
Abstract
This is the first prospective study aiming to quantify the effectiveness and safety of escitalopram monotherapy initiation where therapeutic drug monitoring (TDM) was used to achieve the therapeutic reference range (TRR) of plasma concentration. PsyCise-E (NCT05210140) was a hospital-based study conducted in Belgrade, Serbia, involving 92 outpatients with a baseline Hamilton Rating Scale for Depression (HAM-D) score higher than 13. The primary endpoint was the relative reduction in HAM-D score from baseline to week eight, with dose personalization based on TDM four weeks after treatment initiation. Patients were categorized into groups: (1) unadjusted (they achieved TRR at 10 mg/day), (2) adjusted (their dose was adjusted to achieve TRR) and (3) inadequate (they did not reach TRR). Safety was assessed by the occurrence of adverse drug reactions (ADRs) and QTc interval prolongation. Most patients required a dose escalation beyond 10 mg/day (71/92), and most patients achieved TRR after eight weeks (79/92). The 55% (95% CI: 47–64) reduction in HAM-D scores did not correlate with escitalopram plasma concentrations and did not differ between groups; however, response and remission rates were significantly higher in patients who achieved TRR by week four. The incidence of ADRs (47/92) increased by 3.2% (0.1–6.3) per ng/ml escitalopram, with no significant differences between the groups. QTc prolongation of 5.5 ms (1.8–9.3) did not correlate with plasma concentration and did not differ between groups. While TDM-guided dosing likely only marginally improved escitalopram effectiveness, it increased treatment safety as TDM-guided dose escalation did not lead to ADRs or QTc prolongation.
Similar content being viewed by others

Introduction
Escitalopram is a frequently prescribed antidepressant1,2 with one of the most favorable effectiveness and safety profiles among the 21 most commonly prescribed antidepressants3. In the acute treatment of depressed patients, treatment with escitalopram at a fixed dose of 10 mg/day, a fixed dose of 20 mg/day and a flexible dose of 10–20 mg/day was consistently superior to placebo after eight weeks in three randomized controlled trials (RCT)4,5,6. The effect size was about 3.5 points change in the Montgomery and Åsberg Depression Rating Scale (MADRS), with no significant difference in treatment effectiveness between 10 mg/day and 20 mg/day escitalopram4. Common adverse drug reactions (ADRs) which are highly dose dependent2,4,5 include: insomnia, ejaculatory dysfunction, nausea, increased sweating, fatigue, somnolence, decreased libido, and anorgasmia2,4,5,6. Moreover, a randomized cross-over study with escalating multiple dosing in healthy volunteers has shown that escitalopram prolongs the QTc interval in a dose-dependent manner7, which has prompted regulatory authorities to limit the dose of escitalopram to 10 mg/day in patients over 65 years of age and in patients at increased risk of cardiac arrhythmias8,9.
Therapeutic drug monitoring (TDM) refers to the quantification and interpretation of drug concentrations in the blood in order to personalize dosing and optimize pharmacotherapy. The consensus guidelines define the recommended therapeutic reference range (TRR) for escitalopram blood concentration between 15 and 80 ng/ml and provide a level two recommendation for the use of TDM10; this practically means that TDM is recommended for titrating the dose towards the TRR, for specific indications, or for problem solving. However, the relationship between escitalopram blood concentration, treatment effectiveness and tolerability remains unclear11. In particular, a prospective cohort study found an association between escitalopram serum concentration and antidepressant response in 70 depressed patients12 and estimated that the threshold concentration required for treatment response is 20 ng/ml, whereas retrospective studies found no such association13,14,15,16,17. When patients were categorized into groups based on treatment response, two retrospective studies observed higher escitalopram blood concentrations in responders compared to non-responders18 and treatment failure patients19. Both studies concluded that 15 ng/ml of escitalopram on average is required for an optimal response to treatment. However, a much larger retrospective study was unable to replicate this observation and found no significant difference in escitalopram plasma concentrations between clinical responders and non-responders20. Regarding safety, one retrospective study found a positive association between escitalopram blood concentration and the occurrence of dry mouth21, while none of the other retrospective studies conducted to date found a significant association between escitalopram blood concentration and treatment safety parameters13,14,18,19,21,22,23 despite the high dose-dependence of ADRs2,4,5,7. However, it is noteworthy that most of the previous reports were retrospective post-hoc analyzes of data from very heterogeneous patient cohorts with varying degrees of data representativeness. To our knowledge, no study has prospectively investigated the effectiveness and safety of escitalopram treatment initiation based on TDM-guided dose titration with the aim of achieving the currently recommended TRR. Therefore, this prospective cohort study was designed to resolve the uncertainty stemming from inconsistent and predominantly retrospective findings on the role of TDM in guiding escitalopram treatment.
Accordingly, the aim of this study was to quantify the effectiveness and safety of TDM-guided initiation of escitalopram treatment in a cohort of depressed outpatients and to compare it with the results of previously published fixed-dose studies. For the efficacy comparison, the results of the dose-response meta-analysis by Furukawa et al.24 were used, while for the safety comparison, the pharmacovigilance studies presented on the FDA package insert2 and supporting data7 were used.
