
Abstract
Recent developments in super-resolution microscopy have revolutionized the study of cell biology. However, dense tissues require exogenous protein expression for single cell morphological contrast. In the nervous system, many cell types and/or species of interest – particularly humans – are not amenable to genetic modification and exhibit intricate anatomical specializations which make cellular and subcellular delineation challenging. Here, we present a method we call Patch2MAP for full morphological labeling of individual neurons from any species or cell type for subsequent cell-resolved protein analysis at the nanoscale. By combining patch-clamp electrophysiology with epitope-preserving magnified analysis of proteome (eMAP), our method additionally allows for correlation of physiological properties with subcellular protein expression. We applied Patch2MAP to individual spiny synapses in human cortical pyramidal neurons and demonstrated that electrophysiological AMPA-to-NMDA receptor ratios correspond tightly to respective protein expression levels. We further used Patch2MAP to investigate neuron-to-glioma synapses in surgically resected tissue from a human glioblastoma patient. Patch2MAP thus permits combined subcellular functional, anatomical, and protein analyses of any cell, opening new avenues for direct molecular investigation of the human brain in health and disease.
Similar content being viewed by others


Introduction
Super-resolution microscopy enables the investigation of proteins with nanoscopic resolution1. Synthetic gel-based super-resolution imaging techniques, which include eMAP2,3 and ExM (expansion microscopy)4 achieve ultrastructural imaging by coupling physical expansion of tissue with diffraction-limited microscopy. eMAP further provides the opportunity of highly multiplexed proteomic analysis, as processed tissue is amenable to multiple rounds of re-staining2. However, in dense, intermeshed tissue like the brain, exogenous protein expression (via genetically modified animals or viral transduction) is required to reconstruct cellular morphology and assign protein signals to correct subcellular compartments. This limits current super-resolution techniques to genetically-amenable cell types and species: for example, systematic analysis of protein distributions with cellular resolution is not currently possible in human brain tissue. Electron microscopy has previously been employed for ultrastructural analysis of human synapses5,6,7 but has thus far not been able to provide protein content information. Furthermore, full cellular reconstruction of even a single cell is extremely difficult and laborious with EM, especially for human cells. Electron microscopy with immunogold labeling further advances the capabilities of electron microscopy, yet the number of antibodies that can be used at a time, the affinity of the antibodies, the cost, and the very limited throughput has significantly restricted the use of this technique. On the other hand, super-resolution imaging techniques have been used to image synaptic proteins in the human tissue8,9,10. However, these studies lack morphological labeling, precluding the assignment of proteins to cellular and subcellular compartments. This is a critical problem, as it is unclear which cells, which subcellular compartments (i.e. synapses, axons, dendrites), and which proteins produce malfunctions that mediate brain diseases like Alzheimer’s11,12, autism13, epilepsy 14,15 or cancer16 preventing development of effective therapies. We therefore developed Patch2MAP, a method that combines patch-clamp electrophysiology with eMAP for concomitant functional and super-resolution structural, and proteomic investigation of single cells in brain tissue from any species.
Results
Combining patch-clamp electrophysiology with epitope preserving magnified analysis of the proteome (eMAP)
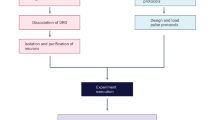
To develop a system for morphological labeling of individual cells amenable to subsequent proteomic analysis, we capitalized on the well-known approach for full anatomical reconstruction via the biocytin-streptavidin complex17. This method consists of acquiring a whole-cell patch-clamp recording of a single cell, filling the patched neuron with biocytin, chemically fixing the brain slice, and utilizing the strong non-covalent bond between streptavidin and biotin to selectively introduce a fluorophore to the filled cell. This approach allows for saturated filling of neurons and morphological reconstruction of fine processes including dendritic spines and axons. To achieve super-resolution proteomic imaging of biocytin-filled cells, we modified the original eMAP protocol (Fig. 1a). We used cortical layer 5 pyramidal cells (L5 PCs) from acute brain slices of adult mice as our model. Initially, we were unable to recover the biocytin distribution and reconstruct the neuron after the tissue-gel hybridization step of eMAP, irrespective of when we introduced the streptavidin-Alexa Fluor 488 (before or after the hydrogel-tissue hybridization). Furthermore, we were unable to recover the morphology of the neuron even after using antibodies that target streptavidin. We reasoned that hydrogel-tissue hybridization may alter the biotin-streptavidin-fluorophore complex. We therefore added an additional step of fixation before the hydrogel-tissue hybridization. We then washed the tissue thoroughly to remove any excess formaldehyde and incubated it in a hydrogel monomer solution containing 30% acrylamide, 10% sodium acrylate, 0.1% bis-acrylamide, and 0.03% VA-044. This process allowed us to preserve the biocytin-streptavidin complex and visualize the morphology of the neuron in the tissue-gel hybrid at an intermediate expansion ratio (Fig. 1b). Using a vibratome, we re-sliced the original 300 μm thick slice (typical for slice physiology) into thinner slices (Fig. 1c) and incubated the resulting slices in a denaturation solution containing 6% SDS (w/v), 0.1 M phosphate buffer, 50 mM sodium sulfite, and 0.02% sodium azide (w/v) in DI water. We then used commercially available antibodies to target synaptic proteins and subsequently expanded the tissue-gel in deionized (DI) water to visualize the ultrastructure of synapses (Fig. 1d, e).

Patch2MAP enables super-resolution morphological and proteomic imaging in neural tissue without exogenous protein expression. a, Schematic of experimental tissue processing pipeline for full morphological labelling of individual neurons and subsequent cell-delineated super-resolution proteomic analysis. b, Left column: two-photon z-stack of a mouse L5 PC filled via somatic patch pipette with Alexa-488 and biocytin in an acute slice. Middle column: confocal z-stack of the same neuron at the stage of intermediate expansion. Strepatvidin Alexa-fluor 488 reveals biocytin. Right column: overlay of the two z-stacks. c, After intermediate expansion, the 300 μm-thick acute brain slice is resliced into 4 thinner slices. Confocal z-stacks show the 4 slices which contain the neuron from b. d, Confocal z-stacks of 3x-expanded soma and branches from the neuron in b and highlighted in c. e, Magnified view of basal branch from d. Green arrowheads indicate example spines shown to the right of the branch. From left to right: image of the cell-filling biocytin channel stained with Strepatvidin Alexa-fluor488, presynaptic protein Bassoon stained with anti-guinea pig-Alexa405 (blue), NMDAR subunit GluN1 stained with anti-mouse-Alexa555 (yellow), and AMPAR subunit GluA1 stained with anti-rabbit-Alexa647 (red). Scale bars correspond to the physical dimensions of the tissue at the time of the imaging without accounting for the level of tissue expansion.
