
Abstract
Biomass has attracted considerable interest in the field of energy storage because of its outstanding characteristics, such as superior charge transport ability, affordability, eco-friendly, structural versatility, and renewability. Moreover, biomass-derived activated carbons, combined with pseudocapacitive materials, have recently gained popularity as efficient electrochemical components for energy storage devices. In this study, activated carbon was prepared from pinecone flowers as the biomass precursor through a process involving alkaline treatment and pyrolytic carbonization in an inert gas environment. Subsequently, CuCo2Se4 nanoparticles (CCS) were synthesized on the Pine Fruit Activated Carbon (PFAC) surface via a hydrothermal method. CCS serves as a pseudocapacitive material to compensate for the volumetric and gravimetric performance limitations of carbon. The resulting PFAC@CCS composite provides efficient pathways for electrolyte ion diffusion and rapid electron transfer when employed as electrode materials. This nanocomposite was evaluated as a potential electrode for supercapacitors. The prepared electrode, leveraging its unique characteristics, exhibits a significantly enhanced specific capacity of 639.55 F g−1 at a current density of 1 A g−1, demonstrating excellent rate capability and commendable cycling stability. Furthermore, a hybrid supercapacitor was constructed using PFAC@CCS as the positive electrode and PFAC as the negative electrode. This device demonstrates exceptional energy storage performance, delivering an energy density of 65.41 Wh kg−1 while operating at a power output of 16.53 W kg−1, with only a minimal 4.5% decline in capacity following 5000 continuous charge-discharge cycles at 25 °C temperature.
Similar content being viewed by others

Introduction
With the advancement of electric vehicles and portable electronic devices, coupled with rising energy costs and the rapid depletion of fossil fuel resources, the development of energy storage systems to meet the urgent needs of society has become imperative. Batteries and supercapacitors are two prominent forms of electrochemical energy storage devices (EESDs), as they both store and convert energy through ion intercalation and migration mechanisms, which have been extensively studied in the past1. However, each EESD possesses unique qualities and characteristics that may render it more suitable for specific applications. Significant differences are observed in the Ragone plot between batteries and supercapacitors, highlighting the distinct characteristics of these two energy storage systems. Batteries can deliver energy densities (Ed) ranging from 150 to 500 Wh/kg; however, due to slower energy transfer rates and reduced electrical power, they face limitations in providing high-speed energy delivery2. To maintain higher energy efficiency, their discharge times typically exceed 10 min or even up to 1 h. In contrast, supercapacitors, characterized by higher power densities (Pd), can fully release their energy in less than 10 s, offering output powers between 10 and 20 kW kg−13,4. These features, along with long operational lifespans, ease of construction, rapid charge-discharge dynamics, high-speed capabilities, quick rechargeability, low costs, and safe performance, make supercapacitors suitable for a variety of applications5.
The development of hybrid supercapacitors (HSCs) combines the advantages of both battery and supercapacitors technologies, offering advantages such as high-power density, high reversibility, and high charging and discharging rates. Battery-type electrodes store energy through faradaic charge transfer, which leads to bulk redox reactions at the electrodes, providing higher specific capacitance and energy density. However, there are problems such as low conductivity, poor cycling performance, limited voltage windows, and low charging rates that prevent their widespread use. Therefore, the development of new electrodes with high electrical conductivity and large surface area is necessary to increase the capacitance and energy density. So far, numerous research has been conducted on carbon cathode materials such as activated carbon, carbon nanotubes, graphene, and porous carbon. Despite noteworthy progress, the energy density of HSCs has not yet reached the desired level. In addition, there are complex production processes, high manufacturing costs, and environmental concerns associated with carbon nanotubes and graphene. Also, the use of fossil fuels as precursors for these materials is problematic from a sustainability perspective6. In this regard, biomass activated carbons have great potential to replace traditional carbon sources in energy storage systems due to their easy production and abundant availability of natural resources7.
In recent years, activated carbons derived from natural biomass sources have garnered significant attention due to their abundant availability, low cost, non-toxic nature, and renewability, in contrast to fossil fuels8,9. Additionally, biomass-derived activated carbon possesses unique features such as tunable surface chemistry, an interconnected porous framework, and abundant functional groups, which contribute to excellent electrical conductivity and enhanced charge storage capacity. Therefore, the exploitation of cost-effective, environmentally friendly, and high-performance carbon materials is highly desirable. In this context, carbons derived from natural biomass sources like coconut shells10 rice husks11 bamboo biochar12 litchi shells13 apricot shells14 and peanut shells15 have attracted considerable attention due to their large surface area, porosity, and cost-effectiveness8.
Despite the superior electrical conductivity and electrochemical stability of biomass-derived activated carbons, several challenges impede their immediate commercial application. Their narrow operating voltage and limited energy density necessitate combining them with pseudocapacitive materials (such as transition metal derivatives or conductive polymers) and two-dimensional substances, leading to the formation of novel composite electrodes. Designing these composite electrodes has been explored as a promising strategy to address the limitations of individual materials16.
Composite electroactive materials prepared using biomass-derived activated carbons and nanostructured substances like transition metal oxides and hydroxides are highly desirable to meet the urgent need for green and sustainable supercapacitors17. The combination of double-layer materials with redox-active substances significantly enhances energy and power density due to rapid and reversible Faradaic reactions facilitated by redox-active species. Furthermore, the highly conductive activated carbon matrix provides numerous nucleation sites for embedding these redox-active and two-dimensional materials, thereby increasing electroactive sites and conductivity18. The robust framework of biomass-derived activated carbon limits structural degradation of active redox species during continuous cycling processes, ensuring extended cycle life for these composite electrodes. Additionally, the interaction between functional groups on the surface of porous biomass carbons and redox-active/2D materials improves structural stability and charge transfer kinetics at the electrode/electrolyte interface. Consequently, energy storage performance, including energy density, can be enhanced by modifying electrode materials..
