
Abstract
The safety and tolerability of histone deacetylase inhibitors (HDACIs) were always a matter of concern. This study aimed to explore the global observational adverse drug reaction (ADR) profiles of the HDACIs: vorinostat, belinostat, panobinostat, pracinostat, entinostat, romidepsin, bufexamac and sodium phenylbutyrate. This study focussed on the investigation whether associations between HDACIs’ ADR profiles and their physicochemical and pharmacological properties exist and how these associations may be harnessed to contribute to safer clinical use. The ADRs of HDACIs were curated from the World Health Organisation (WHO) VigiAccess database (1976–2024). Pharmacology data was curated from the chemical database of bioactive molecules with drug-like properties, European Molecular Biology Laboratory (ChEMBL). Physiochemical and pharmacokinetic properties were curated from PubChem, Drug Bank, ChemDraw and FDA new drug application documents for the HDACIs studied. In total, n = 12,779 ADRs were reported for the selected HDACIs, with n = 15/27 of the system organ class (SOC)-related ADRs showing statistical significance (X2, P < .05). Vorinostat accounted for n = 4,225 of the total ADRs, which may be explained by (1) its extensive use in the clinic and (2) it possessed the most clinically achievable off-target pharmacological interactions (n = 9). This included potent inhibition of the human-ether-à-go-go-related gene (hERG) ion-channel (IC50 = 322 nM vs. cmax = 1,200 nM), which is likely a contributing factor to the highest rate of cardiac ADRs observed (n = 146, P < .05). Musculoskeletal ADRs associated with vorinostat (n = 58, P < .05) can be ascribed to its unique inhibition of HDAC4 (IC50 = 540 nM vs. cmax = 1,200 nM). The gastrointestinal (GI) ADRs of panobinostat (n = 488, P < .05) can partly be related to HDAC3 inhibition (IC50 = 2.0 nM vs. Cmax = 8.0 nM). The absence of HDAC9 inhibition was unique to entinostat and may explain its beneficial drug reaction in depression (n = 0 cases). Thrombocytopenia observed with vorinostat, panobinostat and romidepsin may stem from dual inhibition of HDAC1 and HDAC2 and their higher volume of distributions (Vd). Despite all HDACIs having a similar mechanism of action, their unique pharmacology and differing physicochemical and pharmacokinetic properties landscape results in varying ADR profiles.
Similar content being viewed by others
Introduction
Adverse drug reactions (ADRs) refer to unintended responses to a drug related to any dose, and account for up to 5% of hospital admissions worldwide, at a cost to the Europe Union of 79 Bn EUR per annum1,2. ADRs can be classified by the DoTS and Rawlin-Thompson guidelines (Table S1)3,4. ADRs continue to be a challenge for healthcare systems, often prolonging hospital stays and complicating treatment5 and contribute to or cause 1 in 5 hospital admissions of patients with cancer6,7.
Histone deacetylase inhibitors (HDACIs) are a class of drugs, used to treat diseases, including cancer8,9. Histone deacetylase (HDAC) enzymes control gene expression and can change the acetylation state of histone proteins, which are responsible for organising DNA in the nucleus10. HDAC enzymes participate in several biological processes including cell development, differentiation, proliferation, and apoptosis11. There are 18 HDAC enzymes which can be grouped into 4 major classes12. Class I (HDAC1, 2, 3 and 8) are typically found in the nucleus, class IIa (HDAC4, 5, 7 and 9) and class IIb (HDAC6 and 10) typically shuttle between the nucleus and cytoplasm, and class IV (HDAC11) is typically found in the nucleus. HDACs belonging to classes I, II and IV are zinc-dependent enzymes, and are often referred to as the classical HDACs12,13. Whereas the class III HDACs (includes sirtuins 1–7) are structurally distinct and are nicotinamide adenine dinucleotide (NAD+) dependent enzymes13.
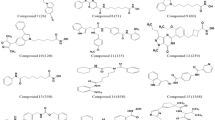
HDACIs have three key structural elements: a Zinc-binding group (ZBG), a cap group and a hydrophobic linker chain (Fig. 1 and Figure S1)14,15. The ZBG of the HDACI interacts with Zn2+ within the HDAC deacetylase domain, disrupting enzyme activity8,15. HDACIs can be grouped based on their chemical structures and their type of ZBG15. Hydroxamic acid HDACIs have a hydroxamate ZBG, benzamide derivatives have a distinct benzamide ZBG, cyclic peptides bind to Zn2+ via a thiol functional group and short-chain fatty acids derivatives feature a carboxyl ZBG16,17. Generally, hydroxamic acid derivatives are amongst the more potent HDACIs with fast-on/fast-off kinetics, whereas benzamides are considered less potent with slower binding kinetics18. Both function as bidentate Zn2+ binders18. Cyclic peptides bind moderately to Zn2+, in a monodentate manner and short-chain fatty acids typically have the weakest zinc-binding interactions14,19.

The general structure for HDACIs; colour coding; red square for cap group, blue arrow for hydrophobic linker group and yellow circle for zinc-binding group. The zinc-binding group chelates the Zn2+ within the HDAC enzyme active site, inhibiting HDAC activity.