Patch2MAP enables super-resolution morphological reconstruction and multiplexed protein imaging of human neurons
To evaluate the applicability of Patch2MAP to human neurons, we acquired fresh human cortex from patients undergoing neurosurgical resection for treatment of epilepsy18,19,20,21. Blocks of cortical tissue were maintained in slicing aCSF, microdissected, and then sliced as was done for mouse cortex. We performed whole cell patch-clamp recordings of human cortical layer 2/3 pyramidal cells (L2/3 PCs) (Fig. 2a) and followed the same steps as in mice: fixation, gelation, and intermediate expansion followed by reslicing (Fig. 2b, c). Patch2MAP effectively produced bright and highly specific morphological and proteomic signals in human neurons and synapses (Figs. 2d, e, f and g and 3). Furthermore, we showed that eMAP-processed human tissue gel can endure destaining and restaining of different proteins (Figs. 2g and 3), demonstrating the suitability of this approach for the investigation of multiple proteins in each ultrastructural compartment. Thus, for the first time, Patch2MAP enables the concomitant ultrastructural investigation of human neuron morphology and protein distribution.

Patch2MAP enables super-resolution morphological and multiplexed proteomic imaging of human synapses. a, Two-photon z-stack and example somatic voltages in response to step current injections of a human L2/3 PC filled via somatic patch pipette with Alexa-488 and biocytin in an acute slice. b, Confocal z-stack of the same neuron at the intermediate expansion stage. Streptavidin Alexa-488 is used to show biocytin intracellular fill. C, Confocal images after re-slicing of the processed human brain slice into 4 thinner slices that contain the neuron shown in b. d, Confocal z-stacks of 3x expanded dendritic branch highlighted in a, b, c. e, Example basal branch from a different human neuron. Dendritic spine of interest indicated by green box. f, Magnified view of dendritic spine from e. g, Multi-round staining images of a Patch2MAP processed human tissue. Biocytin channels from both rounds were used as the reference for image registration. Round 1 (top): Bassoon (blue), GluN1 (yellow), and GluA1 & GluA2 (red). Round 2 (bottom): Shank3 (green), α-synuclein (magenta), and PSD-95 (cyan). Scale bars correspond to the physical dimensions of the tissue at the time of the imaging without accounting for the level of tissue expansion.

Examples of multi-round Patch2MAP imaging of human neocortical synapses. Biocytin channels (left) from both rounds were used as references for image registration. Round 1: Bassoon (blue), GluN1 (yellow), and GluA1 & GluA2 (red). Round 2: Shank3 (green), α-synuclein (magenta), and PSD-95 (cyan). Scale bars correspond to the physical dimensions of the tissue at the time of the imaging without accounting for the level of tissue expansion.
Correlation of physiological properties with subcellular protein expression in human neurons
We took advantage of Patch2MAP to directly test how physiological properties correspond to subcellular protein expression. We targeted two classes of glutamate receptors, AMPA and NMDA receptors, at single spiny synapses in human cortical pyramidal neurons. AMPA and NMDA receptors play key roles in excitatory synaptic transmission and plasticity in the mammalian brain22,23,24,25. Despite more than 40 years of rigorous investigation, it is still unclear how the number of receptor proteins for a given synapse relates to functional synaptic strength. Previous studies attempting to relate spine morphology to function have shown that the size of a dendritic spine predicts the size of its PSD26,27,28. Using electron microscopy (EM) and immunogold labeling of AMPA and NMDA receptors, the relationship between spine volume and synaptic strength has been further investigated29,30,31. Yet the limited numbers of these studies, the small sizes of the samples, and concerns regarding immunogold antibody affinity have made it challenging to draw definitive conclusions about the structure-function relationship. A recent study utilized a combination of slice electrophysiology and high-resolution electron microscopy to demonstrate that synapse size increases in proportion to strength, offering reasonably concrete evidence of the correlation between synapse size and strength32. However, the relationship between synaptic strength to receptor number was not addressed. Towards that end, non-stationary fluctuation analysis coupled with single spine 2P glutamate uncaging has been utilized to estimate the number of functional AMPA receptors in spines33,34.
Here, we used whole cell patch-clamp recording and rapid two-photon glutamate uncaging to measure functional AMPA and NMDA receptor properties35,36,37,38,39 at individual spiny synapses in human neurons. The response of a given spine to glutamate uncaging relies on the uncaging laser power, as well as the uncaging location relative to the synapse of interest and the local delivery of the caged glutamate compound40. Additionally, spine neck resistance and dendritic filtering also affect the amplitude of synaptic responses recorded in the soma41. To account for these potential confounds, we measured the AMPA-to-NMDA receptor ratio (AMPA: NMDA) instead of absolute amplitude. AMPA: NMDA provides a measurement that is largely independent of uncaging parameters and dendritic filtering35,36,42. We activated individual spines at a given dendritic branch with glutamate uncaging under control conditions in current-clamp mode to produce approximately physiologically-sized unitary uEPSPs (Fig. 4a). We then perfused the slice with DNQX and Mg2+-free aCSF and repeated the uncaging at the same spines (Fig. 4a, c, h). This allowed us to create a ratio of mostly AMPAR-mediated uEPSP amplitude to mostly NMDAR-mediated uEPSP amplitude. After the recording, we processed the tissue for subsequent super-resolution imaging, staining the tissue gel hybrid for GluA1 and GluA2 AMPA receptor subunits and the GluN1 NMDA receptor subunit. After ~ 3x expansion of the tissue (Extended Data Fig. 1) we identified the dendritic branch containing the spines of interest and computed the AMPA: NMDA fluorescence signal intensity for every spine that was activated by glutamate uncaging (Fig. 4a, d, h, Extended Data Fig. 2). We found that the functional AMPA: NMDA correlates strongly and significantly with the protein AMPA: NMDA (Fig. 4e, i, j, Extended Data Figs. 2, 3 and 4; n = 76 spines, 11 cells, 4 human patients; Pearson correlation coefficient = 0.65, P-value = 1.6e-10). Our results validate that protein content can be reliably inferred by the signal intensity measured with Patch2MAP. Furthermore, protein content predicts synaptic transmission strength in human cortical neurons.