Due to their unique physicochemical and electrochemical properties, metal oxides have been the focus of research and development as redox electrodes in batteries and supercapacitors19. Various metal oxides have demonstrated excellent electrochemical performance as electrodes for supercapacitor applications, exhibiting high specific capacitance, outstanding cycle life, and superior energy density. The multivalent characteristics of metal oxide materials offer highly reversible redox reactions and good ion adsorption properties at the electrode/electrolyte interface. However, a limiting issue for metal oxides is poor electrical conductivity and significant volumetric contractions during continuous charge/discharge cycles. Recent research has focused on combining metal oxides with biomass-derived activated carbons as a means to overcome the disadvantages of metal oxides20.
Spinels, due to their unique physicochemical and electrochemical properties, have attracted significant attention as redox electrodes in batteries and supercapacitors. Among various spinels, copper cobaltite (CuCo₂O₄) stands out as a promising electrode material for electrochemical applications. This is attributed to its low cost, non-toxicity, and the synergistic effects between copper and cobalt species, which enhance electrochemical activity and conductivity21. .
Nevertheless, the electrical transport efficiency of spinels falls short of its predicted potential, proving inadequate for fast electron movement and enhanced charge storage in supercapacitors. To overcome this drawback, scientists have investigated replacing oxygen with sulfur and selenium. Selenium, in particular, exhibits outstanding metallic characteristics, reduced electronegativity, superior electrical conductivity, and an electron conduction rate of 1 × 10⁻³ s m⁻¹. Moreover, bimetallic selenides exhibit enhanced electrochemical behavior compared to their oxide counterparts, attributed to shorter bond distances during anion exchange processes, leading to faster electronic transfer1,22.
Inspired by the aforementioned studies, in this work, we utilized pine fruit as a biomass source to prepare activated carbon through alkaline treatment and pyrolysis methods. Subsequently, CuCo₂Se₄ nanoparticles (CCS) were evenly deposited on conductive PFAC frameworks, improving the synergistic effect within PFAC@CCS composite structures. When used as electroactive components in supercapacitors, these materials demonstrated superior energy storage capabilities compared to individual PFAC or CCS. Such green and efficient hybrid electrodes demonstrate significant potential in energy storage applications.
Experimental details
All experimental research and field studies involving plant materials were conducted in accordance with institutional, national, and international guidelines and legislation. The pinecones used in this study were collected from pine trees in the public park of Isfahan University of Technology where no specific permission was required under national legislation. The pinecones used in this study were collected from mature Pinus brutia (Calabrian pine, also known as Turkish pine) trees. The first author identified the species, Farshad Tavakoli, based on morphological characteristics and standard botanical references (FLORA IRANICA Book). A voucher specimen has been deposited in the Herbarium of the Faculty of Natural Resources, Isfahan University of Technology, under voucher number 18,431. Also, all chemicals used in this study were of analytical grade and used without further purification. Hydrochloric acid (HCl), manganese nitrate hexahydrate (Cu(NO₃)₂·3 H₂O), cobalt nitrate hexahydrate (Co(NO₃)₂·6 H₂O), and potassium hydroxide (KOH) were purchased from Merck (Germany). Sodium borohydride (NaBH₄), sodium selenite (Na₂SeO₃), N-methyl-2-pyrrolidone (NMP), polyvinylidene fluoride (PVDF), carbon black, and potassium hydroxide (KOH) were obtained from Sigma-Aldrich (USA). Ethanol (96%) was purchased from Bidestan Co. (Qazvin, Iran) and was used for the washing steps. Deionized water was used throughout the synthesis and washing procedures.
Synthesis of activated carbon from pine fruit
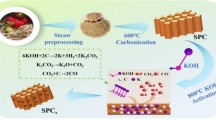
Activated carbon materials were prepared through alkaline treatment and carbonization of pine fruit (Fig. 1(step 1)). Initially, collected pine fruit was thoroughly washed with ethanol and deionized water (DI) to eliminate impurities and dust. After cleansing, they were air-dried at room temperature, followed by oven drying at 70 °C for 18 h. The hardened material was then mechanically ground into a fine powder using a mixer. For the alkaline treatment, an aqueous KOH solution was prepared by dissolving approximately 5.0 g of potassium hydroxide flakes in 80 mL of DI water. Next, 0.5 g of pine fruit powder was introduced into the KOH solution and subjected to heating with continuous magnetic stirring for 10 h. Residual solids were separated via centrifugation, thoroughly washed, and left to dry overnight at 80 °C. The obtained KOH-treated material underwent carbonization at 900 °C for 2 h under a steady argon gas stream to produce activated carbon. The temperature increase was controlled at 5 °C per minute. To eliminate potassium salts, the resulting carbon was neutralized using a 1 mol L−1 HCl solution and rinsed repeatedly with deionized water. Afterward, it was centrifuged and dried overnight at 70 °C, yielding purified PFAC powder.
Fabrication of PFAC@CCS composites
PFAC@CCS composites were prepared using a simple low-temperature hydrothermal method (Fig. 1(step 2)). To synthesize these composites, first, 0.0625 mmol of copper (II) nitrate trihydrate and 0.125 mmol of cobalt (II) nitrate hexahydrate were added to 60 mL of distilled water under magnetic stirring. Subsequently, 0.05 g of sodium selenite (Na₂SeO₃) was introduced into the reaction mixture and stirred for another 15 min to allow selenium incorporation. Subsequently, 0.10 g of sodium borohydride (NaBH₄) was introduced into the prepared solution, followed by continuous stirring for 30 min at ambient temperature until a uniform dark red mixture formed. Then, 0.190 g of PFAC was incorporated and vigorously stirred for an additional hour. The obtained solution was transferred inside a steel autoclave lined with Teflon and subjected to 180 °C (356 °F) for 12 h. After cooling to room temperature, a dark precipitate formed, which underwent multiple filtration steps to eliminate impurities before being rinsed with deionized water. Ultimately, the PFAC@CCS composite powder was acquired by drying at 70 °C (158 °F) for 6 h. As a reference, pure CCS was synthesized using the same approach, excluding PFAC powder.