Results from clinical trials have demonstrated the effectiveness of HDACIs, resulting in licensing across multiple jurisdictions13,18,20,21,22,23,24 (Table S2). This includes the phase II BELIEF trial which established a 26% overall response rate, with 13 complete responses, in patients taking belinostat monotherapy for refractory peripheral T-cell lymphomas25. The PANOROMA-1 phase III trial showed that adding panobinostat to dexamethasone and bortezomib increased median overall survival to 40 months versus 36 months for a placebo, or dexamethasone and bortezomib in patients with refractory multiple myeloma26. Belinostat and panobinostat were subsequently approved by the FDA in 2014 and 2015, respectively.
However, pan-HDACI’s, for example sodium valproate, underscore a potential challenge of non-selective HDAC inhibition amongst other potential mechanisms of action (MoA). Pan-HDACIs are non-selective, targeting multiple HDAC isoforms, which can result in ADRs27,28. Sodium valproate is associated with side effects, including hepatotoxicity, osteoporosis, and neurotoxicity29. Most concerning is its teratogenicity, and its associations with congenital and developmental ADRs30. Thus, women of child-bearing age, prescribed sodium valproate are now enrolled on a pregnancy prevention plan (PPP) in the United Kingdom31. Approximately 20,000 babies in the United Kingdom have been born with foetal valproate syndrome, and 1 in 10 babies were born with spina bifida after in utero exposure to sodium valproate32,33. The challenges of treatment with pan-HDACIs are compounded by the fact that many cancers overexpress specific HDAC isoforms. For example, HDAC1, 2 and 6 are overexpressed in peripheral T-cell lymphoma and HDAC5 and 7 in acute-myeloid leukaemia34,35. Therefore, HDAC inhibition therapy continues to be a promising strategy to improve benefit vs. risk profile in these patients36.
Clinical trials have revealed the main side effects linked to using HDACIs are myelosuppression, cardiotoxicity, and diarrhoea37. However, there is a limited understanding of their ADR profiles beyond carefully controlled clinical trials; in a ‘real-world’-setting both patient and environmental factors are often more varied and complex. A comprehensive understanding of the toxicities associated with HDACIs is crucial to ensure their safe use in clinical practice.
Underpinning this requires a clear understanding of all 18 HDAC isoforms. Though, some isoforms in particular HDAC1 and 2 have been more extensively studied than others (HDAC 3–8, 11–18)38,39,40. There are few HDAC9 selective inhibitors in clinical trials, and the role of HDAC9 during tumorigenesis is still largely unclear41. Similarly, few published studies have focused on developing HDAC10 selective inhibitors, despite the growing understanding of its role in polyamine deacetylase and thus drug-resistant cancer biology42. The HDAC selective inhibitor RGFP109 has shown potential for temozolomide resistance in glioblastoma cell lines in preclinical trials, though no advance to clinical trials have been made at the time of writing43. Therefore, there is an ongoing need to focus research on HDACIs, as part of a broader effort to develop effective therapies that are also better tolerated by patients.
The majority of HDACIs are not licensed in the United Kingdom, suggesting concerns remain relating to their safety, tolerability and efficacy (Table S2). Identifying causal links between HDAC isoforms and ADRs could facilitate the development of more selective HDACIs, improving patient safety and therapeutic outcomes. An extensive literature search revealed a single pharmacovigilance study using the FDA Adverse Event Reporting System (FAERS) on 6 HDACIs44. Herein, this study will be the first to examine global real-world data using the WHO VigiAccess dataset to investigate the pharmacovigilance of all HDACIs and ADRs resulting. It will offer insights into the safety of HDACIs from a global standpoint to facilitate their safer use in clinical practice.
This study will focus specifically on 8 HDACIs; vorinostat, belinostat, panobinostat, pracinostat, entinostat, romidepsin, bufexamac and sodium phenylbutyrate13,18,20,22,23,24,25,26,37 (Table S3).
Aims and objectives
-
To explore the ADR data of the HDACIs vorinostat, belinostat, panobinostat, pracinostat, entinostat, romidepsin, bufexamac and sodium phenylbutyrate using the WHO VigiAccess database.
-
Determine the unique physicochemical and pharmacological properties for each HDACI studied.
-
Determine links between the ADR profiles of these HDACIs and their physicochemical and pharmacological properties.
-
Postulate potential mechanisms to rationalise any side effect differences.
Methods
The HDACIs, vorinostat, belinostat, panobinostat, pracinostat, entinostat, sodium phenylbutyrate, romidepsin and bufexamac were studied based on the inclusion and exclusion criteria of this research (Table 1 and Table S4).
Physiochemical properties
The physicochemical parameters calculated were molecular weight (Da), total polar surface area (tPSA), pKa, hydrogen bond donors (HBDs), hydrogen bond acceptors (HBAs), cLog10P, cLog10D7.4 and if the drug was a p-glycoprotein substrate. Calculated molecular weight, tPSA and cLog10P were calculated using Perkin Elmer ChemDraw version 22.0. The pKa, HBDs, HBAs and whether the HDACIs were p-glycoprotein substrates were curated and analysed using Drug Bank data searches and cross-validated with primary literature45. cLog10D7.4 was curated from the chemical database of bioactive molecules with drug-like properties, European Molecular Biology Laboratory (ChEMBL)46. pIC50 was determined by the negative log10 of the median IC50 of its single homosapien protein target. Lipophilic Ligand Efficiency (LLE) was calculated using the following formula: pIC50 – log10P. An LLE > 5 is linked to fewer off-target interactions thus reducing the risk of ADRs47.