Protein content measured with Patch2MAP predicts synaptic transmission strength at human synapses. a, Right: two-photon image of a human spine filled via somatic patch pipette with Alexa-488 and biocytin in an acute slice. Middle: individual (grey) and average (colored) voltage traces recorded in current clamp mode at the soma in response to glutamate uncaging at the spine in aCSF (left) and after washing in Mg2+- free aCSF with 20 µM DNQX (right). Right: confocal images of the same spine after 3x expansion, from left to right: image of the cell-filling biocytin channel stained with Streptavidin Alexa-fluor 488, AMPAR subunit GluA1 and GluA2 stained with anti-rabbit-Alexa647 (red), and NMDAR subunit GluN1 stained with anti-mouse-Alexa555 (yellow). b, Confocal z-stack of a human PC at the intermediate expansion stage. Branch of interest is indicated by green box. c, Left: 2-photon image of the branch from b during whole cell patch-clamp. Numbers indicate the uncaging locations at individual spines. Right: example averaged voltage traces for spines 3,5 &7 in response to glutamate uncaging in control aCSF (red) and after washing in Mg2+-free aCSF with 20 µM DNQX (yellow). d, Right: confocal image of the same branch in c after ~ 3x expansion through Patch2MAP processing. Left: AMPAR subunit GluR1 and GluR2 stained with anti-rabbit-Alexa647 (red) and NMDAR subunit NR1 stained with anti-mouse-Alexa555 (yellow) superimposed on cell-filling biocytin channel stained with Streptavidin Alexa-fluor 488 for spines 3,5 &7. e, Mean AMPAR/NMDAR uEPSP ratio versus AMPAR/NMDAR signal intensity ratio. Numbers correspond to the spines shown in c, d and e. The data are fit with a line of slope 0.4735; r: correlation coefficients, P: P value. f, As in b for another human PC. g, Top: 2-photon image of the branch from f in acute slice, Bottom: confocal image of the same branch after ~ 3x expansion. h, As in c & d for example spines 1,2 & 3 from g. i, As in e. The data are fit with a line of slope 0.16576. j, Population correlation of mean AMPAR/NMDAR uEPSP ratio with normalized AMPAR/NMDAR signal intensity ratio (n = 76 spines, 11 cells, 4 humans). The data are fit with a line of slope 0.33. (r) correlation coefficients, (P) P value. Scale bars correspond to the physical dimensions of the tissue at the time of the imaging without accounting for the level of tissue expansion.
Investigation of synapses in human brain cancer cells
Glioblastoma (GBM) is the most common primary brain tumor in adults. It exhibits poor prognosis even with maximal therapy43. Interestingly, glioblastomas synaptically integrate into surrounding neuronal networks by forming electrochemical synapses44,45. Recent studies have shown that neuronal activity promotes tumor growth46,47,48 and that tumors themselves can remodel the activity and function of neural circuits49. However, it remains a major challenge to understand GBM disease mechanisms, since glioblastomas exhibit a profuse cellular heterogeneity in their composition50,51 and each cell’s contributions cannot be studied in isolation. Towards this goal, many animal models have been engineered to recapitulate the human disease, but each of them captures only a subset of glioblastoma manifestations in the human brain. Patch2MAP is uniquely positioned to investigate both the function and molecular composition of human GBM on a single cell level embedded in the complex native tumor environment.
We therefore acquired freshly resected cortical tissue from a patient with glioblastoma (Fig. 5a) undergoing surgical treatment. The patient was administered 5-ALA preoperatively, which is metabolized to fluorescent protoporphyrin IX (PpIX) specifically in tumor cells and allows intra-operative delineation of tumor borders from surrounding brain52. We prepared a 5-ALA labeled tissue block arising from the upper border of the tumor for Patch2MAP analysis. We identified tumor cells using two-photon imaging. Tumor cells exhibited 5-ALA-mediated fluorescence, which was nucleus excluded (Fig. 5b). Under visual guidance, we performed patch-clamp whole-cell current-clamp recording from the tumor cell (Fig. 5c). The voltage recording revealed spontaneous slow oscillatory activity (Fig. 5c), as well as faster depolarizations resembling synaptically-driven electrical activity (Fig. 5c inset). Recent studies have highlighted the presence of neuron-to-glioma synapses in human GBM with transcriptomic and pharmacologic experiments arguing for the existence of AMPARs44,45. To test this hypothesis and directly visualize synaptic receptors on human resected GBM tissue, we processed the patched and filled tumor cell through Patch2MAP and stained for AMPARs (GluA1&GluA2), NMDARs (GluN1) and Bassoon. During super-resolution confocal imaging, we observed synapses on the surface of the tumor cell of interest that contained both AMPAR and NMDAR receptors (Fig. 5d, e). Calcium influx plays a central in the growth and invasiveness of glioblastoma, orchestrating processes which normally take place during the development of the nervous system16. Calcium permeable AMPARs of neuron-to-glioma synapses have been proposed to facilitate this calcium influx in tumor. Here, we show that neuron-to-glioma synapses, in the tumor cell we imaged, have both AMPAR and NMDAR receptors, proposing a new potential mechanism of how neuronal activity can induce calcium influx in the tumor cells and promote tumor growth. This is an example of how Patch2MAP can be used to investigate diseased human tissue.

Patch2MAP reveals synaptic protein organization in human GBM cells similar to neurons. a, MRI-FLAIR image of a patient with GBM who received 5-aminolevulic acid (5-ALA) preoperatively. b, Two-photon single-plane image of acute brain slice from the border of the tumor from the patient in a containing labeled cancer cells (cell of interest indicated with white arrow). Note that the nucleus is not filled by 5-ALA. c, Two-photon z-stack and example spontaneous somatic voltages of labeled cancer cell from b filled via somatic patch pipette with Alexa-488 and biocytin. Spontaneous oscillations (upper trace) and magnified faster membrane depolarizations (bottom trace, red inset). d, Confocal z-stacks of re-sliced and expanded cancer cell from b and c. e, Synaptic nanoclusters formed on the membrane of the cancer cell are consistent with functional synapses. They contain NMDA receptors (GluN1, yellow), AMPA receptors (GluA1 & GluA2, red) and Bassoon (blue). Scale bars correspond to the physical dimensions of the tissue at the time of the imaging without accounting for the level of tissue expansion.
Discussion
We introduce a new method to perform super-resolution proteomic microscopy at identified subcellular structures without genetic modification by coupling patch-clamp electrophysiology with eMAP. Previous work has combined patch-clamp physiology with STORM imaging in rodent brain, revealing protein distributions in presynaptic boutons53. In contrast with Patch2MAP, STORM requires a highly specialized microscope set up, photo-switchable fluorophores, and complicated post imaging processing. Furthermore, reconstructing the full neuron or cell is highly challenging with STORM. Our approach provides a generally applicable framework to visualize subcellular morphology and protein content of neurons and non-neuronal cells without exogenous protein expression, using conventional confocal microscopy.