Methods
The outpatient recruitment and follow-up were conducted between August 2020 and September 2023 at the Institute of Mental Health in Belgrade, Serbia. The procedures performed during the study protocol implementation are described in detail in the study protocol NCT05210140 (https://www.clinicaltrials.gov/study/NCT05210140) and schematized in Fig. 1A. The study protocol was approved by the Hospital Ethics Committee (approval number: 2083/1). All procedures involving human participants were conducted in accordance with the ethical standards of the institutional and national research committees and with the 1964 Helsinki declaration and its later amendments. Informed consent was obtained from all participants included in the study. The study report was written following the STROBE guidelines (https://www.strobe-statement.org/).

Effectiveness and safety of escitalopram treatment guided by dose personalization based on quantification of drug plasma concentration. (a) Schematic representation of study protocol; patients who achieved and maintained TRR with 10 mg/day of escitalopram throughout the trial were labeled as unadjusted dose group; patients who required dose adjustment at week 4 to achieve TRR were labeled as adjusted dose group; and patients who failed to achieve or maintain TRR at week 8 even after dose personalization were labeled as inadequate drug level group. (b) Hamilton Rating Scale for Depression (HAM-D) score decreased by 55% (95% CI: 47–64%, p < 0.001) from baseline to week 8, without difference in HAM-D score reduction between groups (p > 0.1). (c) The relative change in HAM-D score from baseline did not correlate with escitalopram plasma concentration (p > 0.1) at week 8. (d) Hamilton Rating Scale for Anxiety (HAM-A) score decreased by 52% (95% CI: 43–62%, p < 0.001) from baseline to week 8, without difference in HAM-A score reduction between groups (p > 0.1). (e) Clinical Global Impression of Severity (CGI-S) score decreased by 48% (95% CI: 39–54%, p < 0.001) from baseline to week 8, without difference in CGI-S score reduction between groups (p > 0.1). (f) Of 92 patients, 56 responded to treatment and 43 achieved remission; more responders (p = 0.0074) and remitters (p = 0.036) belonged to the unadjusted dose group compared to inadequate drug level group. (g) After week 4 and week 8 of escitalopram treatment, 39 and 47 out of 92 patients reported adverse drug reactions, respectively, without difference between groups (p > 0.1). (h) Frequency distribution of reported adverse drug reactions at week 8 is depicted from the most to the least frequent. (i) Escitalopram plasma concentration increased the probability for the occurrence of ADRs after week 8 by 3.2% (95% CI: 0.1–6.3%, p = 0.041) (j) QTc interval from baseline to week 8 was prolonged by 5.5 ms (95% CI: 1.8–9.3 ms, p = 0.0041), without differences in QTc interval prolongation (p > 0.1) between groups. In addition, (k) the QTc interval prolongation was not correlated with escitalopram plasma concentration (p > 0.1). Results are presented as bar charts, scatter plots and line graphs with annotated estimated marginal means ± 95% CI.
Study participants
The study inclusion criteria were: (1) age between 15 and 65 years, (2) a score of 14 or more on the Hamilton Rating Scale for Depression – 21 items (HAM-D)25, (3) initiation of escitalopram therapy, and (4) signed informed consent from the patient (or legal guardian in case of minors). The exclusion criteria were: (1) severe liver dysfunction (abnormal AST/ALT ratio); (2) severe renal dysfunction (abnormal creatinine clearance); (3) dementia; (4) psychotic disorder; (5) perceived high risk of suicide; (6) drug dependence; or (7) treatment with strong CYP2C19 enzyme inhibitors omeprazole, lansoprazole, pantoprazole, rabeprazole, cimetidine, moclobemide, fluoxetine, fluvoxamine, isoniazid or chloramphenicol. The criteria for study discontinuation were: (1) occurrence of intolerable side effects, (2) patient’s decision to no longer participate in the study, (3) escitalopram levels below 5 ng/ml in the second week of follow-up, indicating poor treatment adherence, or (4) study protocol violation.
Exposure and measurements
Sociodemographic data included sex and age; cardiac parameters included blood pressure, heart rate, and QTc interval duration based on electrocardiograms; and psychometric data included the HAM-D score, the Hamilton Rating Scale for Anxiety (HAM-A) score26 and the Clinical Global Impression of Severity Scale (CGI-S)27 score. At follow-up visits, patients were also assessed using the Clinical Global Impression of Improvement Scale (CGI-I)27 and the UKU side effect rating scale28. Psychometric data at baseline, at four-week follow-up, and at eight-week follow-up visits were collected by a trained rater who remained blinded to both dose and plasma concentrations after baseline visit. Two weeks after starting treatment with 10 mg/day escitalopram, patients’ escitalopram plasma concentrations were measured using a previously validated method based on chromatography and mass spectrometry29. After four weeks of treatment with the recommended dose of 10 mg/day escitalopram, dosing was personalized based on TDM results with the aim to ensure that the recommended TRR (15–80 ng/ml) is achieved. Patients continued on 10 mg/day if plasma concentrations were between 25 and 50 ng/ml. If concentrations fell outside this range, doses were adjusted to 5 mg/day (for > 50 ng/ml), 15 mg/day (for 15–25 ng/ml), or 20 mg/day (for < 15 ng/ml). The 25–50 ng/mL interval was pre-specified in the study protocol as a “no adjustment” range, guided by the TRR for escitalopram10 and the FDA bioequivalence criterion of 80–125% of the target exposure30. This principle was applied to account for expected day-to-day variability in plasma concentrations due to differences in absorption, timing of drug intake before sampling, or occasional missed doses. The lower bound of 25 ng/mL ensures that concentrations remain above the TRR lower limit of 15 ng/mL, while the upper bound of 50 ng/mL ensures concentrations remain well below the TRR upper limit of 80 ng/mL in case of substantial daily fluctuations in drug exposure. Clinicians were informed about plasma levels in relation to the TRR, and dose adjustment guidelines were provided to assist clinical decision-making. At the end of the study in the eighth week, additional TDM was performed to assess whether the escitalopram plasma concentration was within the TRR after the dose adjustment (Fig. 1A).