Selected physicochemical properties increase penetration through the blood-brain barrier (BBB), specifically: a molecular weight of < 450 Da, < 6 HBD, < 2 HBA, a neutral or basic drug, tPSA of < 90 Å2, cLog10D7.4 between 1 and 3, and not a p-glycoprotein substrate. Risk of penetration of the BBB increases with the more requirements met48.
Pharmacokinetic properties
The pharmacokinetic properties calculated were Cmax (peak plasma concentration), Tmax (time to reach peak plasma concentration), CYP450 isoform metabolism where applicable, total clearance, half-life (t1/2), volume of distribution (Vd) and plasma protein binding (PPB). Cmax values were curated from FDA monographs for vorinostat, romidepsin and sodium phenylbutyrate and converted to nM49,50,51. PPB and CYP450 isoform inhibition was gathered from the Drug Bank, Electronic Medicines Compendium (EMC), and PubChem45,52,53. The remaining pharmacokinetic data was curated using Boolean logic in the Reaxys and Web of Science databases by searching the ‘drug name’ together with the ‘parameter required’54,55,56,57,58. To the best of our knowledge and after extensive literature searches, no Cmax for bufexamac has been reported (Table 2).
Target affinity
The ChEMBL database was used to collect all reported bioactivity data for single homosapien protein targets for each HDACI. A median IC50 data point was calculated if a single homosapien protein target had > 1 IC50 value reported under analogous experimental conditions46. This was performed by calculating the pIC50 using the following formula: pIC50 = -log10(IC50 in Molar units).
ADR reporting
ADR data was retrieved from the World Health Organisation (WHO) VigiAccess data set (https://www.vigiaccess.org/) on the 14.11.24 for the HDACIs studied59. ADRs reported for vorinostat (2006-), belinostat (2008-), panobinostat (2009-), pracinostat (2015–2022), entinostat (2011–2023), sodium phenylbutyrate (2002-), romidepsin (2009-) and bufexamac (1976–2022) were extracted, cleaned, and analysed using Excel version 24.11 for Microsoft 365.
Statistical analysis
Excel version 24.11 for Microsoft 365 was used to perform the chi-squared tests on the ADR data for each system organ class (SOC), to determine the statistical significance of the ADR signal (P <.05). ADR at the SOC-level (where reports, n < 5) were excluded from the chi-squared test60.
Ethical approval
Ethical approval by the School of Pharmacy sub-ethics committee was not required, as all the data retrieved is publicly available and fully anonymised.
Results
Chemical data
The chemical data for the HDACIs is displayed in Table 2. The most lipophilic drug was romidepsin (cLog10P = 3.44) and was also the most potent (pIC50 = 9.52). Pracinostat and bufexamac had LLEs < 5, (3.99 and 2.60 respectively); this suggests they may have more off-target interactions.
Vorinostat, pracinostat, entinostat, sodium phenylbutyrate and bufexamac are the most likely to penetrate the BBB, having all met 5/7 requirements. Though all the HDACIs had < 6 HBDs, none met the > 2 HBAs limit. Only entinostat and sodium phenylbutyrate were bases, the rest were acids. The tPSA of belinostat (95.50), entinostat (105.81) and romidepsin (142.7), all exceeded > 90Å. The cLog10D7.4 of sodium phenylbutyrate (−0.04) was the only one outside the desired range of 1–3, correlating with its notably low Vd (0.2 L). Romidepsin was the heaviest HDACI (540.69 Da), with a molecular weight > 450 Da and overall was the least likely to penetrate the BBB, meeting only 1/7 requirements. This aligns with cerebrospinal (CSF) measurements in primates which showed romidpesin CSF exposure was 2% after IV administration61.
Entinostat had the longest half-life (52 h), reflected in its once weekly dosing, whereas belinostat had the shortest (1.1 h) which is dosed daily (Table 2). Romidepsin and vorinostat both had the highest Tmax (4 h), whereas belinostat was the lowest 0.42 h. Sodium phenylbutyrate had the highest PPB (98%) compared to vorinostat with the least (72%).
Target affinity
The pharmacological data for the HDACIs are displayed in Table 3. Panobinostat was the most potent towards HDAC1 (0.3 nM), HDAC2 (13 nM) and HDAC3 (2.0 nM) vs. Cmax=8 nM. Panobinostat, vorinostat and romidepsin had additional, clinically achievable, off-target interactions with BRD4, though panobinostat was the most potent (5.0 nM vs. 27 nM vs. 36 nM, respectively). Entinostat uniquely potently inhibited HDAC9 (0.5 nM vs. Cmax=49 nM), but potential off-target interactions with coagulation-factor III and CYP450 3A4(8,000 nM and 730 nM, respectively) were above the Cmax. Similarly, belinostat was the only potent inhibitor of HDAC5 (76 nM vs. Cmax=220 nM) but potential off-target interaction with ASPH were above the Cmax. Pracinostat was able to clinically inhibit HDAC1 (64 nM), HDAC2 (290 nM) and HDAC6 (260 nM) vs. Cmax =940 nM, but potential off-target interactions (n = 2) were above the Cmax. The pan-HDACI vorinostat showed the greatest number of off-target interactions (n = 9), though only (n = 4) were clinically achievable with the most potent being for FGFR1 (IC50 = 12 nM vs. Cmax=1,200 nM). Sodium phenylbutyrate did not clinically inhibit any proteins based on its Cmax (IC50 > Cmax), and no Cmax was reported for bufexamac.