Although significantly more affordable than other techniques like EM and STORM, Patch2MAP still requires antibodies, which can be costly, access to fresh tissue, which is often limited, and specialized expertise to integrate electrophysiology, which adds complexity. Throughput is another limitation—whereas genetic manipulations (which are not feasible in humans) can label thousands of neurons simultaneously, and EM can capture every cell in a given tissue block, Patch2MAP typically yields detailed information for only a handful of cells per experiment (with an upper limit of dozens of neurons per slice when fully optimized). Despite this low throughput, however, Patch2MAP uniquely enables direct linkage of electrophysiological data with protein content at subcellular resolution. Additionally, broad whole-slice protein analysis also remains possible in Patch2MAP, as with other expansion microscopy techniques. As with all fluorescence-based imaging, reported protein abundance can vary between experiments. While this limitation is most pronounced when different antibodies or lots are used across experiments, standardized workflows — including consistent antibody sourcing, aliquoting to minimize freeze–thaw cycles, and batch controls — can further reduce variability. Despite these limitations, the unique combination of functional and molecular information that Patch2MAP provides opens new opportunities to link electrophysiological properties with protein expression.
We link biophysical properties to protein profiles at the level of individual synapses and further show that signal intensity of labeled proteins measured through Patch2MAP can be reliably used to infer the protein concentration. Being able to visualize proteins at the nanoscale in human tissue can accelerate scientific discovery in the field of human neurobiology, where effective treatments are largely missing. We used resected human GBM tissue to visualize, for the first time in human tissue, the molecular nanostructure of neuron-to-glioma synapses. Targeting AMPAR has shown promise in the treatment of glioblastoma preclinically and in early clinical studies54. Understanding the complete synaptic repertoire of neuron-to-glial synapses has the potential to identify new and specific clinical targets for treatment. GBM is only one example where the complex tumor environment creates a highly heterogeneous cell population. This feature heavily relies on the healthy brain-tumor interactions. Other diseases such as dementias, autism and psychiatric disorders arise and manifest in similar ways, via an interaction of many parameters, necessitating the study of neuronal and synaptic dysfunction in the intact human nervous system.
Our method enables the ultrastructural cell-delineated proteomic analysis of any neuron of any species, including humans, and paves the way for further studies in brain health and disease. While animal models are key for understanding human disease mechanisms and probing potential therapies55there are significant challenges to creating useful models of human neurological disorders due the divergence of human brain organization and function56,57,58. Patch2MAP allows for the direct investigation of the human brain and the underlying mechanisms of neurological diseases.
Materials and methods
Animals
Human
Human (Homo sapiens) tissue was acquired through collaboration with the Massachusetts General Hospital (MGH) and the Brigham and Women’s Hospital (BWH). For MGH, tissue was obtained as ‘discarded tissue’ from neurosurgical patients in accordance with protocols approved by the Massachusetts General Hospital Internal Review Board (IRB). Patients consent to surgery and a subset of the resected tissue was considered discarded tissue. Under MGH’s IRB-approved protocol, such discarded tissue was available for this specific research project for use without explicit patient consent. For BWH, the protocol was approved by the IRB, and patients provided consent prior to the surgery. At both institutions, non-essential samples were extracted by the supervising neuropathologist per protocol.
Patients were women or men aged 23–71 years. Additional patient information is included in Extended Data Table 1. Samples were not allocated to distinct experimental groups and information about the patient was not available until after data acquisition and analysis.
Mouse
All animal procedures were done in compliance with the NIH and Massachusetts Institute of Technology Committee on Animal care guidelines. We used C57BL/6 mice (Charles River Laboratories). Male and female mice were used in approximately equal numbers for all experiments at 8–10 weeks of age. Mice were kept on a 12-hour light/dark cycle and had unrestricted access to food and water.
As applicable, all methods described in this study adhered to the ARRIVE guidelines (Animal Research: Reporting of In Vivo Experiments).
Brain slice preparation
Human slice preparation
Resected human tissue was considered discarded tissue after being examined by neuropathologists whose main objective was to ensure there was adequate tissue for diagnostic purposes. Neocortical tissue was obtained from the temporal and frontal lobe in patients undergoing resection for medically-intractable epilepsy. The neocortical tissue displayed no known abnormalities at the level of MRI scans, gross inspection, and subsequent microscopic examination as part of the standard neuropathologic assessment of the tissue. GBM tissue was obtained from a patient who received 5-aminolevulic acid (5-ALA) preoperatively. 5-ALA is metabolized to fluorescent protoporphyrin IX (PpIX) specifically in tumor cells. The tumor was identified intra-operatively by 5-ALA induced PpIX fluorescence. Patients undergoing resective surgery were primarily maintained under general anesthesia with propofol and remifentanil or sufentanil. Some cases utilized inhaled anesthetics, such as isoflurane or sevoflurane. For induction of general anesthesia, paralytic agents including rocuronium or succinylcholine as well as fentanyl were typically used. Resection usually occurred within 90 min of the start of the procedure.
After resection, tissue was placed in ice-cold cutting solution containing (in mM): sucrose 165, sodium bicarbonate 25, potassium chloride 2.5, sodium phosphate 1.25, calcium chloride 0.5, magnesium chloride 7, glucose 20, HEPES 20, sodium pyruvate 3, and sodium ascorbate 5, 295–305 mOsm, equilibrated with 95% O2 and 5% CO2. Samples were transported in sealed conditions for ~ 20 min before being transferred to freshly oxygenated solution. Pia and surface blood vessels that would obstruct slicing were removed. Slicing was performed with a Leica VT1200S vibratome in ice-cold cutting solution. 300 μm-thick slices were incubated for ≥ 30 min at 36 °C in recovery solution containing (in mM): sodium chloride 90, sodium bicarbonate 25, potassium chloride 2.5, sodium phosphate 1.25, calcium chloride 1, magnesium chloride 4, glucose 20, HEPES 20, sodium pyruvate 3, and sodium ascorbate 5, 295–305 mOsm, equilibrated with 95% O2 and 5% CO2. Slices were then stored at room temperature until use. Incubation solutions were replaced every ∼8 h and recordings were performed up to 34 h after slicing. GBM tissue was not sliced since the thickness of the resected piece was already less than 500 μm. GBM tissue was otherwise treated the same as the other human resected cortical tissue.
Mouse slice preparation
Coronal brain slices (300 μm) containing the primary visual cortex (V1) were prepared from 8- to 10-week-old C57BL/6 mice. Animals were deeply anesthetized with isoflurane prior to decapitation. The brain was removed and sliced with a vibratome (Leica) in ice-cold slicing solution containing (in mM): sucrose 90, NaCl 60, NaHCO3 26.5, KCl 2.75, NaH2PO4 1.25, CaCl2 1.1, MgCl2 5, glucose 9, sodium pyruvate 3, and ascorbic acid 1, saturated with 95% O2 and 5% CO2. Slices were incubated in artificial cerebrospinal fluid (aCSF) containing (in mM): NaCl 120, KCl 3, NaHCO3 25, NaH2PO4 1.25, CaCl2 1.2, MgCl2 1.2, glucose 11, sodium pyruvate 3, and sodium ascorbate 1, saturated with 95% O2 and 5% CO2 at 35.5 °C for 25–30 min and then stored at room temperature.