Schematic representation of the preparation process of the PFAC@CCS composites. First step, the preparation of the PFAC through an alkaline treatment and carbonization of pine fruit. Finally, coating PFAC composites with CCS using a simple hydrothermal method at low temperature.
Structural characterization
The morphology and structure were surveyed by a field emission scanning electron microscope (FE-SEM, Quanta FEG 250, FEI, USA) equipped with EDX, and Raman spectra were collected using a TakRam N1-541, with a 532 nm laser. Fourier-transform infrared (FTIR) spectra were recorded by a Thermo Nicolet Avatar 370 FTIR spectrometer. Surface area and porosity measurements were performed using the BET method (BELSORP MINI II) after degassing the samples at 200 °C for 12 h. The crystal phases and surface chemistry were characterized by Philips X’pert diffractometer (PANalytical, Cu Kα, λ = 0.178 nm), X-ray photoelectron spectroscopy (XPS) with a monochromatic Cu-Kα as the X-ray source, respectively.
Electrochemical measurements
Before the fabrication electrodes, nickel foam (1 × 1 cm) underwent a 7-min immersion in 1 mol L−1 HCl to eliminate surface impurities. Afterward, it was carefully washed with deionized water and dried under flowing nitrogen gas. To prepare the electrode slurry, N-methyl-2-pyrrolidone (NMP) acted as a solvent, dispersing 80% electroactive material alongside 10% carbon black and 10% poly (vinylidene fluoride) (PVDF). The resulting blend was thoroughly homogenized using an agate mortar. This uniform slurry was then coated onto nickel foam and subjected to vacuum drying at 60 °C for 12 h, ensuring full solvent removal. A similar process was followed for PFAC@CCS composites and CCS active materials. The active material’s mass was precisely measured at 2 ± 0.1 mg. For electrochemical characterization, a three-electrode system was utilized, where nickel foam coated with active material functioned as the working electrode, platinum wire served as the counter, and a mercury oxide (Hg/HgO) electrode acted as the reference. Capacitance was assessed in a 3 mol L−1 KOH electrolyte solution. Cyclic voltammetry (CV), galvanostatic charge-discharge (GCD), and electrochemical impedance spectroscopy (EIS) measurements were performed using BioLogic-SP-300 and Autolab PGSTAT302N electrochemical systems.
Further, asymmetric supercapacitors were fabricated with PFAC@CCS as the cathode and activated carbon (AC) as the anode, employing an aqueous 3 mol L−1 KOH electrolyte. Negative electrodes consisted of AC powder (80%), acetylene black (10%), and polytetrafluoroethylene (10%) dispersed in NMP before being applied to nickel foam, following previously established methods. These electrodes underwent vacuum drying at 70 °C for 20 h. The negative-to-positive electrode mass ratio (m⁻/m⁺) was determined using the charge balance equation (Q⁻ = Q⁺) to enhance efficiency23.
The specific capacitance (Cs) of electrode materials and asymmetric supercapacitor (ASC) devices is calculated using the following equation:
Here, (Cs) refers to the specific capacity, expressed in either F g−1. The discharge current is represented by I (A), while ∆t denotes the discharge duration in seconds. Furthermore, m signifies the mass of the active material in grams. The factor of 2 in Eq. (2) accounts for the symmetric two-electrode configuration of the supercapacitor cell, where both electrodes are assumed identical. Since the measured capacitance reflects the total cell performance, it is doubled to estimate the specific capacitance of a single electrode. To determine the energy density (E) and power density (P), the following equations were employed:
Results and discussion
Physicochemical characterization
The pore structure of the synthesized PFAC was analyzed using N2 adsorption/desorption isotherms, as shown in Fig. 2(a). The isotherm revealed a typical type-IV behavior according to IUPAC classification. At lower relative pressures (~ 0.1 to 0.4), a sharp increase in adsorption was observed, which can be attributed to micropores. A hysteresis loop observed between relative pressures of 0.4 and 0.9 indicated the presence of mesopores, a result of capillary condensation. Additionally, a slight spike near a relative pressure of 1 signaled the existence of macropores. Consequently, the Brunauer–Emmett–Teller (BET) surface area of the PFAC was measured to be 762.94 m² g−1, indicating a relatively high surface area. The pore size distribution profile of the PFAC@CCS sample (refer to the inset of Fig. S2) closely resembles that of pristine PFAC, indicating that the internal mesoporous channels of the carbon framework remain unobstructed after composite formation. Nitrogen adsorption–desorption analysis yielded a BET specific surface area of 104.49 m² g⁻¹ for the PFAC@CCS hybrid. This value is lower than that of the unmodified PFAC23. Raman spectroscopy was also used to validate the materials, as shown in Fig. 2(b). The observed broad peaks at 1353 cm⁻¹ and 1590 cm⁻¹ correspond to the D-band and G-band, indicative of the disordered and graphitic structures within PFAC (relative intensity between ID and IG bands is equal to 0.95 and for PFAC@CCS is equal to 1.02). Additionally, the EDX spectrum in Fig. 2(c) confirmed the presence of Cu, Co, Se and C elements in sample PFAC@CCS and the presence of C, O elements in sample PFAC (Fig. 2(f)).
The phase and crystallinity of the prepared samples were examined using X-ray diffraction (XRD) analysis. Figure 2(d) displays two key peaks at approximately 25.6° and 43.7°, which are attributed to the (002) and (101) planes of graphitic carbon (JCPDS card no. 00-056-0159). After the deposition of CCS on PFAC, new peaks emerged at 11.3°, 22.7°, 33.45°, and 59.9°, matching the (003), (006), (101), and (110) planes of rhombohedral CCS (JCPDS card no. 01-074-4767).

