISC/UFBA: nº 53844121.4.1001.5030; and Rio de Janeiro, INI/Fiocruz: nº 53844121.4.3001.5240, ENSP/Fiocruz: nº 53844121.4.3001.5240, and SMS/RJ nº 53844121.4.3002.5279). Written informed consent was obtained from participants aged ≥ 18 years. They signed those aged 12 to 17; written assent and parental or guardian consent were also obtained.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Dantas, A.C.S., Oliveira, H.B.M., Pereira, T.T.S. et al. Analysis of the prevalence, spread, and mutation patterns of SARS-CoV-2 variants: data from COVID-19 testing expansion strategy study in salvador, Northeast Brazil. Sci Rep 15, 31866 (2025). https://doi.org/10.1038/s41598-025-17691-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-025-17691-x