Recording aCSF containing (in mM): sodium chloride 120, potassium chloride 3, sodium bicarbonate 25, sodium phosphate monobasic monohydrate 1.25, calcium chloride 1.2, magnesium chloride 1.2, glucose 11, sodium pyruvate 3, and sodium ascorbate 1, 302–305 mOsm, saturated with 95% O2 and 5% CO2.
Patch2MAP protocol
Patch-clamp recording and Biocytin filling
Patch-clamp recordings were performed from the soma of pyramidal neurons at 34–36 °C in recording aCSF containing (in mM): sodium chloride 120, potassium chloride 3, sodium bicarbonate 25, sodium phosphate monobasic monohydrate 1.25, calcium chloride 1.2, magnesium chloride 1.2, glucose 11, sodium pyruvate 3 and sodium ascorbate 1, 302–305 mOsm, saturated with 95% O2 and 5% CO2.
An Olympus BX-61 microscope with infrared Dodt optics and a 60x water immersion lens (Olympus) was used to visualize cells. A two-photon laser scanning system (Prairie Technologies Ultima) with an ultrafast pulsed laser beam (Mai Tai DeepSee lasers) was used to identify tumor cells labeled with 5-ALA-induced PpIX fluorescence at two-photon wavelength of 880 nm. The fluorescence formed a rim around the nucleus of the tumor cells as reported previously45. For GBM tissue, Dodt gradient contrast imaging was used to match the fluorescent image of the tumor cell to it’s brightfield counterpart. Whole-cell current-clamp recordings were performed in bridge mode with a Dagan BVC-700 amplifier with bridge fully balanced. Current and voltage signals were filtered at 10 kHz and digitized at 20 kHz. Patch pipettes were prepared with thin-wall glass (1.5 O.D., 1.1 I.D.). Pipettes had resistances ranging from 3 to 7 MΩ and the capacitance was fully neutralized prior to break in. Series resistances ranged from 7 to 17 MΩ. The intracellular solution contained (in mM): potassium gluconate 134, KCl 6, HEPES buffer 10, NaCl 4, Mg2ATP 4, NaGTP 0.3, phosphocreatine di(tris) 14, 0.1 Alexa 488 (Invitrogen), 5.2 Biocytin (Invitrogen).
During the patch-clamp recording biocytin diffused from the patch pipette to the neuron. A recording time of at least 10 min was sufficient to completely fill the axon and the dendrites of large pyramidal neurons in mouse neocortical layers 5 and human neocortical layers 2 and 3. Longer times may be needed for larger neurons or for studies performed at room temperature. Upon completion of the electrophysiological recording, outside-out patch configuration was established by slowly retracing the recording pipette in voltage clamp mode under visual control. The capacitance and input resistance were monitored and loss of capacitive transients with the collapse of the current responses to a straight line ensured resealing of the cell membrane.
The tissue was then processed through our modified protocol for eMAP2 which allowed recovery of biocytin signals for subsequent morphological reconstruction.
Fixation and biocytin labelling
300 μm thick slices were transferred in 15 ml Falcon tubes containing 10 ml of 4% PFA in phosphate-buffered saline (PBS) at 4 °C and incubated overnight. Slices were incubated in 50 ml Falcon tubes containing 40 ml of PBS for at least 24 h at 4 °C. Slices were transferred subsequently into 48-well plates containing 200 µl of washing solution (PBS containing 0.1% (w/v) Triton X-100(PBST) and 0.02% (w/v) sodium azide) on 37 °C shaker for 4 h. PBST was at least once exchanged before adding 20 µl of Streptavidin, Alexa Fluor™ 488 Conjugate (S32354, Thermo Fisher, 0.2% (w/v)) and slices were incubated on 37 °C shaker overnight. Slices were washed 4 times for 10 min each in PBST. This washing process was repeated twice with an interval of 2 h and slices were transferred to 4% PFA in phosphate-buffered saline (PBS) at 4 °C and incubated for 8–12 h. Slices were switched to PBS at 4 °C for at least 36 h before performing confocal imaging with excitation at 488 nm to image biocytin filled-neurons.
Gelation
Slices were incubated in eMAP hydrogel monomer solution (30% acrylamide (A9099, MilliporeSigma, St. Louis, MO, USA), 10% sodium acrylate (408220, MilliporeSigma), 0.1% bis-acrylamide (161 − 0142, Bio-Rad Laboratories, Hercules, CA, USA), and 0.03% VA-044 (w/v) (Wako Chemicals, Richmond, VA, USA) in PBS) at 4 °C overnight. Then, slices were carefully placed between two glass slides using Blu-Tack adhesive (Bostik, Essendon Fields, Victoria, Australia) and with the help of ~ 290 μm thick homemade spacers. The empty space was filled with additional hydrogel monomer solution. The glass slide setup was then placed inside a 50 ml Falcon tube, which was subsequently placed inside Easy-Gel (LifeCanvas Technologies, Cambridge) with nitrogen gas at 37 °C for 3 h. After gelation, the cartridge was disassembled, and excess gel was trimmed from around the lateral edges of the sample using a razor blade. The resulting tissue-gel hybrid sample was put in PBS with 0.02% sodium azide at 37 °C shaker overnight for hydration.
Re-slicing
The tissue gel hybrid was first expanded by 1.7x. Confocal imaging was performed to identify the neuron and to determine slicing orientation. A thin layer of Krazy Glue (Elmer’s products, inc. 460 Polaris Parkway Westerville, OH 4308) was applied on a 10 mm non-orienting specimen disc (14048143399, Leica Biosystems, Germany). An 300 μm thick slice was placed (neuron closer to the slicing surface) on the glue in the specimen disc and light pressure is applied with a cover slip to make sure uniform application of glue. The tissue gel hybrid was then resliced using a vibratome (VT1200, Leica Biosystems, Germany) in 75 μm (44 μm original) thick slices. From the resulting slices, the ones that contain the neuron of interest are further trimmed with a razor blade to final dimensions of ~ 3 mm*3 mm*75µm.
Denaturation and immunostaining
The slice was incubated in a denaturation solution [6% SDS (w/v), 0.1 M phosphate buffer, 50 mM sodium sulfite, and 0.02% sodium azide (w/v) in DI water] at 37 °C shaker for 6 h. Subsequently, the samples were transferred in washing solution (PBS containing 0.1% (w/v) Triton X-100 (PBST) and 0.02% (w/v) sodium azide) at 37 °C shaker overnight. The washing solution was exchanged at least once before adding the primary antibodies. The slice was incubated with primary antibodies (typical dilution 1:20) in PBST at 37 °C shaker overnight, followed by washing in PBST at 37 °C for 6 h with 4 solution exchanges. The same process was repeated for secondary antibodies (typical dilution 1:10).