Outcomes
The primary endpoint for treatment effectiveness was the reduction in the severity of depressive symptoms, measured as a relative decrease in the HAM-D score from baseline to the eighth week of follow-up. Secondary outcomes included reduction in anxiety symptom severity, measured as a relative decrease in HAM-A scores from baseline to week eight, and changes in global symptom severity and improvement. Global symptom severity was measured by the relative decline in CGI-S scores from baseline to week eight, while global symptom improvement was measured by a CGI-I score of less than four at week eight, indicating at least minimal improvement from baseline. In addition, response rate was measured as the proportion of patients in whom the HAM-D score had decreased by 50% or more from baseline to eight weeks, and remission rate was calculated as the proportion of patients who had a HAM-D score of less than eight at week eight. Safety of treatment was assessed by the proportion of patients experiencing escitalopram-induced ADRs at weeks four and eight, as assessed by the UKU scale, and by calculating the change in QTc interval from baseline to week eight using electrocardiograms. For between-group statistical comparison, the study cohort was divided into three groups: (1) the unadjusted dose group, consisting of patients who achieved and maintained TRR throughout the study with a 10 mg/day dose of escitalopram; (2) the adjusted dose group, consisting of patients who required a dose adjustment at week four to achieve TRR; and (3) the inadequate dose/drug concentration group, consisting of patients who did not achieve or maintain TRR after eight weeks of treatment.
Statistical data analysis
The statistical analysis was carried out using IBM SPSS 20, while the diagrams were created using GraphPad Prism 8.0.1 software. All quantitative data, such as the decrease in HAM-D score, were analyzed using a two-way mixed analysis of variance (ANOVA), with visit as a within-subjects factor and subgroup as a between-subjects factor. LSD post-hoc tests were then performed to assess the statistical significance of differences between groups. All categorical data, including the proportion of patients with ADRs, were analyzed with the chi-square test to assess differences between groups or with binary logistic regression using escitalopram plasma level at week eight, sex, age, and HAM-D baseline score as independent variables. The Person test and Spearman test were used for correlation analyzes for normally and non-normally distributed data, respectively. The p-values below 0.05 were interpreted as a statistically significant difference between the compared groups.
Comparison of study outcomes with previously published randomized clinical trials
To estimate the influence of the placebo effect on the main outcome of the study and to compare it with the results from previously published placebo-controlled, randomized clinical trials of fixed doses of escitalopram, the studies selected from the meta-analysis by Furukawa et al.24 were reanalyzed. The relative reduction in symptom severity for the placebo, 10 mg/day escitalopram, and 20 mg/day escitalopram arms, measured as the percentage change in HAM-D or MADRS scores from baseline to week eight, was meta-analyzed and compared with the primary outcome of the present study. To estimate the relative reduction in symptom severity for each RCT, the Taylor expansion method31 was used to divide the absolute change in symptom severity at eight weeks and the baseline symptom severity score. The calculated relative reductions in symptom severity and their respective standard errors for each trial were then entered into RevMan 5.4 software (Cochrane Collaboration, Foster City, USA) and analyzed using the generic inverse variance option and random effect to obtain grand means for each arm. The grand means of the meta-analysis were then plotted together with the results of the present study and compared by visual inspection. To compare the results regarding the safety of escitalopram treatment between this and previous studies that focused on fixed doses of escitalopram2,7 the incidence of distinct ADRs and QTc interval prolongation were compared by visual inspection.
Results
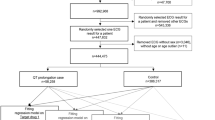
Of the 140 patients enrolled, 92 completed the study protocol (Fig. 1A), while 6 participants (4.3%) were excluded due to plasma concentrations below 5 ng/mL measured at week 2 (Fig. 2). The details on cohort status at each phase and reasons for non-participation are presented on the study flowchart (Fig. 2). The mean age of the cohort was 33 years, the mean baseline HAM-D score was 21, and 61/92 participants were women. After eight weeks, 19 patients maintained TRR throughout the study at the escitalopram dose of 10 mg/day, 13 patients did not achieve or maintain TRR, and 60 patients required a dose adjustment; including 26 patients receiving 15 mg/day, 33 patients receiving 20 mg/day and one patient receiving 5 mg/day between week four and week eight. No significant differences in age, gender, HAM-D score or HAM-A score at baseline were observed between the study groups. The baseline data for the entire cohort and individual groups are shown in Table 1.

