
Abstract
Noug (Guizotia abyssinica) is an economically important oilseed crop in Ethiopia that contributes significantly to local edible oil production and is a good protein source in animal feed. Despite its agronomic importance, the molecular basis of key agronomic traits, such as self-compatibility, photoperiod sensitivity, and oil biosynthesis, remains poorly understood due to the limited availability of genomic resources. To bridge this knowledge gap, we conducted extensive transcriptome profiling of 30 phenotypically diverse noug genotypes through RNA sequencing and de novo assembly. Our analysis generated 409,309 unigenes with an N50 of 584 bp, representing an extensive transcriptomic resource currently available for this crop. A total of 2,547 differentially expressed genes (DEGs) were identified, among which 409 were particularly associated with fatty acid metabolism pathways. Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) analyses revealed significant enrichment in lipid metabolism, stress response, and floral development pathways. Notably, many transcription factor families, such as bHLH, MYB, and WRKY, were differentially expressed between early- and late-flowering genotypes and high- and low-oil varieties, suggesting their regulatory roles in these traits. Transcriptome assembly revealed 58,852 putative transcription factors distributed in 51 families. This study provides fundamental genomic resources for marker-assisted breeding to improve productivity, oil quality, and stress resistance. The identified candidate genes present new opportunities for this underutilized yet agronomically valuable crop through modern biotechnological approaches.
Similar content being viewed by others
Introduction
Noug (Guizotia abyssinica) is an economically important oilseed crop primarily cultivated in Ethiopia and India, contributing significantly to local edible oil production1,2. In addition to its value in human consumption, noug seeds are also used in the United States and Europe to feed birds, particularly finches. The crop is diploid with 2n = 30 chromosomes3 and it relies on a strict outcrossing reproductive method that heavily depends on honeybee pollination4,5,6. Despite its economic importance, where 30% of the country’s oilseed production and 26% of the produced oil are retained for home consumption7 it is economically less explored than other oilseeds, such as soybean or sunflower8. While its agronomic potential is evident9 a lack of genomic tools has been a constraining factor for molecular analysis of mechanisms regulating key traits such as self-compatibility, photoperiod sensitivity, and oil biosynthesis.
Unlike well-studied oil seeds such as sunflower, noug lacks a reference genome, and limited transcriptomic resources are available. Although it originated and has been domesticated in Ethiopia10 and molecular marker studies have confirmed its high genetic diversity within and among populations and its wild relatives11,12,13,14,15,16 functional genomic data are scarce. This is critical because honeybee pollination, strict outcrossing habits, and variability in fatty acid constitution make breeding difficult.
Molecular marker-based studies have confirmed the wide genetic base of this crop, which aligns with trait diversity in locally adapted landraces6,17,18,19. This diversity supports the potential for breeding programs targeting desirable traits such as oil content, fatty acid composition, self-compatibility, days to maturity, and photoperiod response. Noug seeds generally contain 25–56% oil by weight, with an average oil content of approximately 35% 6,17–19. Oleic acid (C18:1) and linoleic acid (C18:2) dominate noug oil, comprising more than 90% of its fatty acid profile19,20. Although linoleic acid enhances nutritional value, its high levels reduce oxidative stability, limiting shelf-life and food applications21,22. Conversely, a relatively high oleic acid content enhances thermal stability, making it favorable for high-temperature cooking and biodiesel use19,23.
Consequently, this study examined the transcriptomic variation underlying key agronomic traits in noug to identify candidate genes for marker-assisted breeding. Hence, RNA-seq analysis of 30 diverse genotypes was conducted with the following three primary objectives: (1) to generate the first comprehensive de novo transcriptome assembly for noug; (2) to identify DEGs associated with target traits, including self-compatibility, photoperiod sensitivity, and oil biosynthesis; and (3) to annotate metabolic pathways and transcription factors and metabolic pathways potentially associated with fatty acid metabolism and the stress adaptation response.
Results
Transcriptome sequencing and assembly
RNA-seq analysis generated 1.9 billion raw reads in 30 noug genotypes, with 64.5 million high-quality reads per genotype (Supplementary Table S1). Data quality assessment revealed that the average G + C content and Phred score of the raw reads met the quality criteria (G + C > 45%, Q30 > 94%, and average quality score > 36). Thus, the transcriptome dataset is considered suitable for downstream analysis of the transcriptome. Following stringent quality filtering (Phred score ≥ 30) and adapter trimming with Cutadapt v2.10, the resulting 1.82 billion clean reads were de novo assembled via Trinity v2.1.1 with default parameter settings (k-mer size = 25, min_contig_length = 200), resulting in 561,322 transcripts that coalesced into 409,309 unigenes after redundancy removal (Table 1). Length distribution analysis revealed a bimodal pattern, with many sequences (71.8%) falling within the range of 200–500 bp (403,196 transcripts) and a significant proportion of longer sequences (9.3% >1 kb, 10,609 contigs > 2 kb). The assembly showed robust metrics, including a maximum contig length of 13.6 kbp, a mean length of 497.9 bp, and an N50 of 590 bp, comparable to those of other oilseed crop transcriptomes. The N50 (584 bp) exceeds that of sunflower (390 bp), indicating robust assembly. While the unigene count (409,309) is high, it reflects noug’s heterozygosity and diversity, consistent with other complex de novo assemblies24,25. Putative TFs (58,852) were identified via strict PlantTFDB criteria (E-value < 1e-10, coverage > 50%, identity > 40%); fragmentation may inflate this estimate, requiring functional validation. The overall transcriptome assembly spanned 279.5 Mb, with comprehensive coverage of the expressed genome of noug (Fig. 1).

Unigene sequence length distribution. Unigene length is presented on the x-axis, whereas the number of unigenes in each range is given on the y-axis.
Genotypic expression diversity and clustering patterns
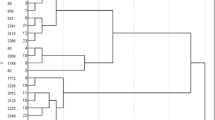
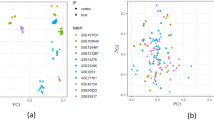
Principal component analysis of the normalized expression data for all 409,309 unigenes revealed biologically meaningful differences between genotypes. The first two principal components accounted for 36.1% of the total transcriptional variation (PC1: 22.4%; PC2: 13.7%), as depicted in Fig. 2A. UPGMA clustering based on Euclidean distances identified five dominant clusters (I-V) with distinct expression profiles (Fig. 2B). Specifically, Group-10 genotypes presented the most differentiated expression profiles, forming a separate cluster (II) with longer average lengths than the remaining groups. In contrast, Group-7 genotypes were heterogeneously classified into three distinct groups (I, II, V), suggesting strong underlying transcriptional plasticity despite phenotypic similarity. These patterns are consistent with previously reported genetic differences in noug populations6,17,18,19 and provide novel insights into the expression-level variation underlying agronomic traits. The observed transcriptional variation found in Groups 7 and 10 indicates the potential for selective breeding to capitalize on this natural variation.