The following primary antibodies were used: GluN1: Anti-NMDAR1 (SYSY 114011), GluA1: Anti-AMPAR1 (SYSY 182003), GluA2: Anti-AMPAR2 (SYSY 182103), Bassoon: Anti-Bassoon (SYSY 141004), Shank3: Anti-SHANK3 (ThermoFisher Scientific PA5-34383), Alpha-synuclein: Anti-alpha Synuclein (ThermoFisher Scientific AHB0261), PSD-95: Anti-PSD-95 (Alomone Labs APZ-009-GP). The following secondary antibodies were used: Goat Anti-Guinea pig IgG H&L (Alexa Fluor® 405) (Abcam ab175678), Goat anti-Mouse IgG (H + L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor Plus 555 (ThermoFisher Scientific A32727), Goat anti-Rabbit IgG (H + L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor Plus 647 (ThermoFisher Scientific A32733). The final concentrations for the above-mentioned antibodies are: 1/24 mg/ml for GluN1, GluA1, GluA2, Bassoon, Shank3, Alpha-synuclein, 1/30 mg/ml for PSD-95 and 2/23 mg/ml for anti-guinea pig 405, anti-mouse 555, anti-rabbit 647.
Mounting and image acquisition
Before each imaging session the slice was incubated in DI water for 1 m to reach final expansion (3x). Then the expanded specimen was placed between a petri dish and a glass-bottom Willco dish (HBSB-5030; WillCo Wells, Amsterdam, The Netherlands) using glass coverslips as spacers. To prevent the samples from drying up during image acquisition, the void space around the samples was filled with DI water. Subsequently, the samples were imaged using a Zeiss LSM 900-AS microscope system using a 63 × 1.2 NA water immersion objective or a Leica TCS SP8 microscope system using a 63 × 1.2 NA water immersion objective.
Multi-round staining
Previously stained tissues were incubated in denaturation solution for 6 h at 37 °C, followed by 10 m at preheated 95 °C denaturation solution to remove bound antibodies. Tissue specimens were then washed in PBST at 37 °C overnight. PBST was exchanged at least twice. The samples were imaged on the microscope to confirm the complete loss in the signal from the antibodies. Tissue was then immunostained as described above.
Expansion factor measurement
To measure the expansion factor, we imaged the same cells at 3 time points: first, during 2-photon imaging of patched cell, then at intermediate expansion of the gel-tissue hybrid, and finally after final expansion (13 human slices). For each cell we calculated the mean expansion ratio from at least 3 measurements among preserved neuronal structures along the x, y dimensions.
Glutamate uncaging
A two-photon laser scanning system (Prairie Technologies Ultima) with dual galvanometers and two ultrafast pulsed lasers beams (Mai Tai DeepSee lasers) were used to simultaneously image and uncage glutamate. One path was used to image Alexa 488 at 920 nm. The other path was used to photolyse MNI-caged L-glutamate (Tocris) at 720 nm. Stock MNI-glutamate solutions (50 mM) were freshly diluted in Mg2+ -free aCSF to 10 mM and a Picospritzer (General Valve) was used to focally apply the MNI-caged L-glutamate via pressure ejection through a large glass pipette above the slice. Laser beam intensity was independently controlled with electro-optical modulators (model 350 − 50; Conoptics). Emitted light was collected by GaAsP photomultipliers. Uncaging dwell time was 0.2 ms. A passive 8X pulse splitter in the uncaging path was used to reduce photodamage59,60. Experiments were not further analyzed if diffuse signs of photodamage were detected (increase in basal fluorescence, loss of transient signals, and/or persistent depolarization).
Measurement of AMPA-to-NMDA receptor ratio at individual spines
Uncaging locations were positioned in close vicinity of spines (< 0.5 μm) from the tip of individual spine heads in the radial direction. 6–12 spines were individually stimulated at each branch. The uncaging stimulus was delivered in each spine separately (using a stimulus interval of 500 ms). Unitary uEPSPs at each spine were evoked 20 to 40 times and responses were averaged. Mg2+ free aCSF containing 20 µM DNXQ was then washed on for at least 15 min. The same uncaging protocol was repeated at the same spines. Care was taken to maintain the initial uncaging locations and focal plane throughout the experiment.
Measurement of uEPSP amplitudes
The AMPAR-mediated and NMDAR-mediated uEPSP amplitude for each spine was measured offline using custom-written MATLAB code. Briefly, the peak amplitude of each recorded uEPSP was measured during the peak window (50 ms post glutamate uncaging). The width of the window was chosen with consideration of both AMPAR and NMDAR dynamics. Baseline potential was calculated as the average voltage in the 50 ms preceding the evoked uEPSP and was then subtracted from the measurement of the peak uEPSP to provide the individual uEPSP amplitude. Individual uEPSP amplitudes (20–40) were averaged to provide AMPAR-mediated and NMDAR-mediated uEPSPs amplitude for each spine.
Image analysis
Identification of spines of interest
The identification of spines of interest is based on: (1) gross morphology (identification of the branch) and (2) spine imaging (identification of the spines). Both are achieved by comparing the 2-photon (physiology step) and confocal (expansion step) images in terms of geometry, branching points, and relative location to other branches/spines. Examples for identifying the same branches before and after expansion are shown in Figs. 1 and 2, and for identifying the same spines before and after the expansion are shown in Fig. 4c, d, g. The spines, which do not have an unambiguous match in the expanded tissue (Extended Data Fig. 3b, c) are excluded (see also Correlation analysis).
Measurement of signal intensity in AMPAR and NMDAR channels
Dendritic spines were analyzed in the biocytin channel using Fiji software. We used a custom-written macro code that first applies a median blur (2 pixels) in the biocytin image and then converts the tip of individual spines to binary masks by thresholding the resulting biocytin image. The rest of the analysis was performed using custom-written MATLAB code. Each image had 4 channels (bassoon, biocytin, NMDAR, AMPAR) and multiple z-slices (z-step of 0.66 μm). The signal intensity in each of the four channels was calculated at every z-slice as the intensity difference of the mask containing the structure of interest and the background. The background intensity was calculated by using the same mask but at random x-y locations of the image to account for the effect of size in the measured intensity among dendritic protrusions. The z-slices that contained the spine of interest were defined as the z-slices where the biocytin intensity signal for a given spine was greater than the median biocytin signal intensity for the same spine from every z-slice + 1 SD. The signal intensity for the AMPAR and NMDAR channels for a given spine was calculated as the sum of the signal intensity in all the z-slices that contained the respective spine, while the controls (Extended Data Fig. 2) were calculated by 1000 random combinations of the same number of slices that did not contain the respective spine.