Spectroscopic characterization of PFAC@CCS composites, PFAC, and CCS. (a) Nitrogen adsorption-desorption isotherms, accompanied by the BJH pore size distribution curve (shown in the inset) for the PFAC sample. (b) Raman spectral analysis of the PFAC material. (c) EDX spectrum illustrates the elemental composition of the CCS sample. (d) XRD patterns comparing PFAC@CCS composites with the CCS sample, with the reference diffraction pattern of CuCo₂Se₄ displayed at the bottom. (e) FT-IR spectra recorded for PFAC@CCS composites and the PFAC sample. (f) EDX spectral data corresponding to the PFAC sample.
FTIR analysis was conducted to investigate and identify the surface functional groups present in activated carbon derived from pinecones, as well as its modified form incorporating CuCo₂Se₄ nanoparticles, within the spectral range of 400–4000 cm⁻¹. The corresponding results are illustrated in Fig. 2(e). As observed in the FTIR spectrum, both unmodified and nanoparticle-modified activated carbon exhibit absorption peaks at approximately 3437 and 1627 cm⁻¹, though with varying intensities. The peak detected at 3444 cm⁻¹ corresponds to the OH stretching vibrations found in phenolic, alcoholic, and carboxylic functional groups. Additionally, a broad band centered around 1000 cm⁻¹ is generally associated with C–O stretching vibrations in ether and alcohol functional groups. The presence of quinones and lactones is confirmed by peaks appearing at 1560 cm⁻¹ and 1695 cm⁻¹. Furthermore, the stretching vibrations attributed to C–O bonds within volatile species and carboxyl acids are evident at 1168 cm⁻¹ and 1107 cm⁻¹. Lastly, the characteristic peaks observed at 1662 cm⁻¹ and 1561 cm⁻¹ correspond to C = C stretching vibrations.
The FTIR spectrum of the synthesized CCS compound (refer to the inset of Fig. S3) revealed absorption bands within the 500–1000 cm⁻¹ region, which are characteristic of metal–selenium (M–Se) vibrational modes. It is plausible to assign the detected peaks in CuCo₂Se₄ to Cu–Se and Co–Se stretching vibrations. Such assignments align with known IR behavior of chalcogenide materials, where M–Se covalent interactions commonly appear between 400 and 700 cm⁻¹. Therefore, the presence of these bands supports the formation of the spinel-type CuCo₂Se₄ structure with proper M–Se bonding.
The elemental mapping images (Fig. 3) demonstrated a uniform distribution of Cu, Co, Se across the PFAC surface, indicating a successful incorporation of CCS nanoparticles. Combining the conductive carbon with pseudocapacitive CCS enhances the material’s energy storage capabilities compared to individual phases or components.

Elemental mapping images of the (a) PFAC, (b) CCS, (c) PFAC@CCS composites.
To evaluate the relative carbon and inorganic content in the final PFAC@CuCo₂Se₄ composite, estimated the carbon-to-inorganic ratio based on the initial synthesis parameters. In addition to stoichiometric estimation, elemental analysis was carried out using EDS (Energy-Dispersive X-ray Spectroscopy), which confirmed the coexistence of C, Cu, Co, and Se in the composite. While EDS provides semi-quantitative data, the elemental ratios observed were in good agreement with the designed input ratios23.
Furthermore, Raman spectroscopy was employed to examine structural changes in the carbon framework. A notable increase in the I_D/I_G ratio in the PFAC@CuCo₂Se₄ spectrum, compared to pristine PFAC, indicates increased disorder and successful incorporation of the inorganic phase.
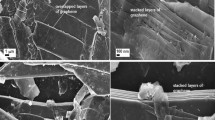
The surface morphology of PFAC, CCS, and the PFAC@CCS nanocomposite was analyzed using field-emission scanning electron microscopy (FE-SEM), as shown in Fig. 4. The PFAC sample (Fig. 4a) displays a well-developed three-dimensional (3D) interconnected porous framework, which is preserved even after alkaline activation and pyrolysis. Upon hydrothermal treatment with CCS precursors, the resulting PFAC@CCS composite (Fig. 4b and d) demonstrates a uniform dispersion of CCS nanoparticles anchored onto the PFAC surface. The 3D carbon network remains structurally intact after the hydrothermal process, indicating good stability of the PFAC scaffold. For comparison, Fig. 4c presents the morphology of CCS synthesized in the absence of PFAC, showing discrete nanoparticles with average diameters in the range of 20–30 nm. The anchoring of CCS nanoparticles onto the PFAC structure, clearly observed in Fig. 4d, confirms the successful formation of the composite through interfacial interaction.

Microscopic characterization of the PFAC, CCS, and PFAC@CCS composites. The wide and magnified FESEM images of (a) the PFAC sample. (b and d) the PFAC@CCS composites. (c) the CCS sample.
Electrochemical performance of PFAC@CCS electrodes in three-electrode cell setup
To assess the energy storage efficiency and electrochemical behavior of the fabricated electrodes, a three-electrode configuration was implemented in a 3.0 mol L⁻¹ KOH electrolyte. Several electrochemical methods, such as cyclic voltammetry (CV) and galvanostatic charge-discharge (GCD) tests, were conducted to examine the PFAC@CCS electrode’s performance. The corresponding results are illustrated in Fig. 5.
Figure S1-a† displays the CV profiles of substrate (Ni foam), CCS and PFAC@CCS electrodes, measured under identical voltage limits (0.00 to 0.60 V) at a scan rate of 20 mV s⁻¹ for comparison. The results demonstrate that the enclosed area of the PFAC@CCS electrode’s CV curve is significantly larger compared to the CCS electrode, signifying improved faradaic activity and increased charge storage capacity. Conversely, the CV response of Ni foam exhibits an almost negligible integrated area compared to the other two electrodes, highlighting its minimal role in energy retention. In cyclic voltammetry, a larger integrated area under the CV curve corresponds to a higher charge storage capacity, reflecting enhanced electrochemical performance. Elemental composition analysis (Fig. 2(f)) revealed that the PFAC sample consists of approximately 84% carbon and ~ 16% oxygen. The relatively high carbon content ensures good electronic conductivity and contributes to electric double-layer capacitance (EDLC). Meanwhile, the oxygen-rich surface functionalities (such as hydroxyl, carbonyl, and carboxyl groups) are known to induce additional pseudocapacitive charge storage through fast surface redox reactions. This dual contribution is reflected in the electrochemical data: the CV curves display slight deviations from ideal rectangular shapes, and the GCD profiles show nonlinear charge–discharge behavior, both of which are characteristic of pseudocapacitive participation. Hence, the coexistence of EDLC and pseudocapacitance arising from oxygen groups enhances the overall charge storage performance of PFAC-based electrodes.