The overall gene expression-based cluster analyses depict the relationships among the 30 genotypes: (A) principal component analysis (PCA) scatter plot and (B) unweighted pair group method with arithmetic mean (UPGMA) dendrogram.
Comprehensive functional annotation
BLAST searches were conducted on 409,309 unigenes in six major databases. The multi-database annotation pipeline successfully assigned putative functions to 211,945 unigenes (51.8% of the total) with significant hits (E-value cutoff = 1e-5) in at least one of the databases, with detailed breakdowns shown in Table 2.
Among the unigenes annotated in the NR database, 87.6% had homology to protein sequences of the Asteraceae family, with Helianthus, Cynara, and other Asteraceae species accounting for 74.6%, 11.8%, and 1.2%, respectively (Fig. 3A and B). Among the unigenes with significant hits against Helianthus species, 99.9% were against H. annuus, reflecting their close phylogenetic relationship. The remaining 12.4% of the annotated unigenes were homologous to diverse plant families, including Leguminaceae (1.5%), Poaceae (0.9%), Solanaceae (0.9%), and Brassicaceae (0.8%) (Fig. 3A and C).

Pie charts displaying the number of unigenes that had significant BLAST hits against the sequences of Asteraceae and other plant families in the NCBI nonredundant protein database (NR): the number of unigenes annotated against Asteraceae species versus those annotated against species of other plant families; (A) further classification of the unigenes annotated against Asteraceae species; (B); further classification of the unigenes annotated against non-Asteraceae species (C). The percentage value beside each category indicates the proportion of unigenes annotated in the NR database for that category.
Gene Ontology classification assigned 115,216 unigenes (28.1%) to three major categories: the biological process (BP), cellular component (CC), and molecular function (MF) classes (Fig. 4). The BP class was dominated by cellular (41,388) and metabolic (39,616) processes; the CC class was enriched for membrane (35,641), cell (25,795), and organelle (16,849) terms; and the MF class was predominantly associated with binding (64,432) and catalytic (55,812) activities. The KEGG pathway analysis mapped 29,795 unigenes (19.6% annotated) to 161 metabolic and regulatory pathways (Fig. 5), with higher representation of lipid metabolism (3,162 unigenes), signal transduction (3,680), carbohydrate metabolism (3,013), and amino acid metabolism (3,340).

Gene Ontology (GO) annotations of noug (A) expressed genes and (B) significantly differentially expressed genes to different functional categories of biological process (BP), cellular component (CC), and molecular function (MF) GO classes.

Kyoto Encyclopedia of Genes and Genomes (KEGG) annotation of noug unigenes into different sub-pathways of the five KEGG pathway classes: cellular processes, environmental information processing, genetic information processing, metabolism, and organismal systems.
The comprehensive TF analysis identified 58,852 putative transcription factors in the PlantTFDB spanning 51 families (Supplementary Figure S1), with bHLH (5,365; 9.1%), MYB-related (3,978; 6.8%), and LBD (3,888; 6.6%) being the most prevalent. These annotations provide crucial functional context for interpreting differential expression patterns.
There were significant matches between transcription factor genes (TFs) from 162 plant species and the unigenes, with lettuce (Lactuca sativa), radish (Raphanus sativus), and wild tomato (Solanum pennellii), being the top three, accounting for 4,015 (6.8%), 3889 (6.6%), and 3,579 (6.1%), respectively (Supplementary Figure S1). Only 612 unigenes (1.0%) were significantly associated with sunflower (Helianthus annuus) transcription factors.
Differential gene expression analysis
Ten different pairwise comparisons were made to determine the DEGs and significant DEGs between each pair (Table 3). Pairwise comparisons were selected to contrast groups with divergent traits: Group-2 (reference: high self-seed set, intermediate flowering) vs. Groups 1,3,4,5,7 (varying self-compatibility/oil); Group-7 (very late flowering) vs. Group-8 (very early flowering) for maturity; and Group-7/8 (photoperiod-sensitive) vs. Group-10 (photoperiod-insensitive). Comparative expression analysis via DESeq26,17,18,19 identified 2,330 DEGs with FDRs < 0.1, including 1,781 significant DEGs (FDR < 0.05, log2FC > 1) (Table 3; Supplementary Table S2). The number of significant DEGs varied substantially between group comparisons, ranging from 43 (Group-2 vs. Group-1) to 572 (Group-2 vs. Group-4).
Hierarchical clustering revealed eight distinct gene expression clusters (A-H) and nine genotype clusters (1–9) with characteristic patterns across genotypes (Fig. 6). Clusters D and E contained many genes whose expression was upregulated in the genotypes of Group-2 but downregulated in the genotypes of the other nine groups. A similar pattern was observed in cluster F, where many genes were upregulated in Group-10 but downregulated in the other nine genotypes. Notably, clusters D and E were upregulated in Group-2 (self-compatible), cluster F was specifically upregulated in Group-10 (photoperiod insensitive), and cluster G was downregulated in late-flowering genotypes.

A heatmap depicting the expression patterns of 1,781 unigenes that were significantly differentially expressed among the 30 noug genotypes (forming eight clusters; clusters A to H), and the genotypes were grouped into nine clusters along the y-axis (Clusters 1 to 9).
Venn diagram analysis (Fig. 7) revealed shared and unique DEG sets across trait comparisons, suggesting specialized and pleiotropic genetic regulation. The significant DEGs were also compared by grouping the ten pairs of groups (Table 3) into two categories, each with five pairs (Fig. 7).

Venn diagrams illustrating the number of DEGs that were upregulated or downregulated across different compared group pairs: (A) comparing the first five group pairs (category-1) for upregulated genes; (B) comparing the first five group pairs (category-1) for downregulated genes; (C) comparing the second five group pairs (category-2) for upregulated genes; and (D) comparing the second five group pairs (category-2).
Volcano plot analysis highlighted several significant DEGs between Group-2 versus Group-1 and Group-7, Group-7 versus Group-8, and Group-10 (Fig. 8). Group-2 and Group-1 differ in self-seed set levels; hence, the significant DEGs between them include genes relevant to self-compatibility. The significant DEGs between Group-2 (intermediate flowering time) and Group-7 (very late flowering time) included those related to flowering time. Similarly, the significant DEGs between Group-7 (very late maturing) and Group-8 (very early maturing) included genes related to flowering time. The DEGs of Group-7 vs. Group-10 were related to flowering time and photoperiod sensitivity.

Volcano plots of significantly differentially expressed genes for (A) Group-2 vs. Group-1, (B) Group-2 vs. Group-7, (C) Group-7 vs. Group-8, and (D) Group-7 vs. Group-10. Each dot corresponds to a gene. In the two groups compared, dots in green denote upregulated genes, dots in red denote downregulated genes, and dots in black denote genes that were not significantly differentially expressed between the groups compared.