Z-score normalization of signal intensity
In order to pool the data of all cells in a single plot (Fig. 4j, Extended Data Fig. 3e), we z-scored the AMPAR: NMDAR signal intensity among each subset of cells, which were treated with the same antibodies. The polyclonal antibodies used in our experiments (GluA1 subunit,182 003 Sysy and GluA2 subunit,182 103 Sysy) are a complex mixture of several antibodies and commercial vials differ from each other. Normalization allows to account for differences of the inherent signal intensity of secondary antibodies.
Correlation analysis
We analyzed 11 dendritic branches (91 spines and 11 cells from 4 humans) where we performed the uncaging experiment and then reconstructed the full length of the dendritic segments with Patch2MAP. From the 2-photon data, 4 spines (4/91) were excluded because they showed signs of photodamage. From the eMAP data, 5 spines (5/91) were excluded because the super-resolution morphological imaging revealed that 2 spines instead of 1 were targeted by the 2-photon glutamate uncaging, and 6 spines (6/91) were excluded because the uncaging location did not correspond to a distinct spine in the expanded branch (Extended Data Fig. 3). The signal intensity for those was calculated either in the photodamaged spine or in one of the two spines or in the adjacent dendritic branch (shaft synapses). These exclusions did not change the results presented in the main figures (Extended Data Fig. 3e).
References
Sahl, S. J., Hell, S. W. & Jakobs, S. Fluorescence nanoscopy in cell biology. Nat. Rev. Mol. Cell Biol. 18, 685–701 (2017).
Park, J. et al. Epitope-preserving magnified analysis of proteome (eMAP). Sci. Adv. 7, 1–14 (2021).
Ku, T. et al. Multiplexed and scalable super-resolution imaging of three-dimensional protein localization in size-adjustable tissues. Nat. Biotechnol. 34, 973–981 (2016).
Chen, F., Tillberg, P. W. & Boyden, E. S. Optical imaging. Expansion microscopy. Science 347, 23 (2015).
Molnár, G. et al. Human pyramidal to interneuron synapses are mediated by multi-vesicular release and multiple docked vesicles. Elife 5, 24 (2016).
Boldog, E. et al. Transcriptomic and morphophysiological evidence for a specialized human cortical GABAergic cell type. Nat Neurosci 21, 78 (2018).
Shapson-Coe, A. et al. A connectomic study of a petascale fragment of human cerebral cortex. BioRxiv (2021).
Kay, K. R. et al. Studying synapses in human brain with array tomography and electron microscopy. Nat. Protoc. 8, 752 (2013).
Micheva, K. D. et al. Distinctive structural and molecular features of myelinated inhibitory axons in human neocortex. eNeuro 5, 1–12 (2018).
Park, J. et al. Integrated platform for multi-scale molecular imaging and phenotyping of the human brain. BioRxiv (2023).
Wang, J., Gu, B. J., Masters, C. L. & Wang, Y. J. A systemic view of alzheimer disease - Insights from amyloid-β metabolism beyond the brain. Nat. Rev. Neurol. 13, 612–623 (2017).
Götz, J., Bodea, L. G. & Goedert, M. Rodent models for alzheimer disease. Nat. Rev. Neurosci. 19, 583–598 (2018).
Lord, C., Elsabbagh, M., Baird, G. & Veenstra-Vanderweele, J. Autism spectrum disorder. Lancet 392, 508–520 (2018).
Barker-Haliski, M. & Steve White, H. Glutamatergic mechanisms associated with seizures and epilepsy. Cold Spring Harb Perspect. Med. 5, 1–15 (2015).
Finnema, S. J. et al. Imaging synaptic density in the living human brain. Sci. Transl. Med. 8, 1–10 (2016).
Mancusi, R. & Monje, M. The neuroscience of cancer. Nature 618, 467–479 (2023).
Liu, F., Zhang, J. Z. H. & Mei, Y. The origin of the cooperativity in the streptavidin-biotin system: a computational investigation through molecular dynamics simulations. Sci. Rep. 6, 753 (2016).
Testa-Silva, G. et al. High bandwidth synaptic communication and frequency tracking in human neocortex. PLoS Biol 12, (2014).
Berg, J. et al. Human neocortical expansion involves glutamatergic neuron diversification. Nature 598, 151–158 (2021).
Beaulieu-Laroche, L. et al. Allometric rules for mammalian cortical layer 5 neuron biophysics. Nature 600, 274–278 (2021).
Beaulieu-Laroche, L. et al. Enhanced dendritic compartmentalization in human cortical neurons. Cell 175, 643–651e14 (2018).
Henley, J. M. & Wilkinson, K. A. Synaptic AMPA receptor composition in development, plasticity and disease. Nat. Rev. Neurosci. 17, 337–350 (2016).
Meldrum, B. S. Glutamate as a neurotransmitter in the brain: review of physiology and pathology. J. Nutr. 130, 1007–1015 (2000).
Malenka, R. C. & Nicoll, R. A. NMDA-receptor-dependent synaptic plasticity: multiple forms and mechanisms. Trends Neurosci. 16, 521–527 (1993).
Huganir, R. L. & Song, I. Regulation of AMPA receptors during synaptic plasticity. TRENDS Neurosci. 25, 578–588 (2002).
Arellano, J. I., Benavides-Piccione, R., DeFelipe, J. & Yuste, R. Ultrastructure of dendritic spines: correlation between synaptic and spine morphologies. Front. Neurosci. 1, 74 (2007).
Harris, K. M., Jensen, F. E. & Tsao, B. Three-dimensional structure of dendritic spines and synapses in rat hippocampus (CA 1) at postnatal day 15 and adult ages: implications for the maturation of synaptic physiology and long-term potentiation. J. Neurosci. 12, 754 (1992).
Harris, K. M. & Stevens, J. K. Dendritic spines of CA1 pyramidal cells in the rat hippocampus: serial electron microscopy with reference to their biophysical characteristics. J. Neurosci. 9, 785 (1989).
Kharazia, V. N. & Weinberg, R. J. Immunogold localization of AMPA and NMDA receptors in somatic sensory cortex of albino rat. J. Comp. Neurol. 412, 292–302 (1999).
Kharazia, V. N., Phend, K. D., Rustioni, A. & Weinberg, R. J. EM colocalization of AMPA and NMDA receptor subunits at synapses in rat cerebral cortex. Neurosci. Lett. 210, 475 (1996).