A stacked bar chart representing the number of ADRs across the main system organ classes for the HDAC inhibitors studied, alongside the P-value for each SOC. Data for pracinostat was omitted due to the low number of reported ADRs; the full breakdown of ADR profiles, including pracinostat, is available in Table S5. Abbreviations: SC, subcutaneous; Pregnancy related ADRs, pregnancy, puerperium & perinatal ADRs; Admin, Administration; GI, gastrointestinal; P-value, probability value.

A pie chart displaying the percentage contribution of each HDAC inhibitor to the total number of ADRs, to the nearest whole number. The pie chart highlights the proportional representation of each HDACIs ADRs within the overall dataset. Full data, including absolute values for each SOC for each HDAC inhibitor are available in Table S5.
ADRs
ADRs were found to be statistically significant across 15/27 SOCs (p <.05) for the HDACIs (Table S6). General disorders & administration site conditions had the highest number of total ADRs (n = 1,801), though not statistically significant. Vorinostat led with the most ADRs (~ 33% of all HDACIs ADRs) and provided high ADRs across the SOCs (n = 18/27) including GI disorders (n = 410), infections & infestations (n = 417) and blood & lymphatic disorders (n = 365), all reaching statistical significance (P <.05). Intriguingly, Fig. 2 highlights bufexamac had almost a six-fold increase in skin and subcutaneous tissue disorders compared to vorinostat (656 vs. 114 reports, respectively). Sodium phenylbutyrate was the only drug with any reports (n = 5) in the pregnancy, puerperium & perinatal conditions. Overall pracinostat had the least ADRs reported (n = 76), with reports of (n = 0) in 11 SOCs, accounting for only ~ 1% of total HDACIs ADRs.
A visual inspection of Figs. 2 and 3 highlights there were higher incidences of ADRs with the hydroxamic acids and cyclic peptides, compared to the benzamide and short-chain fatty acid derivatives (Figure S1). This was expected, as hydroxamic acid-based ZBGs typically have higher binding affinities for Zn2+ within the HDAC active site and can uniquely chelate Zn2+ in a bidentate manner, essentially the hydroxamic acid group being able to attach to the Zn2+ in two places resulting in a stronger bonding network18,19. The lack of selectivity can result in chelation with HDAC isoforms and other zinc-dependent metalloproteins (i.e. carbonic anhydrase or aminopeptidase) within the body, contributing to its ADR profile62,63. Similarly, cyclic peptides have more complex structures compared to benzamide and short-chain fatty acid derivatives (Figure S1), thus are more likely to interact extensively with surrounding amino-acids within the HDAC active site as well as other non-HDAC proteins in the body15,64.
Discussion
Blood and lymphatic system disorders
Vorinostat (n = 365), panobinostat (n = 323) and romidepsin (n = 286) had the highest related blood and lymphatic system ADRs (P <.05). Further analysis revealed (n = 125), (n = 142) and (n = 113) reports of thrombocytopenia with vorinostat, panobinostat and romidepsin, respectively. Given they all target HDAC1 and 2 at clinically achievable levels, their inhibition of HDAC1 and 2 is likely to contribute to the increase in thrombocytopenia seen with these HDACIs. Dual inhibition of HDAC1 and HDAC2 in mice models resulted in thrombocytopenia by causing megakaryocyte apoptosis; differentiation of megakaryocyte-erythrocyte lineages is crucial for producing red-blood cells and platelets65. These HDACIs, except for belinostat, demonstrated the highest Vd thus greater concentrations of the drug can get into the bone-marrow, enhancing their effect on megakaryocytes66.
Furthermore, only vorinostat, panobinostat and romidepsin showed potent, off-target interactions with the BRD4 protein. The inhibition of BRD4 is commonly associated with haematological ADRs, including thrombocytopenia67. Approximately, 15% of patients enrolled in the phase I study of the BRD4 inhibitor AZD5153 experienced grade 3 or higher thrombocytopenia, making it amongst the most reported haematological side effect68.
The HDACIs used for the treatment of blood cancers (Figs. 2 and 3) had higher incidences of ADRs. The lowest ADRs were recorded for entinostat (n = 21), sodium phenylbutyrate (n = 17) and bufexamac (n = 13), consistent with their primary indications being unrelated to the blood and lymphatic system. Therefore, the mechanism of HDAC1 and HDAC2 inhibition plus the off-target BRD4 inhibition shared by vorinostat, panobinostat and romidepsin specifically could explain the higher incidences of thrombocytopenia observed, despite also being used for haematological cancers65,67,68.
Cardiac disorders
Vorinostat had the highest reports of cardiac ADRs (n = 146, P <.05), which was correlated to its potent inhibition of the human-ether-à-go-go-related gene (hERG) ion-channel (IC50 = 320 nM vs. Cmax = 1,200 nM). The hERG ion-channels are vital for cardiomyocyte repolarisation, and thus inhibition of these channels has been associated with ventricular arrythmias, known as torsades de pointes69,70,71. Vorinostat was the only HDACI to report any cases of torsades de pointes, totalling (n = 7) further supporting the correlation between hERG and the ADR profile.
Musculoskeletal and connective tissue disorders
Higher incidences of musculoskeletal and connective tissue disorders were reported with vorinostat (n = 81, P <.05), potentially attributed to its potent inhibition of HDAC4 (IC50 = 540 nM vs. Cmax = 1,200 nM). HDAC4 is crucial for skeletal muscle regeneration, mice with HDAC4 knock-out suffered perinatal deaths due to abnormal skeletal formation72,73.