Validation of differentially expressed genes
Eight candidate DEGs representing key traits were validated by qRT-PCR in 18 genotypes spanning four phenotypic groups. Strong concordance was observed between RNA-seq and qRT-PCR results (R²=0.89, P < 0.001; Supplementary Table S7). For example, TRINITY_DN97581_c0_g3_i2 (oil biosynthesis) showed consistent upregulation in high-oil genotypes (log2FC = 2.1, qRT-PCR ΔΔCt = -3.4 cycles; P = 0.003). The high correlation between log2FC and ΔΔCt values (R²=0.94, P < 0.001) confirms the reliability of our transcriptome analysis.
Annotation of significantly differentially expressed genes
The annotation of significant DEGs between pairs of groups in different databases revealed their functional roles (Figs. 9, 10 and 11). The annotation in the GO database revealed many GO terms associated with significant DEGs between each pair of groups (Fig. 4B; Supplementary Table S4-A to J). The significant DEGs were annotated with 26, 25, and 21 terms from the BP, CC, and MF GO classes (Fig. 9), 148 DEGs across ten pairs of groups enriched for 41 KEGG pathways belonging to 14 KEGG pathway classes (Fig. 10; Supplementary Table S5-A to J), and 50 TF family proteins associated with significant DEGs across the ten group pairs (Fig. 11; Supplementary Table S6-A to J). The most frequent hit TF family was bHLH, whereas the most frequent hit plant species was Lactuca sativa (Fig. 11; Supplementary Figure S1).

A horizontal bar graph of Gene Ontology (GO) annotations showing the number of genes significantly differentially expressed between pairs of groups to different GO terms of the biological process (BP), cellular component (CC), and molecular function (MF) GO classes. Each GO term corresponds to one to four group pairs. Note: Group pairs are abbreviated: e.g., G2_G1 refers to significant DEGs for Group-2 vs. Group-1.

A horizontal bar graph of Kyoto Encyclopedia of Genes and Genomes (KEGG) annotations showing the number of significantly differentially expressed genes between pairs of groups to different enriched KEGG pathways of various KEGG pathway classes. Each KEGG pathway corresponds to one to six group pairs. Note: Group pairs are abbreviated: e.g., G2_G1 refers to significant DEGs for Group-2 vs. Group-1.

A horizontal bar graph showing transcription factors (TFs) corresponding to significantly differentially expressed genes between different pairs of groups. Each TF corresponds to one to ten group pairs. The number of significant DEGs for the corresponding group pair is given in the graph. Note: Group pairs are abbreviated: e.g., G2_G1 refers to significant DEGs for Group-2 vs. Group-1.
Significantly differentially expressed genes and self-compatibility
Four self-compatible (Group-1 to Group-4) and six self-incompatible genotypes were contrasted in this study (Table 4), with a focus on gene expression differences. Between Group-2 (high self-seed set) and Group-4 (very low self-seed set), considerable DEGs related to lipid metabolism, phosphorus metabolic processes, and plastid functions were present within the BP and CC categories of the GO. The MF category highlighted catalytic and oxidoreductase activities, with 155 and 36 DEGs, respectively (Fig. 9). Pathway analysis via the KEGG database revealed significant enrichment in four key metabolic processes associated with self-compatibility: flavonoid biosynthesis (ko00941), plant circadian regulation (ko04712), isoquinoline alkaloid production (ko00950), and terpenoid-quinone biosynthesis (ko00130). The TFs bHLH, WRKY, and NAC were overrepresented among the DEGs, suggesting their regulatory role in self-fertilization.
A comparative study of Group-2 (high self-seed set) and Group-1 (low self-seed set) identified DEGs involved in pollen and seed development, including genes involved in sporopollenin biosynthesis and pollen wall assembly. KEGG enrichment revealed riboflavin metabolism and diterpenoid biosynthesis (Fig. 10), whereas TF analysis revealed regulators such as BBR-BPC, NAC, and B3. Interestingly, these TFs were differentially expressed in Group-2 vs. Group-4, supporting the likelihood of their involvement in self-compatibility. Other comparisons between self-compatible (Group-2) and self-incompatible genotypes (Group-5 and Group-7) identified DEGs related to small molecule metabolism, protein kinase activity, and photosynthesis-related processes, with MYB-related, bHLH, and ARF TFs playing major roles.
This study revealed that self-compatible genotypes possess specific metabolic and gene expression profiles of regulatory genes compared with self-incompatible genotypes. The most important results are the involvement of flavonoid and terpenoid metabolic pathways, pollen development genes, and some TFs (e.g., bHLH, WRKY, NAC) in self-fertilization (Fig. 11). These findings constitute the foundation for studying the molecular mechanisms of self-compatibility, which can be directly translated to improve the reproductive efficiency of crops.
Significantly differentially expressed genes and earliness
Concerning earliness, Group-7 (very late) is distinct from Group-8 (very early). The GO annotation of significant DEGs for Group-7 vs. Group-8 revealed that the most frequent terms were carboxylic acid metabolic process (GO:0000967), oxoacid metabolic process (GO:0097576), and organic acid metabolic process (GO:0000966) of the BP GO class, with six DEGs each (Fig. 9; Supplementary Table S4). Under the MF GO class, catalytic activity (GO:0003824) was the most frequent term, with 21 DEGs. KEGG annotation revealed that 15 of the 64 significant DEGs for this pair were related to 29 KEGG pathways (Supplementary Table S5). Five of these pathways, including RNA polymerase (ko03020), were enriched (P < 0.05). According to the PlantTFDB annotations, 16 of the 64 DEGs were annotated with 12 TF family proteins. The three most frequent DEGs were bHLH, ERF, and LBD, with 16, 10, and 8 DEGs, respectively (Fig. 11). NF-YA was the only TF that was differentially expressed between Group-4 and Group-5, but not between Group-2 and Group-4.
Significantly differentially expressed genes and oil and oleic acid contents
On average, Group-1, Group-4, and Group-5 had higher oil and oleic acid contents than Group-2. The significant DEGs between Group-2 and Group-1 were annotated with 33 terms belonging to the MF GO class. Among these DEGs, TRINITY_DN82849_c0_g2_i2 was associated with diacylglycerol O-acyltransferase activity (GO:0004144) and acylglycerol O-acyltransferase activity (GO:0016411). However, other genotypes whose oil and oleic acid contents differ have inconsistent expression patterns for this gene. One of the nine TFs associated with significant DEGs between Group 2 and Group 1 was WRKY, which is known to regulate lipid biosynthesis.
Among the DEGs significantly upregulated in Group-2 compared with Group-4, TRINITY_DN97581_c0_g3_i2 was annotated with several terms of the three GO classes, including lipid metabolic process (GO:0006629; BP) and acyl-carrier-protein desaturase activity (GO:0045300; MF). Furthermore, this gene was upregulated in Group-2 compared with Group-5 and was annotated with several GO terms, including the fatty acid metabolic process (GO:0006631; BP). This DEG was annotated with pathways for fatty acid biosynthesis (ko00061) and unsaturated fatty acid biosynthesis (ko01040) in the KEGG database. Fatty acid biosynthesis was the second most enriched pathway for Group-2 vs. Group-4, which included four DEGs. The upregulation of the TRINITY_DN97581_c0_g3_i2 gene in high-oil genotypes suggests a role in lipid biosynthesis. Another DEG upregulated in Group-2 compared with Group-4 was TRINITY_DN105918_c1_g3_i1. This DEG was annotated with different KEGG pathways, including lipid metabolic process (GO:0006629; BP) and linoleic acid metabolism (ko00591). Among the TFs associated with the significant DEGs between Group-2 and Group-4 were bHLH, ERF, FAR1, MYB-related, and WRKY, which were previously reported to play a role in lipid biosynthesis.