Takumi, Y., Ramírez-León, V., Laake, P., Rinvik, E. & Ottersen, O. P. Different modes of expression of AMPA and NMDA receptors in hippocampal synapses. Nat. Neurosci. 2, 618–624 (1999).
Holler, S., Köstinger, G., Martin, K. A. C., Schuhknecht, G. F. P. & Stratford, K. J. Structure and function of a neocortical synapse. Nature 591, 111–116 (2021).
Matsuzaki, M. et al. Dendritic spine geometry is critical for AMPA receptor expression in hippocampal CA1 pyramidal neurons. Nat. Neurosci. 4, 1086–1092 (2001).
Tanaka, J. I. et al. Number and density of AMPA receptors in single synapses in immature cerebellum. J. Neurosci. 25, 412 (2005).
Lafourcade, M. et al. Differential dendritic integration of long-range inputs in association cortex via subcellular changes in synaptic AMPA-to-NMDA receptor ratio. Neuron 2022, 1–15. https://doi.org/10.1016/j.neuron.2022.01.025 (2022).
Yaeger, C. E., Vardalaki, D., Brown, N. J. & Harnett, M. T. Dendritic compartmentalization of input-specific integration and plasticity rules across cortical development. bioRxiv 02.02.478840 (2022).
Malinow, R. & Malenka, R. C. AMPA receptor trafficking and synaptic plasticity. Annu. Rev. Neurosci. 25, 103–126 (2002).
Magee, J. C. & Cook, E. P. Somatic EPSP amplitude is independent of synapse location in hippocampal pyramidal neurons. Nat. Neurosci. 3, 895–903 (2000).
Bekkers, J. M. & Stevens, C. F. NMDA and non-NMDA receptors are co-localized at individual excitatory synapses in cultured rat hippocampus. Nature 341, 230–233 (1989).
Ellis-Davies, G. C. R. Two-Photon uncaging of glutamate. Front. Synaptic Neurosci. 10, 1–13 (2019).
Rall, W. Theoretical significance of dendritic trees for neuronal input-output relations. Neural Theory Model. ed. R.F. Reiss Stanf. Univ. Press (1964).
Vardalaki, D., Chung, K. & Harnett, M. T. Filopodia are a structural substrate for silent synapses in adult neocortex. Nature 612, 475 (2022).
Ostrom, Q. T. et al. National-level overall survival patterns for molecularly-defined diffuse glioma types in the united States. Neuro Oncol. https://doi.org/10.1093/neuonc/noac198 (2022).
Venkatesh, H. S. et al. Electrical and synaptic integration of glioma into neural circuits. Nature 573, 539–545 (2019).
Venkataramani, V. et al. Glutamatergic synaptic input to glioma cells drives brain tumour progression. Nature 573, 532–538 (2019).
Venkataramani, V. et al. Glioblastoma hijacks neuronal mechanisms for brain invasion. Cell 185, 2899–2917e31 (2022).
Venkatesh, H. S. et al. Targeting neuronal activity-regulated neuroligin-3 dependency in high-grade glioma. Nature 549, 533–537 (2017).
Venkatesh, H. S. et al. Neuronal activity promotes glioma growth through neuroligin-3 secretion. Cell 161, 803–816 (2015).
Krishna, S. et al. Glioblastoma remodelling of human neural circuits decreases survival. Nature 617, 963 (2023).
Garofano, L. et al. Pathway-based classification of glioblastoma uncovers a mitochondrial subtype with therapeutic vulnerabilities. Nat Cancer 2, 78 (2021).
Neftel, C. et al. An integrative model of cellular states, plasticity, and genetics for glioblastoma. Cell 178, 745 (2019).
Hadjipanayis, C. G., Widhalm, G. & Stummer, W. What is the surgical benefit of utilizing 5-aminolevulinic acid for fluorescence-guided surgery of malignant gliomas? Neurosurgery 77, 856 (2015).
Dudok, B. et al. Cell-specific STORM super-resolution imaging reveals nanoscale organization of cannabinoid signaling. Nat. Neurosci. 18, 75–86 (2015).
Izumoto, S. et al. Seizures and tumor progression in glioma patients with uncontrollable epilepsy treated with perampanel. Anticancer Res. 38, 253 (2018).
van der Worp, H. B. et al. Can animal models of disease reliably inform human studies?? PLOS Med. 7, e1000245 (2010).
Nestler, E. J. & Hyman, S. E. Animal models of neuropsychiatric disorders. Nat. Neurosci. 13, 1161–1169 (2010).
Rice, J. Animal models: not close enough. Nature 484, S9–S9 (2012).
Hartung, T. Thoughts on limitations of animal models. Park Relat. Disord. 14, 83–85 (2008).
Ji, N., Magee, J. C. & Betzig, E. High-speed, low-photodamage nonlinear imaging using passive pulse splitters. Nat. Methods. 5, 197–202 (2008).
Harnett, M. T., Xu, N. L., Magee, J. C. & Williams, S. R. Potassium channels control the interaction between active dendritic integration compartments in layer 5 cortical pyramidal neurons. Neuron 79, 516–529 (2013).
Acknowledgements
We thank Dae Hee Yun and Kwanghun Chung for sharing their expertise on eMAP, and Lukas Fisher, Marie-Sophie van der Goes, Jakob Voigts, and Courtney Yaeger for constructive criticism of the manuscript.
Funding
Boehringer Ingelheim Fonds Foundation (D.V.). NIH RO1NS106031 (M.T.H.). NIH R01NS113079 (M.T.H.). James W. and Patricia T. Poitras Fund at MIT (M.T.H.)
Author information
Authors and Affiliations
Contributions
D.V. and M.T.H conceived of the project. D.V. designed experiments, performed electrophysiological recordings, processed the fixed tissue, performed super-resolution imaging, analyzed the data, and prepared figures. T.L.D.P. performed fixation, biocytin staining, gelation, denaturation, and immunostaining of the brain slices. G.R.C., B.S.C and M.R. performed surgeries that resulted in human tissue. M.P.F. oversaw removal and parcellation of tissue as well as overall IRB aspects and regulatory aspects of the project with regard to human participants. S.S.C. helped in designing methods for acquiring human tissue and ensured that tissue was collected. M.T.H. supervised all aspects of the project and wrote the manuscript with D.V.
Corresponding author
Ethics declarations
Competing interests
The authors declare competing financial interests: M.T.H. and D.V. are co-inventors on patent application prepared by MIT covering the Patch2MAP technology (U.S. Provisional Patent Application 63/400787).
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Vardalaki, D., Pham, T.L.D., Frosch, M.P. et al. Patch2MAP combines patch-clamp electrophysiology with super-resolution structural and protein imaging in identified single neurons without genetic modification. Sci Rep 15, 34613 (2025). https://doi.org/10.1038/s41598-025-18207-3
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-18207-3