Pregnancy, puerperium & perinatal conditions
Sodium phenylbutyrate was the only HDACI with ADRs in this pregnancy-associated SOC (n = 5), though statistical significance could not be determined. Sodium phenylbutyrate is used for the treatment of urea cycle disorders, conditions that can affect anyone of any age, including women of child-bearing potential74. Pregnancy can be a trigger for urea cycle disorders, supporting the use of sodium phenylbutyrate in this patient group75. In contrast, most other HDACI are primarily used in oncology, where pregnancy is rare owing to patients ages, co-morbidities, and infertility from cancer treatments76. Sodium phenylbutyrate is also the only category C medication used during pregnancy, reflecting limited harm to the foetus whereas the other HDACIs studied are category D, suggesting confirmed foetal harm or lack sufficient research21,50,51,77,78,79. The fact that clinicians are likely to favour using category C drugs in practice and the demographic differences are likely to explain the observed pregnancy-associated ADRs being only associated with sodium phenylbutyrate.
Psychiatric and nervous system disorders
Table 3 reveals entinostat as the only potent HDAC9 inhibitor, (IC50 = 0.5 nM vs Cmax = 49 nM). Research shows that HDAC9 is expressed in the brain and HDAC9 inhibition can induce an anti-depressant like effect80,81. HDAC9 was found to be highly expressed in the hippocampal neurons of mice with chronic resistant stress and depression and concluded HDAC9 inhibition could be a promising treatment strategy for depressive disorders80. Hence, this may be part of the explanation for entinostat having a lack of ADR reports relating to anxiety, depression, or suicide, as a potential beneficial drug reaction (BDR). The other HDACIs studied, which did not inhibit HDAC9 at a clinically achievable level, were associated with at least (n ≥ 1) case of anxiety or depression, with vorinostat and panobinostat, respectively (n = 6 and n = 3 cases, respectively, P <.05). Vorinostat was the only HDACI to report a completed suicide. Whilst this could be associated with its ability to penetrate the BBB (meeting 5/7 requirements) coupled with its particularly low PPB (71%) where more ’free drug’ is available to cross the BBB increasing the risk of psychiatric side effects (Table 2), this finding likely involves various confounding factors including psychiatric co-morbidities and socio-environmental factors, warranting further study.
Vorinostat has also been detected at concentrations > 10 nM in the CSF of paediatric brain tumour patients54. Therefore, it has the potential to clinically inhibit fibroblast growth factor receptor 1 (FGFR1) (IC50 = 12 nM vs. Cmax = 1,200 nM). FGFR1 is a protein essential for hippocampal neuron formation and required for long-term memory consolidation and learning82,83. Inhibition of FGFR1 could alter vital pathways regulated by FGF2, which helps to regulate anxiety and associated mood disorders, and interfering FGFR1 signalling predisposes patients to psychiatric disorders including anxiety and depression, which could potentially be another reason for the high incidences of psychiatric ADRs seen with vorinostat84,85.
Skin and subcutaneous tissue disorders
Bufexamac led with the greatest ADRs relating to the skin and subcutaneous tissue (n = 656), with (n = 214) for contact dermatitis. Bufexamac was withdrawn from the EU market in 2010 due to concerns of allergic contact dermatitis, therefore the detection of this signal in the dataset further reinforces the validity of this approach to signal detection using the WHO VigiAccess database21. The aryl ether functional group unique to bufexamac (Fig. 2), has the potential to form quinone methide metabolites, which can cause skin irritation86,87,88. Coupled with being the only HDACI studied that is applied topically (Table 1), to treat inherently inflammatory skin conditions such as eczema likely explains this much higher incidence of skin ADRs observed21.
Vorinostat (n = 114), romidepsin (n = 67) and belinostat (n = 37) followed bufexamac with the highest ADRs in this SOC, and these 3 HDACIs are primarily used in cancers relating to the skin (Table 3). This patient population is already likely to have compromised skin integrity and other skin malignancies, which may predispose them to greater incidences of associated skin ADRs as a result. Thus, the confounder of indication prevented further mechanistic interpretation.
Specifically, vorinostat reported the highest cases of alopecia (n = 13), correlating to its potent inhibition of FGFR1, given alopecia is also a known ADR of FGFR1 inhibitors89. For example, 49% of patients treated with the FGFR inhibitor pembigatanib experienced grade 1 or 2 alopecia90. However, only (n = 2) cases were reported with romidepsin and sodium phenylbutyrate, respectively, despite having the next highest reports of alopecia, and no reports of alopecia were associated with the other HDACIs, none of which inhibited FGFR1. Thus, the almost 7-fold increase in alopecia cases observed with vorinostat, suggests FGFR1 inhibition may be contributing to this ADR, though this tentative finding requires further investigation to confirm this association.
Infections and infestations
HDACIs are given to immunocompromised patients, who have likely undergone prior chemo or radiotherapy, as HDACIs are usually indicated in the later stages of cancers, when other treatment options have been exhausted91. This inherent vulnerability could explain the high incidences of infections seen across all the HDACIs, totalling (n = 1,048, P <.05), with the highest cases for vorinostat (n = 417), panobinostat (n = 289) and romidepsin (n = 180). Various HDAC isoforms participate in regulating immune responses; HDAC4 encourages type I IFN signalling by interacting with TBK1/IKKε to initiate antiviral immunity, HDAC5 regulates macrophage differentiation and recruitment and HDAC8 preserves the function of natural killer cells, though no clear patterns could be detected92,93,94.