Significantly differentially expressed genes and photoperiod sensitivity
The photoperiod sensitivity was lower in Group-10 than in the other nine groups. Comparative transcriptome analysis of the photoperiod-sensitive (Group-7) and photoperiod-insensitive (Group-10) genotypes revealed that organonitrogen compound metabolism (GO:1901564; 25 DEGs), organelle components (GO:0044422; 20 DEGs), and catalytic activity (GO:0003824; 13 DEGs) were the highly enriched GO terms in the BP, CC, and MF classes, respectively (Fig. 9). Similarly, the GO annotation of significant DEGs between Group-8 (high photoperiod sensitivity) and Group-10 (low photoperiod sensitivity) revealed that cellular metabolic process (GO:0044237), cell (GO:0005623), and metal ion binding (GO:0046872) were the most frequent terms associated with the BP, CC, and MF GO classes, respectively, and occurred in 75, 65, and 56 significant DEGs (Fig. 9).
The KEGG annotation revealed that 56 significant DEGs between Group-7 and Group-10 belong to 64 KEGG pathways (Supplementary Table S5). Among these, seven pathways, to which 16 DEGs were assigned, were enriched (P < 0.05), including tyrosine metabolism (ko00350) and fatty acid degradation (ko00071). For Group-8 versus Group-10, 85 significant DEGs were attributed to 87 KEGG pathways, including circadian rhythm-plant (ko04712). Seven pathways were enriched, to which 20 DEGs were assigned (P < 0.05). These include quorum sensing (ko02024) and fatty acid degradation (ko00071) (Supplementary Table S5; Fig. 10). Among the significant DEGs between Group-7 and Group-10, 98 were associated with 30 different TFs. The three genes with the greatest frequency were MYB-related, bHLH, and B3, with 9, 8, and 8 DEGs, respectively (Fig. 11). In the case of Group-8 vs. Group-10, 110 DEGs were successfully annotated with 34 different TFs. The three most frequent TFs were MYB-related, bHLH, and C3H and included 11, 9, and 7 DEGs, respectively (Fig. 11).
Discussion
RNA sequencing-based transcriptome profiling has emerged as a powerful tool for identifying differentially expressed genes (DEGs) in plant species, providing critical insights into their regulatory processes and functional implications. While extensively utilized in model organisms such as Arabidopsis26 brassicas27,28 and sunflower29 no complete transcriptomic analysis has yet been described for noug (Guizotia abyssinica). Our study fills this gap by characterizing gene expression profiles in noug genotypes, where priority has been assigned to critical agronomic traits such as self-compatibility, oil content, fatty acid composition, days to flowering, and photoperiod sensitivity. Identifying novel unigenes and DEGs further enriches genomic tools and resources for noug and underscores the need for functional validation to elucidate their roles in trait regulation. Although phenotypic groups (Table 4) did not always form distinct clusters in overall expression profiles (Fig. 2B), DEG analysis between groups contrasting for specific traits (e.g., SC vs. SI, early vs. late) successfully identified candidate genes. This suggests trait regulation involves specific transcriptional subnetworks rather than genome-wide shifts. These findings lay a foundation for molecular breeding strategies focused on developing improved cultivars with optimized traits.
Transcriptome assembly and functional annotation
The N50 value, a key metric for assessing transcriptome assembly quality, was significantly greater in G. abyssinica (590 bp) than in H. annuus (390 bp)30 indicating a robust assembly31,32,33. Among the 409,309 unigenes identified, 51.8% (211,945) exhibited significant homology to sequences in public databases (E-value ≤ 1e-5). However, 58.5% lacked matches in the NR database, potentially due to their noncoding nature, short sequence length, or the limited availability of G. abyssinica genomic data. Among the annotated unigenes, 74.6% aligned with H. annuus proteins, whereas only 0.9% matched Asteraceae family proteins, highlighting the underrepresentation of noug in existing databases.
Gene Ontology (GO) analysis annotated 28.1% (115,216) of the unigenes into at least one GO term categorized as biological process (BP), molecular function (MF), or cellular component (CC) categories. Predominant BP terms included cellular and metabolic processes, biological regulation, and response to stimuli, whereas CC terms were enriched in membrane-, cell-, and organelle-related functions. MF annotations were dominated by binding, catalytic, and transporter activities, which is consistent with roles in signal transduction and metabolic regulation34. Further pathway analysis via KEGG revealed that 19.6% (29,795) of the annotated unigenes participated in 161 metabolic and regulatory pathways, with significant representation of lipid, amino acid, and carbohydrate metabolism, which aligns with findings in H. annuus33.
Trait-associated gene expression patterns
Seed setting is a critical developmental stage regulated by genetic and environmental factors that affect seed number, size, and yield potential. In our study, many unigenes were linked to metabolism pathways with significant involvement in lipid, phosphorus, and phosphate-containing compound metabolic processes. Notably, E3 ubiquitin-protein ligases, known to regulate seed development35 were implicated in our dataset. Differentially expressed gene analysis revealed two important genes associated with this trait: DN97095_c2_g1_i7, a CBL-interacting serine/threonine-protein kinase 23 homolog (implicated in ATP binding and protein phosphorylation), and DN79699_c0_g3_i1, a putative guanosine tetraphosphate diphosphokinase RSH1 ortholog of H. annuus involved in nucleotide metabolism. These findings align with studies on the mechanisms of seed setting in Brassica napus36 suggesting conserved regulatory mechanisms.
Early maturity is another adaptive trait for drought escape in arid climates37. Early flowering in Arabidopsis is controlled by complex signaling networks of transcription factors (TFs) and metabolic alterations35,38. Fatty acids play a role in the synthesis of suberin and cutin wax to reinforce cell membrane integrity and the structural barrier against abiotic stresses39. Our findings revealed that the RNA polymerase IV pathway (ko03020) was significantly enriched in early-maturing genotypes. RNA Polymerase IV also takes part in pollen development in Brassica rapa40 where its activity during meiosis influences pollen formation41 and microspore development in Capsella rubella42 suggesting its role in accelerating reproductive development. Functional studies in Capsella rubella have shown that the loss of function of RNA polymerase IV disrupts microspore development42 indicating a direct mechanistic link between flowering time regulation and pollen development. These findings suggest that Pol IV-mediated epigenetic control of reproductive development may be responsible for the early-mature phenotypes observed in noug. Notably, these findings highlight the importance of RNA polymerase IV-mediated regulation to control flowering time adaptation in noug. In addition, bHLH, ARF, and MYB-related TFs were differentially expressed in the early-maturing genotypes. ARFs control auxin-responsive gene expression and influence developmental timing43.