Study flowchart.
Effectiveness and safety of TDM-guided Escitalopram treatment
Of the 92 patients who completed the study, 71 required a dose escalation beyond 10 mg/day at week four to achieve the recommended TRR, and 79 were within the TRR at week eight (Fig. 1A). The effectiveness of TDM-guided escitalopram treatment was measured by changes in HAM-D, HAM-A and CGI-S scores from baseline to eight-week follow-up and by achieving a CGI-I score of less than four at week eight. At week eight, the HAM-D score decreased by 55% from baseline (Fig. 1B), with no correlation observed between the reduction and measured escitalopram plasma concentrations (Fig. 1C). Significant improvement was also measured as a 52% and 48% reduction in HAM-A (Fig. 1D) and CGI-S scores (Fig. 1E), respectively, from baseline to week eight, along with a CGI-I score that was significantly below the 4-point threshold at week eight. No statistically significant differences in symptom improvement were observed among patients in the unadjusted dose group, the adjusted dose group, and the inadequate drug level group, as measured by any of the above outcomes (Fig. 1B, D, E; Table 2). Next, response and remission rates were compared between the study groups. Patients in the unadjusted dose group were significantly more likely to have a response and remission than patients in the inadequate drug level group (Fig. 1F). In conclusion, escitalopram dose adjustment resulted in drug exposure within a TRR in the majority of patients and treatment effectiveness was unrelated to escitalopram plasma concentrations in patients within the TRR.
Escitalopram-induced ADRs according to the UKU side effect rating scale occurred in 39 and 47 of 92 patients at four and eight weeks, respectively (Fig. 1G), with no difference between study groups (Fig. 1H). Table 3 presents the frequency of the most commonly reported ADRs across treatment groups, organized by symptom category. Escitalopram plasma concentration at week eight was associated with an increased likelihood of ADRs, which increased by 3.2% per one ng/mL increase in serum concentration (Fig. 1I). In addition, arterial blood pressure remained stable during the eight-week period, while heart rate decreased by 3.5 beats per minute and the QTc interval was prolonged by 5.5 ms in the entire cohort. The reduction in heart rate and prolongation of the QTc interval did not differ significantly between the study groups (Table 2). In addition, QTc interval prolongation at week eight showed no correlation with escitalopram plasma concentration (Fig. 1J, K). No patient exceeded the clinically significant QTc threshold of 450 ms at week 8 (Table 2). In conclusion, despite the correlation between escitalopram plasma concentration and risk of ADRs seen in fixed-dose trials, TDM-guided dose escalation did not result in a significant increase in the incidence of ADRs and QTc prolongation compared to patients who remained at 10 mg/day throughout the study.
Comparison of effectiveness and safety outcomes with previously published data
Of the 12 RCTs in the study by Furukawa et al.24, 10 were reanalyzed; The SCT-MD-35 trial was excluded because the escitalopram dose was 4 mg/day32, and the NCT00822744 trial was excluded because the effectiveness results for the placebo and escitalopram treatment could not be estimated separately33. All RCTs presented data for the placebo, eight RCTs presented data for the 10 mg/day escitalopram, and six RCTs presented data for the 20 mg/day escitalopram. The majority of trial participants were Caucasian. Five studies used the HAM-D scale and five studies used the MADRS scale. Patients treated with 10 mg/day and 20 mg/day escitalopram showed similar reductions in symptom severity of 52% and 51%, respectively, while patients treated with placebo showed a 43% reduction in symptom severity (Fig. 3A).

Comparison of the effectiveness and safety of escitalopram treatment in cohorts treated with guided and unguided dosing. (a) Meta-analysis comparing the effectiveness of escitalopram treatment between the placebo, 10 mg/day and 20 mg/day arms in fixed-dose randomized controlled trials and the current study using relative symptom reduction on the scale defined as the primary outcome. The placebo arm showed a 43% (95% CI: 38–49%; N = 1,309, p < 0.001), the 10 mg/day escitalopram arm showed a 52% (95% CI: 47–57%; N = 1,355, p < 0.001), and the 20 mg/day escitalopram arm a 51% (95% CI: 46–57%; N = 865, p < 0.001) reduction in symptom severity. (b) Comparison of the incidence of the most common adverse drug reactions between the two fixed-dose studies listed on the FDA drug label for escitalopram and the current study is presented indicating that the incidence of adverse drug reactions was between the incidence observed in patients treated with 10 mg/day and 20 mg/day. (c) Comparison of the observed change in QTc interval between the current study with TDM guided-dose and the FDA randomized, controlled, cross-over, multiple fixed-dose study. The white and gray bars represent the same cohort (N = 113) sequentially exposed to different escitalopram doses, while the red bar represents the current cohort as change from baseline to week 8. The QTc elongation in the present cohort is comparable to the QTc prolongation observed when subjects were treated with 10 mg/day and 20 mg/day. Results are presented as means ± 95% CI.