GI and site of administration disorders
Both GI and administration site disorders are common ADRs of HDACIs (n = 1,307 and n = 1,801, respectively). GI disorders were most pronounced for the acidic drugs; the hydroxamic acid derivatives and cyclic peptides, their acidic pKa could lead to irritation in the gastric mucosa95. Particularly, panobinostat had the greatest reports for GI disorders (n = 488, P <.05). Table 3 highlights that panobinostat is usually co-administered with the corticosteroid, dexamethasone, and proteasome inhibitor, bortezomib: both well-documented for causing GI bleeding and intestinal irritation20,96,97. Panobinostat was the most potent HDAC3 inhibitor (IC50 = 2.0 nM vs. Cmax = 8.0 nM), an enzyme involved in preserving the intestinal epithelial barrier and mucosa integrity, which may explain the higher reports of GI ADRs with panobinostat98.
Both belinostat and romidepsin had the most administration site disorders compared to its reports within other SOCs (n = 151) and (n = 380), respectively. They are primarily administered via the intravenous route (Table 3), and the acidity of the drugs could cause irritation in the veins and surrounding tissue99.
Neoplasms benign, malignant & unspecific
Romidepsin, the only pro-drug amongst the HDACIs studied to treat cancer, requires intracellular activation via glutathione or thioredoxin100. At tumour sites there can be reduced levels of glutathione or thioredoxin resulting in suboptimal activation of romidepsin and less effective treatment, reflected in the (n = 48) cases of malignant neoplasm progressions - the most significant amongst all the HDACIs studied (P <.05)101,102. This tentative observation emphasises the need for more research into the intracellular and biochemical activation of romidepsin in cancer cells, to ensure optimal treatment and reduction of cancer progression.
Healthcare professionals (HCPs) and staff are encouraged to report any ADRs to their respective pharmacovigilance jurisdiction which in turn feeds into the WHO VigiAccess database, though critically causality does not need to be proven59. Therefore, the HDACIs may not be associated with the ADR, but rather this may be due to other co-founders (i.e. polypharmacy, co-morbidities, genetics, and others). Typically, HDACIs are used in the advanced stages of cancer, thus patients are likely to have had prior treatment with chemo/radiotherapy or other platinum-based therapies which might have contributed to or caused the ADR91.
Fundamentally, the spontaneous reporting of ADRs has weaknesses, particularly the risk of under-reporting103. Many factors can impact the reporting of a drug; time on the market, ADR severity, and novelty and indication of the drug104. This is particularly important considering the small number of reports for HDACIs generally. Within this study, ADR reports varied through the years from 1996 to 2024, this coupled with the lack of prescribing data available makes it difficult to standardise the ADRs across the SOCs. Some of these drugs are still in clinical trials and bufexamac has been withdrawn from the market, therefore affecting the number of ADRs generated21.
Despite the pharmacology data being extracted from ChEMBL, it is unlikely it provided every protein interaction possible, therefore interactions with other targets remain unknown.
Conclusion
The development of HDACIs has shown promise as an effective treatment strategy for many cancers, emphasising the need to understand their polypharmacology and associated ADRs. Vorinostat accounted for almost a third of the total ADRs (n = 4,225), with side effects ranging from cardiac, musculoskeletal, gastrointestinal, and psychiatric system organ classes. Vorinostat also had the most off-target interactions (n = 9), including the hERG ion channel (IC50 = 322 nM vs. Cmax = 1,200 nM) translating to the most cardiac ADRs, and the only HDACI with reports of torsades de pointes. Vorinostat also had the greatest cases of anxiety and depression, potentially ascribed to its lack of HDAC9 inhibition and ability to penetrate the BBB, in contrast entinostat, a potent HDAC9 inhibitor (IC50 = 0.5 nM vs. Cmax = 49 nM), reported no cases of anxiety of depression. While vorinostat’s ability to uniquely inhibit HDAC4 (IC50 = 540 nM) this likely explains its contribution of approximately 40% of all HDACIs musculoskeletal ADRs, given its prominent role in skeletal myogenesis. Dual inhibition of HDAC1 and 2 may have contributed to vorinostat, panobinostat and romidepsin’s increased incidences of thrombocytopenia, alongside their higher Vd’s, validating the existing link between increased Vd and thrombocytopenia.
Romidepsin activation as a pro-drug could explain its high incidences of neoplasms. Romidepsin and belinostat, which are administered i.v., showed the most administration site disorders within their own SOCs, but not across all the HDACIs studied, thus these tentative links require further investigation. No clear patterns emerged between the infection ADRs and polypharmacology, likely attributed to the overlapping roles of HDACs within the immune system.
The methodology used in this study was substantiated by reproducing ADR signals linked to HDACIs that were reported from clinical trials, specifically haematological (n = 1,090), infections and infestations (n = 1,048) and gastrointestinal abnormalities (n = 1,307). Integrating chemical and pharmacological data with real-life, global pharmacovigilance datasets provided valuable insights to investigate ADRs associated with HDACIs. The data generated by this powerful approach could be used to predict the ADR profiles of novel HDACIs coming onto the market in the years to come.
Data availability
Data is provided within the manuscript or supplementary information files.