Seed oil accumulation is an essential trait that involves de novo fatty acid synthesis in plastids and triacylglycerol (TAG) biosynthesis and assembly, with lipid degradation modulating energy homeostasis in the endoplasmic reticulum44. Fatty acid degradation, through lipolysis, produces TAG and generates metabolites such as acyl-CoA and acetyl-CoA via β-oxidation, which conserves energy44. Some genotypes have a low oil content because of the frequent degradation of fatty acids. Some of the most upregulated genes in our study were DN46215_c0_g1_i1, the lipid binding upregulated gene, and DN98334_c1_g2_i2, the acyl group transferase activity upregulated gene. Some of the genotypes are related to lipid transport and the oil content in the fatty acid degradation pathway (ko00071). MYB-related, bHLH, C3H, and LBD are the most dominant TF families involved in the regulation of developmental processes as well as metabolism, such as seed size and oil content, in Brassica rapa45,46. Upregulated genes such as DN46215_c0_g1_i1 (lipid binding) and DN98334_c1_g2_i2 (acyltransferase activity) suggest genotype-specific variations in oil content. The fatty acid degradation pathway (ko00071) was prominent among the DEGs, with MYB-related, bHLH, C3H, and LBD TFs playing central roles. MYB TFs, known to regulate lipid metabolism in green algae47 may similarly influence oil biosynthesis in noug. Hence, knowledge of fatty acid biosynthesis and degradation pathways is important in exploring the molecular mechanisms governing the oil content of noug. Crossbreeding genotypes with photoperiod-sensitive, self-incompatible, and high-oil-containing traits (e.g., Ga08-03 and Ga10-06) could yield photoperiod-insensitive, early-maturing cultivars with high seed and oil yields that are suitable for low-altitude cultivation. Further investigation of significant DEGs and TFs will significantly clarify the molecular mechanisms underlying genomic-led breeding for noug.
Photoperiod sensitivity is a critical trait that can be included in the environmental adaptations of plants. However, such adaptation through modification by DNA methylation is heritable, although reversible, and is individually dependent on external stress and developmental stimuli48. For example, IDM1 prevents DNA hypermethylation of homologous genes under stress to increase photoperiod sensitivity through IDM1 activities49. Two-component response regulator-like APRR3 is another process that regulates the photoperiodic flowering response in Arabidopsis thaliana50Cicer arietinum51 and Glycine max52. In addition, E3 ubiquitin-protein ligases are critical regulators of several pathways related to photoperiodism, mediating light responses through photoreceptors, phytohormones, and other signaling networks53. DEG analysis between photoperiod-sensitive and photoperiod-insensitive genotypes revealed that an environmental adaptation-associated gene, DN94708_c2_g1_i11, was significantly upregulated in photoperiod-insensitive genotypes. In addition, the dominant TF families, MYB-related, bHLH, C3H, and LBD, are involved in light signaling and stress responses. MYB-related TFs are most important in plant development and metabolic processes and modulate cell differentiation, the cell cycle, and hormone and environmental responses54,55. MYB and C3H TFs regulate CONSTANS and FLOWERING LOCUS T expression56,57 whereas LBD TFs increase drought tolerance58. This increased photoperiod insensitivity in the Group-10 genotype was presumably a consequence of more rapid induction of MYB-related, C3H, and LBD TFs triggered by environmental cues. Hence, crossbreeding photoperiod-insensitive genotypes (e.g., Ga08-03 and Ga10-06) with high-oil, self-compatible lines could yield early-maturing cultivars suitable for low-altitude cultivation.
Conclusion
Our study presents the first comprehensive transcriptomic analysis of noug (Guizotia abyssinica), identifying 409,309 unigenes and 2,547 DEGs linked to key agronomic traits. Functional analyses revealed enriched pathways related to lipid metabolism and stress response, with bHLH, MYB, and WRKY transcription factors emerging as critical regulators. Notably, E3 ubiquitin ligases, RNA polymerase IV, and CONSTANS-like TFs were associated with flowering time and oil biosynthesis, suggesting targets for breeding climate-resilient cultivars. While 58.5% of the unigenes remain unannotated, this study lays a foundation for future functional studies (e.g., CRISPR-Cas9) and marker-assisted breeding to enhance noug productivity and stress adaptation.
Materials and methods
Plant material
This study utilized 30 phenotypically distinct noug genotypes (Table 4). Comprehensive phenotyping data (means, variances, statistical analyses) and trait images are published in6,17,18,19. With approximately two-thirds derived from breeding populations selected for improved traits, including self-compatibility, early maturation, reduced photoperiod sensitivity, and increased oil/oleic acid content, as described in59. Breeding populations were derived from crosses between Ethiopian landraces (detailed origins in59. Landraces were obtained from the Ethiopian Biodiversity Institute (accession numbers in Table S1 of59. Parents are not included; this study focuses on advanced/segregating material. One-third of the genotypes were selected from landrace populations based on differences in one or more target traits between them and from the breeding populations. Among the 30 genotypes, twelve are self-compatible, although to varying degrees, whereas the other eighteen are strictly self-incompatible. The days to maturity of the genotypes ranged from 120 days (very early types) to 180 days (very late types). Among the 30 genotypes, three were selected from breeding populations capable of flowering when the photoperiod exceeded 12 h. The mean oil content of the source populations was 30–45% of their dry seed weight. The oleic acid content of all the source populations except Ga01-16, Ga02-01, Ga01-02, and Ga02-02 was lower than 13%, although the oleic acid content primarily depends on the environmental temperature. The 30 genotypes were grouped into ten groups based on their similarity in one or more target traits described in Table 4 below.
The 30 genotypes were grouped into ten groups based on their similarity in one or more target traits described in Table 4 below. Grouping was based solely on phenotypic similarity in target traits (Table 4) to enable focused DEG analysis between trait extremes (e.g., high vs. low self-seed set), reducing complexity despite imperfect clustering in overall transcriptomes (Fig. 2B).
Planting and sampling
Seeds of the 30 genotypes were planted in 1.5 L plastic pots filled with soil at the Swedish University of Agricultural Sciences (Alnarp, Sweden). Leaf tissue from each genotype was collected one month after planting, snap-frozen in liquid nitrogen, and stored at − 80 °C until RNA extraction was performed.