The incidence of various ADRs in this cohort was compared to those reported in the FDA safety data from the escitalopram package insert for placebo, 10 mg/day and 20 mg/day2. The incidence of the most common adverse drug reactions in the present cohort was intermediate between those of the 10 mg/day and 20 mg/day fixed-dose groups, although slightly closer to the 10 mg/day group (Fig. 3B). Next, the observed changes in QTc interval were compared with the results of the FDA randomized controlled cross-over trial of escalating multiple doses in healthy volunteers7. The observed mean QTc interval prolongation of 5.5 ms in the TDM-guided cohort studied here was comparable to the mean prolongation of 4.5 ms in the 10 mg/day fixed-dose group and the mean prolongation of 6.6 ms in the 20 mg/day fixed-dose group (Fig. 3C). In summary, the TDM-guided cohort showed a comparable effectiveness and safety profile to the cohorts treated with fixed 10 mg and 20 mg daily doses.
Discussion
While most patients required a dose escalation to achieve TRR, the majority of patients eventually succeeded to achieve it and responded to escitalopram treatment. While escitalopram blood concentration was not associated with a reduction in symptom severity, response and remission rates were higher in the group that achieved and maintained TRR than in the group that did not achieve TRR. Escitalopram blood concentration was associated with a 3.2% increased incidence of ADRs per ng/ml escitalopram in plasma, but not with QTc interval prolongation. In the TDM-guided settings, dose escalation beyond 10 mg/day, which was required in the majority of patients, did not result in significant safety concerns, in contrast to the studies in which the dose was not guided by TDM and in which ADRs and QTc interval prolongation were more pronounced at higher doses.
The placebo effect resulted in a 43% reduction in symptom severity, while the fixed dose of 10 mg/day, the fixed dose of 20 mg/day and the flexible, TDM-guided escitalopram dose reduced symptom severity by 52%, 51% and 55%, respectively. In the study cohort, no significant difference in symptom severity reduction was observed between the unadjusted dose group, the adjusted dose group, and the inadequate drug level group. Furthermore, the absence of a correlation between escitalopram plasma concentration and symptom reduction is consistent with previous studies13,14,15,16,17,20, and suggests that increasing drug exposure beyond the lower limit of the TRR is unlikely to yield additional clinical benefit. Most patients in the present study were within the TRR, and it is likely that escitalopram concentrations above 15 ng/mL do not enhance therapeutic response due to a pharmacodynamic ceiling effect. This interpretation is supported by evidence that serotonin transporter (SERT) occupancy reaches a functional threshold at approximately 8.9 ng/mL (25 nM) of serum escitalopram, a concentration derived from the experimentally determined SERT inhibition constant (Ki = 9.2 nM)34, combined with data showing that cerebrospinal fluid concentrations are approximately one-third of serum concentrations35, as previously operationalized by Jukić et al.36. Moreover, the inherent problem with this and similar studies is a poor signal-to-noise ratio37, as more than one-third of participants responded due to the placebo effect and more than one-third belonged to the group of absolute non-responders to escitalopram, regardless of dose. When interindividual variability in relative HAM-D score reduction was circumvented by binary categorization of patients based on symptom reduction, response and remission rates were higher in the unadjusted dose group than in the inadequate drug level group (Fig. 1F). This confirms the previous observation12 that achieving a TRR improves the effectiveness of escitalopram treatment. Although the benefit of TDM-guided dosing in terms of effectiveness is likely to be marginal in a naturalistic setting, it may still be important for a subgroup of patients who respond to escitalopram but not placebo; however, the study design and limited sample size do not allow firm conclusions to be drawn on this point. Clarifying the effectiveness of TDM-guided dosing would require a randomized trial with a placebo run-in, as discussed in detail in the Limitations section.
The results also underline the frequent need to increase the escitalopram dose beyond 10 mg/day to achieve TRR. However, such a step is associated with safety concerns, as the incidence of ADRs is dose-dependent2,4,5,7 and also blood concentration-dependent, as observed here. Since dosing in this cohort was TDM-guided, patients who were already within the TRR at 10 mg/day were spared the dose escalation that could occur in a naturalistic setting and potentially cause avoidable dose-dependent ADRs. In fact, TDM-guided dose escalation did not lead to an increase in reported ADRs, while in contrast, the ADR incidence in the 20 mg/day fixed-dose group was twice as high as in the 10 mg/day fixed-dose group when the dose was not TDM-guided2. Furthermore, TDM-guided dose escalation did not lead to a significant prolongation of the QTc interval. This suggests that clinically meaningful escitalopram-induced QTc prolongation probably occurs mainly at supratherapeutic escitalopram blood levels7,38, because at non-TDM-guided dosing, the QTc interval was prolonged by 4.5 and 10.7 ms at doses of 10 and 30 mg/day, respectively7. Overall, the TDM-guided treatment approach has the potential to improve the safety of escitalopram treatment, including clinically relevant concerns in patients at higher cardiovascular risk8,9,38.
The results discussed suggest that routine use of TDM for escitalopram monotherapy may not be justified with present evidence. Instead, its clinical utility appears to be limited to situations where non-adherence or rapid metabolism is suspected. In this context, TDM plays a dual role: it enables personalized dose titration while also helping to identify patients with likely poor adherence. In the present cohort, 4.3% of patients had escitalopram plasma levels below 5 ng/mL at week 2, strongly suggesting non-adherence. Noteworthy, patients in this cohort knew that TDM will be performed, which implicates significantly higher incidence of non-adherence in naturalistic settings. Importantly, in the absence of TDM, clinicians may misattribute nonresponse to poor adherence or ultrarapid metabolism, leading to unnecessary dose escalation, which is known to compromise treatment safety. If the patient is actually adherent and within the TRR but simply not pharmacodynamically responsive to the drug, such dose increases may result in supratherapeutic exposure and associated ADRs. This highlights the clinical relevance of TDM in protecting patients from avoidable safety concerns while informing better treatment decisions.