References
Komagamine, J. Prevalence of urgent hospitalizations caused by adverse drug reactions: a cross-sectional study. Sci. Rep. 14 (1), 6058. https://doi.org/10.1038/s41598-024-56855-z (2024).
European Commission. Strengthening pharmacovigilance to reduce adverse effects of medicines. (Accessed 19 December 2024). https://ec.europa.eu/commission/presscorner/detail/de/memo_08_782 (2008).
Calderón-Ospina, C. & Bustamante-Rojas, C. The DoTS classification is a useful way to classify adverse drug reactions: a preliminary study in hospitalized patients. Int. J. Pharm. Pract. 18 (4), 230–235. https://doi.org/10.1111/j.2042-7174.2010.00039.x (2010).
Robinson, M. & Amare, M. Adverse drug reactions. Anaesth. Intensive Care. 24 (3), 210–214. https://doi.org/10.1016/j.mpaic.2022.12.021 (2023).
Suh, D., Woodall, B. S. & Santis, E. R. H. Clinical and economic impact of adverse drug reactions in hospitalized patients. Sage Journ. 34 (12), 1373–1379. https://doi.org/10.1345/aph.10094 (2000).
Lavan, A. H., O’Mahony, D., Buckley, M., O’Mahony, D. & Gallagher, P. Adverse drug reactions in an oncological population: prevalence, predictability, and preventability. Oncologist 24 (9), 968–967. https://doi.org/10.1634/theoncologist.2018-0476 (2019).
Shrestha, S., Shakya, R., Shrestha, S. & Shakya, S. Adverse drug reaction due to cancer chemotherapy and its financial burden in different hospitals of Nepal. Symbiosis 2 (1), 1–7. https://doi.org/10.15226/2476-2431/2/1/00114 (2017).
Shin, H. S., Choi, J., Lee, J. & Lee, S. Y. Histone deacetylase as a valuable predictive biomarker and therapeutic target in immunotherapy for Non-Small cell lung cancer. Cancer Res. Treat. 54 (2), 458–468. https://doi.org/10.4143/crt.2021.425 (2022).
Ganai, S. A., Ramadoss, M. & Mahadevan, V. Histone deacetylase (HDAC) Inhibitors - emerging roles in neuronal memory, learning, synaptic plasticity and neural regeneration. Curr. Neuropharmacol. 14 (1), 55–71. https://doi.org/10.2174/1570159X13666151021111609 (2016).
Gallinari, P., Marco, S. D., Jones, P., Pallaoro, M. & Steinkühler, C. HDACs, histone deacetylation and gene transcription: from molecular biology to cancer therapeutics. Cell. Res. 17 (1), 195–211. https://doi.org/10.1038/sj.cr.7310149 (2007).
Reichert, N., Choukrallah, M. A. & Matthias, P. Multiple roles of class I HDACs in proliferation, differentiation, and development. Cell. Mol. Life Sci. 69 (1), 2173–2187. https://doi.org/10.1007/s00018-012-0921-9 (2012).
Curcio, A., Rocca, R., Alcaro, S. & Artese, A. The histone deacetylase family: structural features and application of combined computational methods. Pharmaceuticals 17 (5), 620. https://doi.org/10.3390/ph17050620 (2024).
Ahmad, B. et al. Investigating potential cancer therapeutics: insight into histone deacetylases (HDACs) inhibitions. Pharmaceuticals 17 (4), 444. https://doi.org/10.3390/ph17040444 (2024).
Geurs, S., Clarisse, D., Bosscher, K. D. & D’Hooghe, M. The Zinc-Binding group effect: lessons from Non-Hydroxamic acid Vorinostat analogs. J. Med. Chem. 66 (12), 7698–7729. https://doi.org/10.1021/acs.jmedchem.3c00226 (2023).
Zhang, L., Zhang, J., Jiang, Q., Zhang, L. & Song, W. Zinc binding groups for histone deacetylase inhibitors. J. Enzyme Inhib. Med. Chem. 33 (1), 714–721. https://doi.org/10.1080/14756366.2017.1417274 (2018).
Li, Y. et al. Zinc-dependent deacetylase (HDAC) inhibitors with different zinc binding groups. Curr. Top. Med. Chem. 19 (3), 223–241. https://doi.org/10.2174/1568026619666190122144949 (2019).
Yue, K. et al. Comparison of three zinc binding groups for HDAC inhibitors – A potency, selectivity and enzymatic kinetics study. Bioorg. Med. Chem. Lett. 70 (1), 1–7. https://doi.org/10.1016/j.bmcl.2022.128797 (2022).
Bondarev, A. D. et al. Recent developments of HDAC inhibitors: emerging indications and novel molecules. Br. J. Clin. Pharmacol. 87 (12), 4577–4597. https://doi.org/10.1111/bcp.14889 (2021).
Porter, N. J. & Christianson, D. W. Structure, mechanism, and Inhibition of the zinc-dependent histone deacetylases. Curr. Opin. Struct. Biol. 59 (1), 9–18. https://doi.org/10.1016/j.sbi.2019.01.004 (2019).
Mu, S. et al. Panobinostat PK/PD profile in combination with bortezomib and dexamethasone in patients with relapsed and relapsed/refractory multiple myeloma. Eur. J. Clin. Pharmacol. 72 (2), 152–161. https://doi.org/10.1007/s00228-015-1967-z (2016).