RNA extraction and quality control
Total RNA was extracted from approximately 100 mg of leaf tissue from each sample via the RNeasy Plant Mini Kit (74904, QIAGEN). Next, DNase treatment was performed on the extract via an Ambion Turbo DNA-Free Kit (AM1907, Thermo Fisher Scientific, CA, USA). The quantity and quality of the extracted RNA were assessed via an Agilent Bioanalyzer 2100 (Agilent Technologies, CA, USA), a NanoDrop ND-1000 spectrophotometer (Saveen Werner, Sweden), and agarose gel electrophoresis. High-quality RNA samples were subsequently sent to CD Genomics (New York, USA) for RNA sequencing and analysis. Upon arrival, further samples were examined on 1% agarose gels for any evidence of degradation or contamination. The samples were then assessed for purity on a spectrophotometer (IMPLEN, CA, USA), the concentration was measured with a Qubit 2.0 fluorometer (Life Technologies, CA, USA), and sample integrity was assessed using the Agilent Bioanalyzer 2100 system (Agilent Technologies, CA, USA) and the RNA Nano 6000 Assay Kit.
Library preparation, clustering, and sequencing
The RNA library of each sample was created from 1.5 mg of RNA via the NEBNext UltraTM RNA Library Prep Kit for Illumina (NEB, USA) according to the manufacturer’s instructions. Index codes were added to the adapter sequences to identify different sequences to their respective samples, as described previously by59. The library fragments were subsequently cleaned via the AMPure XP system (Beckman Coulter, Beverly, USA), which recognizes fragments of insert sizes 150–200 bp long. Following adapter ligation and PCR amplification, the DNA fragments were purified with Beckman Coulter’s AMPure XP system (Beverly, USA). Library quality was assessed using an Agilent Bioanalyzer 2100 system before clusters and paired-end sequencing were generated on the Illumina HiSeq 2500 platform. The index-coded samples were subsequently clustered via the TruSeq PE Cluster Kit v3-cBot-HS (Illumina) according to the manufacturer’s instructions. Finally, the sequences were clustered, and high-quality paired-end reads were generated using the Illumina HiSeq 2500 platform.
Data quality control, de novo transcript assembly, and splicing
The raw sequencing reads were filtered via a series of methods to obtain high-quality data for further analysis. First, adapters and poly-N sequences of the raw reads were removed via in-house Python scripts, and clean reads were obtained. Then, Phred quality scores were calculated for the clean reads, and reads with Phred quality scores < 30 (error rate > 0.1%) were excluded. The Trinity software package (Trinity v. 2.1.1;60 performs de novo transcript assembly of high-quality reads since no reference genome is available for noug. Recent studies highlight key considerations for robust de novo transcriptome assembly, including quality control metrics and parameter optimization24,25. To accomplish this, single-read1 and single-read2 files were created by merging the two read files of the 30 genotypes and then used for transcript assembly and splicing with Trinity60utilizing the parameter max_kmer_cov as 2 and all other parameters as defaults. The analysis of the length distribution of transcripts led to the identification of the longest spliced transcripts of different genes, i.e., unigenes, which were used for various downstream analyses. The unigenes have been deposited at DDBJ/EMBL/GenBank as a Transcriptome Shotgun Assembly project under accession number GJSF00000000 (https://www.ncbi.nlm.nih.gov/nuccore/GJSF00000000). The RNA-seq quality-trimmed raw reads were deposited in the Sequence Read Archive (SRA) under accession number PRJNA763316 (https://www.ncbi.nlm.nih.gov/bioproject/PRJNA763316/).
Gene expression level and differential expression analyses
The gene expression level in each sample was estimated via RNA sequencing via the expectation maximization package (RSEM v.1.2.08;61which is based on read counts determined via the mapping of sequenced paired-end reads onto the assembled transcriptome. DEGs were identified between pre-defined phenotypic groups (Table 4) to target specific trait contrasts despite group heterogeneity in other traits. This prioritizes the discovery of trait-associated candidates over strict group-wide expression differences. By calculating fragments per kilobase pair per million reads (FPKM, the abundance of each gene was determined, and transcripts with FPKM values greater than 0.5 were regarded as expressed. The DESeq262 R package was used to perform differential expression analysis of the ten groups of genotypes. While biological replication was limited (pooled RNA for Ga01-12 and Ga01-16; single plants for others), DESeq2’s dispersion estimation (fitType = ‘local’) accounts for unreplicated designs to identify candidate DEGs for downstream validation. A gene was considered significantly differentially expressed if it presented a false discovery rate (FDR)-adjusted P value below 0.01 and a log2-fold change (log2FC) above 2. DEGs and genotype groups were evaluated using a two-way hierarchical cluster analysis with the pheatmap63 v.1.0.8 R package. Principal component analysis (PCA) was conducted via the “adegenet”64 package in R to determine the overall relationships among the 30 genotypes. The unweighted pair group method with arithmetic mean (UPGMA) cluster analysis was also performed on the same data based on pairwise Euclidean distance via the “vegan”65 package in R.
Gene function annotation
Gene function annotations of the unigenes were conducted in six major databases to obtain comprehensive gene function information: Universal Protein (UniProt, http://www.ebi.ac.uk/uniprot/;66, Nonredundant Protein (NR; https://www.ncbi.nlm.nih.gov/;67), Kyoto Encyclopedia of Genes and Genomes (KEGG; http://www.genome.jp/kegg/;68, Nucleotide (NT; https://www.ncbi.nlm.nih.gov/), Gene Ontology (GO; http://www.geneontology.org/;69 and the plant transcription factor database (PlantTFDB v.3.0, http://planttfdb.gao-lab.org;70. The functional annotation of differentially expressed genes (DEGs) was performed via major genomic databases. GO terms were mapped via InterProScan, followed by enrichment analysis with topGO (Fisher’s exact test). Transcription factors were identified through a BLAST search against PlantTFDB v3.0 (E-value < 10^-10, query coverage > 50%, identity > 40%).
Validation of DEGs by qRT-PCR
Eight DEGs (2 self-compatibility, 2 photoperiod-related, 1 flowering time, 3 oil biosynthesis) were selected for validation using quantitative reverse transcription PCR (qRT-PCR). Total RNA from 18 representative genotypes (3 per group from Groups 2, 7, 8, and 10) was reverse-transcribed using the SuperScript IV First-Strand Synthesis System (Thermo Fisher). Gene-specific primers (Supplementary Table S7) were designed using Primer-BLAST with melting temperatures ranging from 58 °C to 60 °C. Reactions were performed in triplicate on a QuantStudio 3 system using PowerUp SYBR Green Master Mix (Applied Biosystems), with actin as the reference gene. The ΔΔCt values were calculated and compared to RNA-seq log2FC values.
Data availability
The data presented in this manuscript are included in supplemental tables, and the raw data were submitted to NCBI under the BioProject ID: PRJNA763316 (https://www.ncbi.nlm.nih.gov/bioproject/PRJNA763316).
References
Kandel, H. & Porter, P. Niger (Guizotia abyssinica)(L. f.) Cass. Production in northwest Minnesota. University of Minnesota Extension Service (2002).
Seegeler, C. J. P. Oil Plants in Ethiopia: their Taxonomy and Agricultural Significance (Wageningen University and Research, 1983).
Dagne, K. Meiosis in interspecific hybrids and genomic interrelationships in Guizotia Cass.(Compositae). Hereditas 121, 119–129 (1994).