The CV profiles for the PFAC@CCS and CCS electrodes, recorded at different scanning speeds, are presented in Fig. 5a-b. As anticipated, the cathodic and anodic sweeps in the CV curves correspond to the oxidation-reduction processes involving Co³⁺/Co²⁺ and Cu²⁺/Cu³⁺ in the presence of OH⁻ ions, as illustrated by the equations below:
The PFAC@CCS electrode maintains nearly identical CV characteristics to the CCS electrode, even as the scan rate increases. This stability indicates efficient mass transport, superior electrochemical reversibility, and excellent rate capability for PFAC@CCS electrode. Additionally, with an increase in scan rate, the oxidation peak shifts to a higher potential, while the reduction peak moves toward a lower potential. This phenomenon is probably due to ionic transport limitations within the electrochemically active substance at elevated scan rates, restricting the full execution of reversible faradaic reactions.
To evaluate rate capability and specific capacities, GCD assessments were performed. The GCD profiles of the PFAC@CCS electrode, recorded at different current densities (from 1 to 16 A g⁻¹) within a voltage range of 0.00 to 0.60 V (versus Hg/HgO), are depicted (Fig. 5c). The redox processes involving Cu⁺/Cu²⁺ and Co⁴⁺/Co³⁺ pairs result in nonlinear characteristics in the GCD curves, aligning with the CV findings. Internal resistance, evident as an iR drop in GCD curves, plays a key role in influencing rate performance. The relatively small iR drop across all current densities indicates exceptional electrical conductivity and excellent rate capability.
To assess the electrode’s rate capability, the specific capacitance was measured at different current densities, as depicted in Fig. 5d. The data indicate that the PFAC@CCS electrode attains an impressive capacitance of 639.55 F g⁻¹, which gradually declines with increasing current density. This reduction is due to the limited participation of the electroactive components in the redox processes as the current densities rise. Despite this, the electrode maintains 67.2% of its original capacitance even after a 16-fold rise in current density (ranging from 1 to 16 A g⁻¹), highlighting its exceptional ability to sustain performance under rapid charge-discharge conditions.
Figure 5d presents the rate performance of the CCS electrode. As evident, both its rate capability and specific capacitance across all tested current densities are notably lower in comparison to the values recorded for the PFAC@CCS electrode. To investigate long-term cycling durability, 6000 successive GCD cycles were conducted at different current densities from 12 to 16 A g⁻¹. According to Fig. 5e, the PFAC@CCS electrode shows exceptional durability over cycles, preserving 88.1% of its primary charge storage ability following 5000 consecutive cycles-significantly outperforming the CCS electrode, which experiences a 78.9% capacity loss. To assess the capacitive behavior and charge transport characteristics of the electrodes, electrochemical impedance spectroscopy (EIS) was performed. Figure 5f presents the Nyquist plot, which spans a frequency range from 0.01 Hz to 100 kHz, along with the inset equivalent circuit used to fit the EIS data. The EIS spectra analysis reveals that the PFAC@CCS electrode exhibits a lower internal resistance (0.14 Ω) compared to the CCS electrode (0.31 Ω), indicating enhanced conductivity for the PFAC@CCS structure. The impedance spectrum is characterized by two distinct regions: (i) In the high-frequency range, the semicircular feature reflects the charge transfer resistance (Rct) occurring at the electrode-electrolyte boundary, where the diameter of the semicircle is influenced by the Faradaic reactions of the Cu⁺/Cu²⁺ and Co⁴⁺/Co³⁺ redox pairs. The PFAC@CCS electrode demonstrates a significantly lower Rct than the CCS electrode, suggesting superior conductivity and efficient electron transfer. (ii) The inclined line observed in the segment of the spectrum with lower frequency represents the resistance pertaining to the movement of ions within the electrode material from the electrolyte1,19.
An increase in the slope above 450 is indicative of a purely capacitive response. In lower frequencies, the Bode diagram offers a clearer representation of the system’s capacitive characteristics compared to the Nyquist diagram. Figure 5f displays the Bode diagrams for both PFAC@CCS and CCS electrodes. The PFAC@CCS electrode exhibits a phase angle of approximately 70°, while the CCS electrode shows a phase angle of around 52°. The larger phase angle in the PFAC@CCS electrode suggests that it experiences less resistance to ion diffusion compared to the CCS electrode. The outstanding charge transfer, superior electrical conductivity, and impressive energy storage performance of the PFAC@CCS electrodes can be ascribed to numerous factors, including:
(i) The nanoporosity of pine nuts activated carbon increases ion diffusion in the internal surface areas, leading to more available active sites that enhance electrochemical activity. On the other hand, it maximizes the surface area and reduces the distance over which ions need to diffuse, improving capacitive performance. (ii) Selenium in the spinel structure, along with cobalt and copper, enhances redox reactions, leading to faster electron transfer. Furthermore, the relaxation time constant (τ₀) is a crucial parameter that reflects the speed of response in electrochemical systems. This can be further analyzed through the phase angle versus frequency plots, where τ₀ is calculated as τ₀ = 1/2πf₀, with f₀ being the frequency corresponding to a phase angle of − 45°. According to Fig. 5f, PFAC@CCS has a τ₀ of 0.28 s, which is much smaller than that of CCS (0.99 s), highlighting the faster reaction kinetics of PFAC@CCS. This indicates that the electrochemical kinetics of PFAC@CCS is better than that of single CCS24.