Related to cost-effectiveness, which is of relevance for clinical use and reimbursement, the cost of a single TDM measurement is approximately €20–8039, while one day in the hospital is 4–16 times more expensive than a single TDM10. While to the best of our knowledge, no cost-effectiveness study has been performed specifically for TDM usage in escitalopram monotherapy of depression, TDM can reduce the cost of antidepressant treatment. In particular, treatment cost is reduced by 16% in elderly SSRI users, hospitalization is shortened by 23 days for inpatients treated with citalopram, and treatment failure is prevented by identifying sub-therapeutic concentrations early in the course of treatment10. Furthermore, treatment-resistant depression (TRD) is associated with a substantial economic burden, with TRD patients incurring nearly double the annual costs compared to non-TRD patients; in particular, €15,907 vs. €8,335 per patient per year40. Numerous TRD cases are in fact pseudo-TRD due to sub-therapeutic drug exposure caused by non-adherence, ultrarapid metabolism, or drug–drug interactions; all these factors can be easily identified by performing TDM10,41. Therefore, while it is still unclear whether universal routine TDM for escitalopram is cost-effective, a targeted strategy triggered by specific situations appears economically justified; such situations are: (1) early non-response (2–4 weeks), (2) unexpected adverse effects at low doses, (3) suspected non-adherence, (4) clinically relevant drug–drug interactions, or (5) treatment in special sensitive populations. On the flip-side, situations where TDM is unlikely to add value include (1) stable responders without tolerability issues, (2) late measurements with no planned dose adjustments, or (3) settings where turnaround time is insufficiently fast to improve treatment management. However, unequivocal conclusion about cost-effectiveness of TDM in escitalopram monotherapy of depression for each specific situation would require a dedicated methodological framework and a follow-up cost-benefit analysis.
Limitations
The main limitations of this study are: (1) The limited cohort size of 92 patients meant that certain subgroups were particularly small, most notably the inadequate drug level group, which consisted of 13 patients. This markedly reduces the statistical power to detect subtle but potentially clinically relevant between-group differences, especially for safety outcomes, and increases the likelihood of false-negative findings. Consequently, the results for these subgroups should be interpreted with caution. Future studies with larger and more evenly distributed samples are needed for detection of clinically meaningful effects and quantification of effect sizes. (2) The absence of a randomized comparison substantially reduces the ability to draw firm causal inferences about the added value of TDM-guided dosing. Comparisons with historical fixed-dose data provide some context, but cannot fully account for potential confounding variables or time-related biases. Future adequately powered RCTs with TDM-guided and control group treated as usual are warranted to provide unequivocal evidence regarding the clinical utility of TDM-guided escitalopram treatment. (3) The high placebo response rate and presence of pharmacodynamic non-responders complicate the interpretation of the marginal group-level benefit observed with TDM-guided dosing. As highlighted earlier37, standard antidepressant trials often suffer from poor signal-to-noise ratios due to heterogeneous response patterns, making it difficult to detect robust concentration–response associations. Clinically, this suggests that TDM may not uniformly enhance efficacy across all patients but can offer valuable insights in selected cases, in particular when treatment failure raises questions of adherence, atypical pharmacokinetics, or subtherapeutic exposure. Future studies could benefit from incorporating placebo run-in periods and early response stratification to better isolate pharmacologically responsive subgroups who can benefit from TDM the most, (4) The inclusion criteria i.e., age range of 15–65 years, exclusion of patients with severe somatic comorbidities, use of antidepressant monotherapy, and outpatient treatment setting, limit the generalizability of the findings to the following patient categories: preadolescents, older adults, inpatients, individuals with significant medical comorbidities, and those requiring complex antidepressant polypharmacy. To broaden the generalizability and clinical applicability of findings, future studies should include broader and more diverse populations, including older adults, inpatients, and individuals with relevant somatic comorbidities or on complex pharmacotherapy regimens or focus on these distinct categories, (5) The pharmacogenetic data, such as CYP2C19 and CYP2D6 genotypes were not collected in this study. Variability in escitalopram plasma levels may partly reflect differences in metabolic capacity related to genetic polymorphisms, as well as body weight, renal function, and liver function. Although preemptive genotyping could assist in identifying patients at risk of underexposure or supratherapeutic concentrations, it constitutes a distinct intervention from TDM. Future complementary research should evaluate the potential benefits of integrating preemptive pharmacogenetic profiling with follow-up TDM analysis to improve dose optimization and maximize the utilization of available molecular diagnostic, computational, and treatment monitoring tools.
Conclusion
The introduction of escitalopram monotherapy by TDM-guided dose titration probably only marginally increases the overall effectiveness of treatment, which may still be relevant for a subgroup of patients who are not placebo responders and absolute non-responders. TDM-guided escitalopram treatment improves the safety of escitalopram treatment by preventing unnecessary dose increases beyond the therapeutic reference range, which are associated with the risk of adverse drug reactions and QTc prolongation.