Geleta, M., Asfaw, Z., Bekele, E. & Teshome, A. Edible oil crops and their integration with the major cereals in North Shewa and South welo, central highlands of ethiopia: an ethnobotanical perspective. Hereditas 137, 29–40 (2002).
Geleta, M. Genetic Diversity, Phylogenetics, and Molecular Systematics of Guizotia Cass.(Asteraceae) (Swedish University of Agricultural Sciences, 2007).
Geleta, M. & Bryngelsson, T. Population genetics of self-incompatibility and developing self-compatible genotypes in Niger (Guizotia abyssinica). Euphytica 176, 417–430 (2010).
EIAR, Ethiopian Institute of Agricultural Research. Oilseeds Research Strategy 2016–2023. (2017).
USDA-GAIN. Ethiopia Oilseeds Report Annual 10 (USDA, 2021).
Getinet, A. & Sharma, S. Niger (Guizotia abyssinica (L. f.) Cass.) promoting the conservation and use of underutilized and neglected crops. 5. Institute of Plant Genetics and Crop Plant Research, Gatersleben/International Plant Genetic Resources Institute, Rome, Italy (1996).
Hiremath, S. C. & Murthy, H. Domestication of Niger (Guizotia abyssinica). Euphytica 37, 225–228 (1988).
Dempewolf, H. et al. Establishing genomic tools and resources for Guizotia abyssinica (Lf) Cass.—the development of a library of expressed sequence tags, microsatellite loci, and the sequencing of its Chloroplast genome. Mol. Ecol. Resour. 10, 1048–1058 (2010).
Geleta, M., Bryngelsson, T., Bekele, E. & Dagne, K. Comparative analysis of genetic relationship and diagnostic markers of several taxa of Guizotia cass. (Asteraceae) as revealed by AFLPs and RAPDs. Plant Syst. Evol. 265, 221–239 (2007b).
Geleta, M., Bryngelsson, T., Bekele, E. & Dagne, K. Genetic diversity of Guizotia abyssinica (L. f.) Cass.(Asteraceae) from Ethiopia as revealed by random amplified polymorphic DNA (RAPD). Genet. Resour. Crop Evol. 54, 601–614 (2007a).
Geleta, M., Bryngelsson, T., Bekele, E. & Dagne, K. Assessment of genetic diversity of Guizotia abyssinica (Lf) Cass.(Asteraceae) from Ethiopia using amplified fragment length polymorphism. Plant. Genetic Resour. 6, 41–51 (2008).
Tsehay, S. et al. New Transcriptome-Based SNP markers for Noug (Guizotia abyssinica) and their conversion to KASP markers for population genetics analyses. Genes 11, 1373 (2020).
Terefe, M., Birmeta, G., Girma, D., Geleta, M. & Tesfaye, K. Analysis of genetic diversity and population structure of oilseed crop Noug (Guizotia abyssinica) accessions collected from Ethiopia. Molecular Biology Reports, 1–13 (2022).
Gebeyehu, A., Hammenhag, C., Ortiz, R., Tesfaye, K. & Geleta, M. Characterization of oilseed crop Noug (Guizotia abyssinica) using Agro-Morphological traits. Agronomy 11, 1479 (2021).
Geleta, M. & Ortiz, R. The importance of Guizotia abyssinica (niger) for sustainable food security in Ethiopia. Genet. Resour. Crop Evol. 60, 1763–1770 (2013).
Geleta, M., Stymne, S. & Bryngelsson, T. Variation and inheritance of oil content and fatty acid composition in Niger (Guizotia abyssinica). J. Food Compos. Anal. 24, 995–1003 (2011).
Dagne, K. & Jonsson, A. Oil content and fatty acid composition of seeds of Guizotia Cass (Compositae). J. Sci. Food. Agric. 73, 274–278 (1997).
Wanasundara, U. N. & Shahidi, F. Canola extract as an alternative natural antioxidant for Canola oil. J. Am. Oil Chemists’ Soc. 71, 817–822 (1994).
Dehghani, A. A., Mohammadi, Z. B., Maghsoudlou, Y. & Mahoonak, A. S. Intelligent Estimation of the Canola oil stability using artificial neural networks. Food Bioprocess Technol. 5, 533–540 (2012).
Petros, Y. et al. Developing high oleic acid in Guizotia abyssinica (Lf) cass. By plant breeding. Plant. Breed. 128, 691–695 (2009).
Kumar, V., Sugumaran, K., Al-Roumi, A. & Shajan, A. De-novo transcriptome assembly and analysis of lettuce plants grown under red, blue, or white light. Sci. Rep. 12, 22477 (2022).
Raghavan, V., Kraft, L., Mesny, F. & Rigerte, L. A simple guide to de Novo transcriptome assembly and annotation. Brief. Bioinform. 23, bbab563 (2022).
Duan, K., Willig, C. J., De Tar, J. R., Spollen, W. G. & Zhang, Z. J. Transcriptomic analysis of Arabidopsis seedlings in response to an Agrobacterium-mediated transformation process. Mol. Plant Microbe Interact. 31, 445–459 (2018).
Xing, M. et al. Transcriptome profiling of resistance to fusarium oxysporum f. Sp. conglutinans in cabbage (Brassica oleracea) roots. PloS One. 11, e0148048 (2016).
Xu, H. M. et al. Transcriptome analysis of brassica Napus pod using RNA-Seq and identification of lipid-related candidate genes. BMC Genom. 16, 1–10 (2015).
Fass, M. I. et al. Exploring sunflower responses to sclerotinia head rot at early stages of infection using RNA-seq analysis. Sci. Rep. 10, 13347 (2020).
Liang, C. et al. Identification of differentially expressed genes in sunflower (Helianthus annuus) leaves and roots under drought stress by RNA sequencing. Bot. Stud. 58, 1–11 (2017).
Scaglione, D. et al. Large-scale transcriptome characterization and mass discovery of SNPs in Globe artichoke and its related taxa. Plant Biotechnol. J. 10, 956–969 (2012).
Dlugosch, K. M., Lai, Z., Bonin, A., Hierro, J. & Rieseberg, L. H. Allele identification for transcriptome-based population genomics in the invasive plant Centaurea solstitialis. G3: Genes| Genomes| Genetics 3, 359–367 (2013).
Pegadaraju, V., Nipper, R., Hulke, B., Qi, L. & Schultz, Q. De Novo sequencing of sunflower genome for SNP discovery using RAD (Restriction site associated DNA) approach. BMC Genom. 14, 1–9 (2013).
Bachlava, E. et al. SNP discovery and development of a high-density genotyping array for sunflower. PloS One. 7, e29814 (2012).
Orozco-Arroyo, G., Paolo, D., Ezquer, I. & Colombo, L. Networks controlling seed size in Arabidopsis. Plant. Reprod. 28, 17–32 (2015).
Dąbrowska, G. B., Turkan, S., Tylman-Mojżeszek, W. & Mierek-Adamska, A. Silico study of the RSH (RelA/SpoT Homologs) gene family and expression analysis in response to PGPR bacteria and salinity in brassica Napus. Int. J. Mol. Sci. 22, 10666 (2021).