Investigation of the electrochemical properties of PFAC@CCS and CCS electrode within a three-electrode system. (a) Cyclic voltammetry (CV) curves illustrating the response of the PFAC@CCS electrode at varying scan rates. (b) CV profiles corresponding to the CCS electrode under different scan conditions. (c) GCD curves of the PFAC@CCS electrode recorded at multiple current densities. (d) Performance evaluation of the PFAC@CCS electrode under different charge-discharge rates. (e) Long-term cycling durability of the PFAC@CCS electrode assessed at various current densities. (f) Electrochemical impedance spectroscopy (EIS) analysis, displaying the Nyquist plot of PFAC@CCS and CCS electrodes, accompanied by its equivalent circuit model and the Bode phase angle plot (inset).
Additionally, to better understand the contribution of each component, the CV curves of neat PFAC at various scan rates are provided in the Supporting Information (Fig. S1). The rectangular shape of the curves indicates the EDLC behavior of PFAC, which contrasts with the redox features observed in the PFAC@CCS electrode.
To examine the kinetics of energy storage for the PFAC@CCS electrode material, the power law model was employed. This model establishes a relationship between the scan rate (measured in mV/s) and the current response (i), described by the equation i = a·vb, where i stands for current, v is the scan rate, and a and b are fitting parameters. The exponent b is extracted from the slope of the linear regression of log(i) versus log(v), as expressed in the following formula:
The “b” value shows how the charge storage process happens. If b = 0.5, it means the current is controlled by diffusion. If b = 1, the current comes from capacitive (surface) effects. When b is between 0.5 and 1, both diffusion and capacitive processes contribute25.
For PFAC@CCS (Fig. 6(b)), the b value is 0.5192, which is close to diffusion-controlled behavior. The contributions from diffusion and capacitive storage were also confirmed by Dunn and Trasatti methods26,27. To distinguish between these two mechanisms, a plot of i/v^0.5 versus v^5 commonly known as the Dunn method—is utilized. By fitting a straight line to this plot, the slope yields k₂ and the intercept gives k₁. This approach is illustrated in Fig. 6(c) for the PFAC@CCS sample.

(a) Calculation of the b value for PFAC@CCS, and (b) The k1 and k2 calculation for PFAC@CCS.
Electrochemical performance of the PFAC@CCS//PFAC device
A broad potential window is considered a key factor in achieving higher energy density in supercapacitors (SCs). By integrating a faradaic component in the cathodic section and employing a capacitive element in the anodic part, both the specific capacity and energy density of the system are improved. This increase in energy density is attributed to the cell voltage exceeding the thermodynamic decomposition potential of water. Specifically, the capacitance is enhanced by the electrochemical redox activities happening at the positive electrode, while the elevated energy density is a result of the high potential of capacitor-type carbon electrodes for reversible hydrogen electrosorption. As a result, asymmetric supercapacitors offer superior energy and power densities compared to their conventional counterparts. For assessing the real-world electrochemical behavior of the PFAC@CCS electrode, we designed an asymmetric supercapacitor utilizing PFAC@CCS as the cathodic component and PFAC as the anodic counterpart, denoted as PFAC@CCS//PFAC19.

Electrochemical characterization of the PFAC@CCS//PFAC ASC system. (a) Cyclic voltammetry (CV) analysis of PFAC@CCS and CCS electrode materials in a three-electrode configuration using a 3.0 mol L⁻¹ KOH electrolyte. (b) CV responses of the ASC device under different voltage windows at a fixed scan rate of 20 mV s⁻¹. (c) CV profiles recorded at a range of scan rates. (d) GCD curves obtained across various current densities. (e) Rate performance evaluation of the ASC device. (f) Ragone plot illustrating the energy and power density comparison of the PFAC@CCS//PFAC ASC system against other energy storage systems. (g) Cycling durability of the ASC device tested at 16 A/g under varying temperature conditions.
To assess the electrochemical performance of PFAC@CCS for real-world applications, an asymmetric supercapacitor (ASC) was fabricated, with PFAC@CCS and PFAC acting as the cathodic unit and PFAC functioning as the anodic counterpart. To enhance efficiency and attain the widest operational voltage range, the ideal mass proportion of the two electrodes was established relying on the charge-discharge (CD) characteristics of the PFAC@CCS and PFAC electrodes, as estimated using Eq. (2).
To evaluate the stable operating voltage of the assembled asymmetric device, CV tests were performed across different voltage ranges using a 3 mol L−1 KOH electrolyte at scanning speeds of 20 mV s⁻¹ (Fig. 7b).The results revealed that, due to limitations from oxygen release occurring at specific voltages exceeding 1.70 V, the complete asymmetric supercapacitor maintained a stable operating voltage up to 1.60 V, which is significantly higher than the typical 0.00–1.00 V range observed in conventional symmetric AC capacitors. As a result, a 3 mol L−1 KOH electrolyte and an operating potential window of 1.60 V were selected for all subsequent electrochemical tests. Figure 7b display the CV profiles of the PFAC@CCS//PFAC ASC across different voltage ranges, whereas Fig. 7c presents the CV curve of the PFAC@CCS//PFAC ASCs at varying scan rates from 10 to 200 mV s⁻¹. These results confirm that the charge storage mechanism is attributed to both the electrostatic energy storage from AC and the redox capacitance from PFAC@CCS. Figure 7d present the GCD profiles of the PFAC@CCS//PFAC ASC device subjected to diverse current density, from 1 to 16 A g−1. The non-typical triangular shapes observed in the GCD profiles align well with the CV profiles. Furthermore, the asymmetric device demonstrates excellent reversibility, low internal resistance, and impressive rate capability, even when exposed to high current levels of up to 16 A g−1, as indicated by the consistent symmetrical shapes and minimal peak shifts in the CV and CD profiles. The findings also reaffirm the contribution of both PFAC’s electrostatic capacitance and CCS’s redox capacitance to the charge storage process.