Data availability
The datasets generated and/or analyzed during the current study are available from the corresponding author on reasonable request.
References
ClinCalc. Escitalopram – drug usage statistics. ClinCalc DrugStats Database. https://clincalc.com/DrugStats/Drugs/Escitalopram (2024).
Forest Pharmaceuticals. Lexapro: package insert. https://www.accessdata.fda.gov/drugsatfda_docs/label/2017/021323s047lbl.pdf (2017).
Cipriani, A. et al. Comparative efficacy and acceptability of 21 antidepressant drugs for the acute treatment of adults with major depressive disorder: a systematic review and network meta-analysis. Lancet 391, 1357–1366 (2018).
Burke, W. J., Gergel, I. & Bose, A. Fixed-dose trial of the single isomer SSRI Escitalopram in depressed outpatients. J. Clin. Psychiatry. 63, 331–336 (2002).
Wade, A., Lemming, O. M. & Hedegaard, K. B. Escitalopram 10 mg/day is effective and well tolerated in a placebo-controlled study in depression in primary care. Int. Clin. Psychopharmacol. 17, 95–102 (2002).
Lepola, U. M., Loft, H. & Reines, E. H. Escitalopram (10–20 mg/day) is effective and well tolerated in a placebo-controlled study in depression in primary care. Int. Clin. Psychopharmacol. 18, 211–217 (2003).
US Food and Drug Administration. Revised recommendations for celexa (citalopram hydrobromide) related to a potential risk of abnormal heart rhythms with high doses. FDA Saf. Communication. http://www.fda.gov/drugs/drugsafety/ucm297391.htm (2012).
Medicines and Healthcare products Regulatory Agency. Citalopram and escitalopram: QT interval prolongation. Drug Saf. Update. https://www.gov.uk/drug-safety-update/citalopram-and-escitalopram-qt-interval-prolongation (2014).
Health Canada. Antidepressant Cipralex (escitalopram): updated information regarding dose-related heart risk. Health Canada Advisory. http://www.healthycanadians.gc.ca/recall-alert-rappel-avis/hc-sc/2012/13674a-eng.php (2012).
Hiemke, C. et al. Consensus guidelines for therapeutic drug monitoring in neuropsychopharmacology: update 2017. Pharmacopsychiatry 51, 9–62 (2018).
Eichentopf, L. et al. Systematic review and meta-analysis on the therapeutic reference range for escitalopram: blood concentrations, clinical effects, and serotonin transporter occupancy. Front. Psychiatry. 13, 972141 (2022).
Florio, V., Porcelli, S., Saria, A., Serretti, A. & Conca, A. Escitalopram plasma levels and antidepressant response. Eur. Neuropsychopharmacol. 27, 940–944 (2017).
Kuo, H. W. et al. CYP1A2 genetic polymorphisms are associated with early antidepressant Escitalopram metabolism and adverse reactions. Pharmacogenomics 14, 1191–1201 (2013).
Tadic, A. et al. Randomized controlled study of early medication change for non-improvers to antidepressant therapy in major depression: the EMC trial. Eur. Neuropsychopharmacol. 26, 705–716 (2016).
Leuchter, A. F. et al. Comparative effectiveness of biomarkers and clinical indicators for predicting outcomes of SSRI treatment in major depressive disorder: results of the BRITE-MD study. Psychiatry Res. 169, 124–131 (2009).
Steen, N. E. et al. Serum level of Venlafaxine is associated with better memory in psychotic disorders. Schizophr Res. 169, 386–392 (2015).
Hodgson, K. et al. Genetic differences in cytochrome P450 enzymes and antidepressant treatment response. J. Psychopharmacol. 28, 133–141 (2014).
Hart, X. M. et al. Concentrations of Escitalopram in blood of patients treated in a naturalistic setting: focus on patients with alcohol and benzodiazepine use disorder. Eur. Arch. Psychiatry Clin. Neurosci. 273, 75–83 (2023).
Hart, X. M., Amann, F., Brand, J., Eichentopf, L. & Grunder, G. Low Escitalopram concentrations in patients with depression predict treatment failure: a naturalistic retrospective study. Pharmacopsychiatry 56, 73–80 (2023).
Kasperk, N. et al. Pharmacokinetic correlates of clinical response in a naturalistic sample of escitalopram-treated patients. Expert Rev. Clin. Pharmacol. 17, 247–253 (2024).
Hodgson, K. et al. Exploring the role of drug-metabolising enzymes in antidepressant side effects. Psychopharmacol. (Berl). 232, 2609–2617 (2015).
Kuzin, M. et al. Assessing Pharmacokinetic correlates of escitalopram-related adverse drug reactions. Ther. Drug Monit. 46, 246–251 (2024).
Islam, F. et al. Effects of CYP2C19 and CYP2D6 gene variants on Escitalopram and Aripiprazole treatment outcome and serum levels: results from the CAN-BIND 1 study. Transl Psychiatry. 12, 366 (2022).
Furukawa, T. A. et al. Optimal dose of selective serotonin reuptake inhibitors, venlafaxine, and Mirtazapine in major depression: a systematic review and dose-response meta-analysis. Lancet Psychiatry. 6, 601–609 (2019).