Cattivelli, L. et al. Drought tolerance improvement in crop plants: an integrated view from breeding to genomics. Field Crops Res. 105, 1–14 (2008).
Ruan, Y. L., Patrick, J. W., Bouzayen, M., Osorio, S. & Fernie, A. R. Molecular regulation of seed and fruit set. Trends Plant Sci. 17, 656–665 (2012).
Upchurch, R. G. Fatty acid unsaturation, mobilization, and regulation in the response of plants to stress. Biotechnol. Lett. 30, 967–977 (2008).
Grover, J. W. et al. Maternal components of RNA-directed DNA methylation are required for seed development in brassica Rapa. Plant J. 94, 575–582 (2018).
Walker, J. et al. Sexual-lineage-specific DNA methylation regulates meiosis in Arabidopsis. Nat. Genet. 50, 130–137 (2018).
Zhang, S., Wu, X. Q., Xie, H. T., Zhao, S. S. & Wu, J. G. Multifaceted roles of RNA polymerase IV in plant growth and development. J. Exp. Bot. 71, 5725–5732 (2020).
Finet, C., Berne-Dedieu, A., Scutt, C. P. & Marlétaz, F. Evolution of the ARF gene family in land plants: old domains, new tricks. Mol. Biol. Evol. 30, 45–56 (2013).
Goepfert, S. & Poirier, Y. β-Oxidation in fatty acid degradation and beyond. Curr. Opin. Plant. Biol. 10, 245–251 (2007).
Niu, Y. et al. Deciphering the transcriptional regulatory networks that control size, color, and oil content in brassica Rapa seeds. Biotechnol. Biofuels. 13, 1–20 (2020).
Rajavel, A. et al. Unravelling the complex interplay of transcription factors orchestrating seed oil content in brassica Napus L. Int. J. Mol. Sci. 22, 1033 (2021).
Shi, M., Yu, L., Shi, J. & Liu, J. A conserved MYB transcription factor is involved in regulating lipid metabolic pathways for oil biosynthesis in green algae. New Phytol. 235, 576–594 (2022).
Chen, X., Schönberger, B., Menz, J. & Ludewig, U. Plasticity of DNA methylation and gene expression under zinc deficiency in Arabidopsis roots. Plant Cell Physiol. 59, 1790–1802 (2018).
Qian, W. et al. A histone acetyltransferase regulates active DNA demethylation in Arabidopsis. Science 336, 1445–1448 (2012).
Matsushika, A., Makino, S., Kojima, M. & Mizuno, T. Circadian waves of expression of the APRR1/TOC1 family of pseudo-response regulators in Arabidopsis thaliana: insight into the plant circadian clock. Plant Cell Physiol. 41, 1002–1012 (2000).
Ahmad, B. et al. Genome-wide identification and expression analysis of two component system genes in Cicer arietinum. Genomics 112, 1371–1383 (2020).
Han, X., Wang, D. & Song, G. -q. Expression of a maize SOC1 gene enhances soybean yield potential through modulating plant growth and flowering. Sci. Rep. 11, 1–14 (2021).
Shu, K. & Yang, W. E3 ubiquitin ligases: ubiquitous actors in plant development and abiotic stress responses. Plant Cell Physiol. 58, 1461–1476 (2017).
Chen, Z. et al. MYB transcription factors becoming mainstream in plant roots. Int. J. Mol. Sci. 23, 9262 (2022).
Li, J., Han, G., Sun, C. & Sui, N. Research advances of MYB transcription factors in plant stress resistance and breeding. Plant. Signal. Behav. 14, 1613131 (2019).
Corbesier, L. et al. FT protein movement contributes to long-distance signaling in floral induction of Arabidopsis. Science 316, 1030–1033 (2007).
Cheng, X. et al. Identification of the wheat C3H gene family and expression analysis of candidates associated with seed dormancy and germination. Plant Physiol. Biochem. 156, 524–537 (2020).
Jiao, P. et al. ZmLBD2 a maize (Zea Mays L.) lateral organ boundaries domain (LBD) transcription factor enhances drought tolerance in Transgenic Arabidopsis Thaliana. Frontiers Plant. Science 13 (2022).
Gebeyehu, A., Hammenhag, C., Tesfaye, K., Ortiz, R. R. & Geleta, M. RNA-Seq provides novel genomic resources for Noug (Guizotia abyssinica) and reveals microsatellite frequency and distribution in its transcriptome. Front. Plant Sci. 13, 1073 (2022).
Grabherr, M. G. et al. Trinity: reconstructing a full-length transcriptome without a genome from RNA-Seq data. Nat. Biotechnol. 29, 644 (2011).
Li, B. & Dewey, C. N. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 12, 1–16 (2011).
Anders, S. & Huber, W. Differential expression analysis for sequence count data. Nature Precedings, 1–1 (2010).
Kolde, R. & pheatmap Pretty Heatmaps. R package version 1.0. 12 (2019).
Jombart, T. Adegenet: a R package for the multivariate analysis of genetic markers. Bioinformatics 24, 1403–1405 (2008).
Oksanen, J. et al. The vegan package. Community Ecol. Package. 10, 719 (2007).
Consortium, U. UniProt: a hub for protein information. Nucleic Acids Res. 43, D204–D212 (2015).
Bleasby, A. J. & Wootton, J. C. Construction of validated, non-redundant composite protein sequence databases. Protein Eng. Des. Selection. 3, 153–159 (1990).
Kanehisa, M. & Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 28, 27–30 (2000).
Consortium, G. Vol. 47 D330–D338 (2019).
Jin, J., Zhang, H., Kong, L., Gao, G. & Luo, J. PlantTFDB 3.0: a portal for the functional and evolutionary study of plant transcription factors. Nucleic Acids Res. 42, D1182–D1187 (2014).
Acknowledgements
The authors thank Sida for financing this research. Authors also thank the Department of Plant Breeding, Swedish University of Agricultural Sciences, for the technical support during the study.
Funding
Open access funding provided by Swedish University of Agricultural Sciences. This study was financed by the Swedish International Development Cooperation Agency (Sida) through research and training grants awarded to Addis Ababa University (AAU-SLU Biotech).
Author information
Authors and Affiliations
Contributions
Conceptualization, A.G., M.G., and R.O.; Methodology: A.G., M.G., R.R.V., and C.H.; Software: A.G. and M.G.; Data analysis: A.G., M.G., C.H., and R.O.; Writing original draft: A.G.; Writing, review, and editing: all co-authors; Funding acquisition: R.O., M.G., and K.T.; Supervision: all co-authors. All the authors have read and agreed to the published version of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Gebeyehu, A., Hammenhag, C., Tesfaye, K. et al. Transcriptome analysis identifies genes regulating self-compatibility, flowering time, and oil biosynthesis in Noug (Guizotia abyssinica). Sci Rep 15, 32475 (2025). https://doi.org/10.1038/s41598-025-18728-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-025-18728-x