Two serially connected PFAC@CCS//PFAC ASC device can illuminate parallel blue (10 min), green (20 min), and yellow (15 min) LEDs.
The capacitance values for the devices are computed from the GCD data shown in Fig. 7e. The ASC device that was assembled demonstrates a remarkable specific capacitance, with values ranging from 183.8 F g−1 at 1 A g−1 to 116.2 F g−1 16 A g−1. Additionally, the device retains 60.25% of its initial charge storage after a 16-fold increase in current density, demonstrating excellent electrochemical performance. This outcome confirms its excellent electrochemical performance..
As the current density increases: (i) the ion exchange rate in the carbon-based electrode exceeds that of the pseudocapacitive electrode, leading to charge imbalance between the two ASC electrodes; (ii) the high current density-induced diffusion limitations result in reduced utilization of active material and constraints on the faradic process at the electrodes’ outer surface. Consequently, the gradual reduction in the specific capacity of the ASC is observed as the current rate increases.
The ASC device that was prepared is depicted in the Ragone plots (Fig. 7f). The Ragone plot in Fig. 7(f) provides a direct comparison of the electrochemical performance of the PFAC@CCS//PFAC device with various energy-storage technologies such as conventional supercapacitors, rechargeable batteries, and fuel cells. The calculated energy density of 65.41 Wh kg⁻¹ at a power density of 16.53 W kg⁻¹ positions the device beyond the typical operating range of electrical double layer capacitors, which are generally limited to < 10 Wh kg⁻¹. At the same time, it demonstrates a much higher power capability compared to most battery systems, where the discharge rate is constrained by sluggish ion diffusion and redox kinetics. Therefore, the PFAC@CCS//PFAC system effectively bridges the performance gap between supercapacitors and batteries, delivering both fast charge–discharge capability and appreciable energy storage, which highlights its potential for hybrid and flexible energy‐storage applications.
To assess long-term stability, the device underwent 5,000 uninterrupted GCD cycles at a current density of 16 A g−1 (Fig. 7g). After this process, the PFAC@CCS//PFAC ASC device retained around 95.5% of its initial capacitance at 25 °C temperature, confirming the exceptional endurance of the fabricated electrode materials. As depicted in Fig. 7g, the PFAC@CCS//PFAC ASC device preserves 91.2% of its specific capacitance at 15 °C, even after undergoing 5000 charge-discharge cycles. These observations confirm that the PFAC@CCS//PFAC ASC device maintains stability not only at standard room temperature but also under cold conditions. Importantly, with a further rise in operating temperature, the rate of capacitance loss increases as well. When exposed to an elevated temperature of 40 °C, approximately 82.2% of its original capacitance remains intact following 5000 cycles. The progressive capacitance decline observed over cycling is predominantly due to water evaporation, which gradually restricts ion mobility within the electrolyte9.
To evaluate the real-world applicability, two PFAC@CCS ASC devices were linked sequentially (Fig. 8). This configuration effectively powered multiple light-emitting diodes (LEDs) in yellow, green, and blue colors.
Conclusion
In conclusion, the novel and cost-effective PFAC@CCS composites were successfully fabricated by utilizing biomass as a conductive framework. Through alkali treatment and carbonization, PFAC powder with meso-, micro-, and nano-porous characteristics was easily synthesized from pinecone powder. The conducting HAPC powder was then combined with nickel salts and a hydrolyzing agent, leading to the formation of hierarchical ultrathin PFAC@CCS composites under hydrothermal conditions. When used as electroactive materials in supercapacitors, the composites exhibited a high specific capacity of approximately 1279.1 F g−1 at a current density of 1.0 A g−1, remarkable rate performance (~ 67.2%) following a 16-fold increase in current density, and excellent cycling durability (~ 88.1%) after 6000 cycles. The assembled ASC device showed a promising energy density 65.41 Wh kg−1, impressive power density 16.53 W kg−1, and exceptional cycle longevity with only about 4.5% capacity loss after 6000 charge/discharge cycles at room temperature. The improved energy storage performance of PFAC@CCS can be attributed to the high surface area of PFAC, facilitated ion diffusion in the electrolyte due to metal-selenium bonds, and the synergistic effects between copper and cobalt species that enhance electrochemical activity and conductivity. These exceptional performances highlight the promising potential of PFAC@CCS as an ASC material. This approach presents a highly promising method for utilizing biomass sources to produce carbon-based materials and their composites with transition metal selenides for energy storage devices.
Data availability
No datasets were generated or analysed during the current study.
References
Tavakoli, F., Rezaei, B., Taghipour Jahromi, A. R. & Ensafi, A. A. Facile synthesis of yolk-shelled CuCo2Se4 microspheres as a novel electrode material for supercapacitor application. ACS Appl. Mater. Interfaces. 12, 418–427 (2019).
Zhang, Y., Ma, Q., Wang, S., Liu, X. & Li, L. Poly (vinyl alcohol)-assisted fabrication of Hollow carbon spheres/reduced graphene oxide nanocomposites for high-performance lithium-ion battery anodes. ACS Nano. 12, 4824–4834 (2018).
Liu, H., Liu, X., Wang, S., Liu, H. K. & Li, L. Transition metal based battery-type electrodes in hybrid supercapacitors: A review. Energy Storage Mater. 28, 122–145 (2020).
Yang, S., Wang, S., Liu, X. & Li, L. Biomass derived interconnected hierarchical micro-meso-macro-porous carbon with ultrahigh capacitance for supercapacitors. Carbon N Y. 147, 540–549 (2019).
Sharma, K., Arora, A. & Tripathi, S. K. Review of supercapacitors: materials and devices. J. Energy Storage. 21, 801–825 (2019).
Atchudan, R. et al. Facile synthesis of nitrogen-doped porous carbon materials using waste biomass for energy storage applications. Chemosphere 289, 133225 (2022).
Vinayagam, M., Suresh Babu, R., Sivasamy, A. & de Barros, A. L. F. Biomass-derived porous activated carbon nanofibers from Sapindus trifoliatus nut shells for high-performance symmetric supercapacitor applications. Carbon Lett. 31, 1133–1143 (2021).