Hamilton, M. Development of a rating scale for primary depressive illness. Br. J. Soc. Clin. Psychol. 6, 278–296 (1967).
Hamilton, M. The assessment of anxiety States by rating. Br. J. Med. Psychol. 32, 50–55 (1959).
Guy, W. ECDEU assessment manual for psychopharmacology. In US Department of Health, Education, and Welfare 218–222 (National Institute of Mental Health, 1976).
Lingjaerde, O., Ahlfors, U. G., Bech, P., Dencker, S. J. & Elgen, K. The UKU side effect rating scale: a new comprehensive rating scale for psychotropic drugs and a cross-sectional study of side effects in neuroleptic-treated patients. Acta Psychiatr Scand. Suppl. 334, 1–100 (1987).
Jeremic, A. et al. Validation of a quick and simple chromatographic method for simultaneous quantification of sertraline, escitalopram, risperidone, and Paliperidone levels in the human plasma. Arh Farm. 71, 365–377 (2021).
U.S. Food and Drug Administration. Guidance for industry: Bioavailability and bioequivalence studies submitted in NDAs or INDs — general considerations. https://www.fda.gov/media/88254/download (2014).
Stuart, A. & Ord, J. K. (eds) Kendall’s Advanced Theory of Statistics 6th edn, Vol. 1 (Arnold, 1998).
ClinicalTrials.gov. SCT-MD-35 (NCT00109044). (accessed 6 August 2024). https://clinicaltrials.gov/study/NCT00109044
ClinicalTrials.gov; EU Clinical Trials Register. NCT00822744 / EudraCT 2008-001718-26. (accessed 6 August 2024). https://clinicaltrials.gov/ct2/show/NCT00822744 and https://www.clinicaltrialsregister.eu/ctr-search/trial/2008-001718-26/SK
Koldsø, H. et al. The two enantiomers of Citalopram bind to the human serotonin transporter in reversed orientations. J. Am. Chem. Soc. 132, 1311–1322 (2010).
Paulzen, M. et al. Measuring Citalopram in blood and central nervous system: revealing a distribution pattern that differs from other antidepressants. Int. Clin. Psychopharmacol. 31, 119–126 (2016).
Jukić, M. M. et al. Impact of CYP2C19 genotype on Escitalopram exposure. Am. J. Psychiatry. 175, 463–470 (2018).
Preskorn, S. H. Therapeutic drug monitoring (TDM) in psychiatry (part I): why studies attempting to correlate drug concentration and antidepressant response don’t work. J. Psychiatr Pract. 20, 133–137 (2014).
Faraj, P. et al. Pro-arrhythmic effect of Escitalopram and Citalopram at serum concentrations commonly observed in older patients: a study based on a cohort of 19,742 patients. EBioMedicine 95, 104779 (2023).
Baumann, P. et al. The AGNP-TDM expert group consensus guidelines: focus on therapeutic monitoring of antidepressants. Dialogues Clin. Neurosci. 7, 231–247. https://doi.org/10.31887/DCNS.2005.7.3/pbaumann (2005).
Taipale, H. et al. Healthcare utilization, costs, and productivity losses in treatment-resistant depression in Finland – a matched cohort study. BMC Psychiatry. 22, 484. https://doi.org/10.1186/s12888-022-04115-7 (2022).
Howes, O. D., Thase, M. E. & Pillinger, T. Treatment resistance in psychiatry: state of the Art and new directions. Mol. Psychiatry. 27, 58–72. https://doi.org/10.1038/s41380-021-01200-3 (2022).
Acknowledgements
The authors wish to thank Assoc. Prof. Olivera Vuković and Assist. Prof. Milutin Kostić for their valuable advice and support in specific aspects of the study. The authors are also grateful to Tatjana Pravilović and Maja Stojisavljević for their assistance with blood sample collection and data management during the study.
Funding
Open access funding provided by Karolinska Institute. This work was supported by the Science Fund of the Republic of Serbia grant (PsyCise, project number: 6066800), awarded to MM Jukić.
Author information
Authors and Affiliations
Contributions
M.M.J., N.P.M., and Č.M. conceived and designed the analysis.P.G.V., A.J., M.V., F.M., D.P., Z.P., J.D.Đ., B.P., and B.M. collected the data.Z.P., F.M., B.M., M.I.S., Č.M., N.P.M., and M.M.J. contributed data or analysis tools.P.G.V., B.M., and M.M.J. performed the analysis.P.G.V., M.I.S., and M.M.J. wrote the manuscript.All authors reviewed and approved the final version of the manuscript.
Corresponding author
Ethics declarations
Competing interests
MM Jukić is a cofounder and the executive director of TDM health DOO. M Ingelman Sundberg is a cofounder and executive board chairmen of HepaPredict AB. The other authors reported no conflicts of interest.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Vuković, P.G., Jeremić, A., Vezmar, M. et al. Effectiveness and safety of escitalopram treatment personalized based on therapeutic drug monitoring of drug plasma concentration: a prospective cohort study. Sci Rep 15, 32470 (2025). https://doi.org/10.1038/s41598-025-18517-6
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-18517-6