Reddygunta, K. K. R. & Kumar, B. D. Biomass activated carbon composites and their potential in supercapacitor applications: current trends and future perspectives. Energy Fuels. 38, 10560–10588 (2024).
Yang, G. et al. A low cost, wide temperature range, and high energy density flexible quasi-solid-state zinc-ion hybrid supercapacitors enabled by sustainable cathode and electrolyte design. Nano Energy. 90, 106500 (2021).
Barzegar, F., Bello, A., Dangbegnon, J. K., Manyala, N. & Xia, X. Asymmetric supercapacitor based on activated expanded graphite and pinecone tree activated carbon with excellent stability. Appl. Energy. 207, 417–426 (2017).
Arkhipova, E. A., Novotortsev, R. Y., Ivanov, A. S., Maslakov, K. I. & Savilov, S. V. Rice husk-derived activated carbon electrode in redox-active electrolyte–new approach for enhancing supercapacitor performance. J. Energy Storage. 55, 105699 (2022).
Khuong, D. A., Tsubota, T. & Nguyen, H. N. Comprehensive Understanding of the impact of alkaline-activated bamboo-derived carbon for enhancing supercapacitor performance. J. Phys. Chem. C. 128, 17313–17327 (2024).
Dhakal, G., Kumar, D. R., Sahoo, S. & Shim, J. J. Litchi seed biowaste-derived activated carbon supporting matrix for efficient symmetric and asymmetric supercapacitors. Carbon N Y. 208, 277–289 (2023).
Wang, L., Xie, L., Wang, H., Ma, H. & Zhou, J. Sustainable synthesis of apricot shell-derived hierarchical porous carbon for supercapacitors: A novel mild one-step synthesis process. Colloids Surf. Physicochem Eng. Asp. 637, 128257 (2022).
Shanmugasundaram, E. et al. Peanut shell-derived activated carbon incorporated with nitrogen anode and Cobalt cathode materials (two-in-one strategy) for asymmetric supercapacitor (N-PAC//PVA-KOH//Co-PAC) applications. RSC Sustain. 3, 413–426 (2025).
Nagaraju, G., Cha, S. M. & Yu, J. S. Ultrathin nickel hydroxide nanosheet arrays grafted biomass-derived honeycomb-like porous carbon with improved electrochemical performance as a supercapacitive material. Sci. Rep. 7, 45201 (2017).
Wang, R. et al. NiCo2O4 hexagonal nanoplates/cornstalk-derived porous carbon composites for high-performance supercapacitor electrodes. Energy Fuels. 36, 13256–13265 (2022).
Rawal, S., Joshi, B. & Kumar, Y. Synthesis and characterization of activated carbon from the biomass of saccharum Bengalense for electrochemical supercapacitors. J. Energy Storage. 20, 418–426 (2018).
Ensafi, A. A., Moosavifard, S. E., Rezaei, B. & Kaverlavani, S. K. Engineering onion-like nanoporous CuCo2O4 Hollow spheres derived from bimetal–organic frameworks for high-performance asymmetric supercapacitors. J. Mater. Chem. A. 6, 10497–10506 (2018).
Kamari Kaverlavani, S. et al. Tunable fabrication of Hollow nano sword-like CuCo2O4 derived from bimetal–organic frameworks as binder-free electrodes. ACS Sustain. Chem. Eng. 10, 13310–13318 (2022).
Saleki, F., Mohammadi, A., Moosavifard, S. E., Hafizi, A. & Rahimpour, M. R. MOF assistance synthesis of nanoporous double-shelled CuCo2O4 Hollow spheres for hybrid supercapacitors. J. Colloid Interface Sci. 556, 83–91 (2019).
Abbasi, L., Arvand, M. & Moosavifard, S. E. Facile template-free synthesis of 3D hierarchical ravine-like interconnected MnCo2S4 nanosheet arrays for hybrid energy storage device. Carbon N Y. 161, 299–308 (2020).
Jose, R. & Yang, C. C. Standardization challenges of biomass-based carbon electrodes for supercapacitors. J Alloys Compd, 181012 (2025).
Li, H. et al. Kinetics-favorable ultrathin NiCo-MOF nanosheets with boosted pseudocapacitive charge storage for quasi-solid-state hybrid supercapacitors. Inorg. Chem. 61, 3866–3874 (2022).
Pany, S., Nashim, A., Mohanty, R. & Parida, K. Deciphering the electrochemical kinetics of sulfur vacancy-assisted nitrogen-doped NiCo2S4 combined with sulfur-doped gC3N4 towards supercapacitor applications. Mater. Adv. 6, 1442–1454 (2025).
Nashim, A., Pany, S. & Parida, K. Effect of synthesis methods on the activity of NiO/Co3O4 as an electrode material for supercapacitor: in the light of X-ray diffraction study. RSC Adv. 14, 233–244 (2024).
Pany, S., Nashim, A., Parida, K. & Nanda, P. K. Construction of NiCo2O4/Og-C3N4 nanocomposites: a battery-type electrode material for high-performance supercapacitor application. ACS Appl. Nano Mater. 4, 10173–10184 (2021).
Acknowledgements
The authors extend their gratitude to the Research Council of Isfahan University of Technology (IUT) for their valuable support in facilitating this research endeavor. There was no special funding.
Author information
Authors and Affiliations
Contributions
Farshad Tavakoli: Conceptualization, Methodology, Investigation, and Writing-original draft. Ali (A) Ensafi: Supervision, Methodology, Validation, Writing-review, and editing. (B) Rezaei: Advisor, and Validation.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Tavakoli, F., Ensafi, A.A. & Rezaei, B. Pine fruit activated carbon modified with CuCo2Se4 for the application in high-performance battery-type supercapacitor. Sci Rep 15, 32211 (2025). https://doi.org/10.1038/s41598-025-17938-7
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-17938-7