European Medicines Agency. Bufexamac – referral. (Accessed 20 December 2024). https://www.ema.europa.eu/en/medicines/human/referrals/bufexamac (2010).
Iwata, H. et al. Efficacy and exploratory biomarker analysis of entinostat plus exemestane in advanced or recurrent breast cancer: phase II randomized controlled trial. Jpn J. Clin. Oncol. 53 (1), 4–15. https://doi.org/10.1093/jjco/hyac166 (2022).
Garcia-Manero, G. et al. Phase II randomized Double-Blinded study of pracinostat in combination with Azacitidine in patients with untreated Higher-Risk myelodysplastic syndromes. Am. Cancer Soc. 123 (6), 994–1002. https://doi.org/10.1002/cncr.30533 (2018).
NICE | the National Institute for Health and Care Excellence. (Accessed 20 December 2024). https://www.nice.org.uk/ (2021)
O’Connor, O. A. et al. Belinostat in patients with relapsed or refractory peripheral T-Cell lymphoma: results of the pivotal phase II BELIEF (CLN-19) study. J. Clin. Oncol. 33 (23), 2492–2499. https://doi.org/10.1200/JCO.2014.59.2782 (2015).
San-Miguel, J. F. et al. Overall survival of patients with relapsed multiple myeloma treated with Panobinostat or placebo plus bortezomib and dexamethasone (the PANORAMA 1 trial): a randomised, placebo-controlled, phase 3 trial. Lancet Haematol. 3 (11), 506–515. https://doi.org/10.1016/S2352-3026(16)30147-8 (2016).
Zhang, S. et al. Pan-HDAC (Histone Deacetylase) inhibitors increase susceptibility of thoracic aortic aneurysm and dissection in mice. Arterioscler. Thromb. Vasc Biol. 41 (11), 2848–2850. https://doi.org/10.1161/ATVBAHA.121.316808 (2021).
Bhatti, U. F. et al. Comparative analysis of isoform-specific and non-selective histone deacetylase inhibitors in attenuating the intestinal damage after hemorrhagic shock. Trauma. Surg. Acute Care Open. 4 (1), 1–9. https://doi.org/10.1136/tsaco-2019-000321 (2019).
Sztajnkrycer, M. D. Valproic acid toxicity: overview and management. J. Toxicol. Clin. Toxicol. 40 (6), 789–801. https://doi.org/10.1081/clt-120014645 (2002).
Phillips, B., Evans, I., Skerrett, V. & Jones, A. M. United Kingdom National register study of Anti-Epileptic medications: suspected foetal congenital and Pregnancy-Associated side effects. MedRxiv https://doi.org/10.1101/2024.03.26.24304895 (2024).
Watkins, L. V., Cock, H. R., Angus-Leppan, H. & Shankar, R. Valproate and the pregnancy prevention programme: exceptional circumstances. Br. J. Gen. Pract. 69 (681), 166–167. https://doi.org/10.3399/bjgp19X701897 (2019).
Clayton-Smith, J. et al. Diagnosis and management of individuals with fetal valproate spectrum disorder; a consensus statement from the European reference network for congenital malformations and intellectual disability. Orphanet J. Rare Dis. 14 (1), 180. https://doi.org/10.1186/s13023-019-1064-y (2019).
Cummings, C., Stewart, M., Stevenson, M., Morrow, J. & Nelson, J. Neurodevelopment of children exposed in utero to lamotrigine, sodium valproate and carbamazepine. Arch. Dis. Child. 96 (7), 643–647. https://doi.org/10.1136/adc.2009.176990 (2011).
Lu, G. et al. Update on histone deacetylase inhibitors in peripheral T-cell lymphoma (PTCL). Clin. Epigenetics. 15 (1), 1–19. https://doi.org/10.1186/s13148-023-01531-8 (2023).
Wang, P., Wng, Z. & Liu, J. Role of HDACs in normal and malignant hematopoiesis. Mol. Cancer. 19 (5), 1–21. https://doi.org/10.1186/s12943-019-1127-7 (2020).
Vogl, D. T. et al. Ricolinostat, the first selective histone deacetylase 6 inhibitor, in combination with bortezomib and dexamethasone for relapsed or refractory multiple myeloma. Clin. Cancer Res. 23 (13), 3307–3315. https://doi.org/10.1158/1078-0432.CCR-16-2526 (2019).
Shah, R. S. Safety and tolerability of histone deacetylase (HDAC) inhibitors in oncology. Drug Saf. 42 (2), 235–245. https://doi.org/10.1007/s40264-018-0773-9 (2019).
Durham, B. S., Grigg, R. & Wood, I. C. Inhibition of histone deacetylase 1 or 2 reduces induced cytokine expression in microglia through a protein synthesis independent mechanism. J. Neurochem. 143 (2), 147–256. https://doi.org/10.1111/jnc.14144 (2017).
Yanginlar, C. & Logie, C. HDAC11 is a regulator of diverse immune functions. Biochim. Biophys. Acta Gene Regul. Mech. 1861 (1), 54–59. https://doi.org/10.1016/j.bbagrm.2017.12.002 (2018).
Zhang, Y., Andrade, R., Hanna, A. A. & Pflum, M. K. H. Evidence that HDAC7 acts as an epigenetic reader of AR acetylation through NCoR-HDAC3 dissociation. Cell. Chem. Biol. 29 (7), 1162–1173. https://doi.org/10.1016/j.chembiol.2022.05.008 (2022).
Yang, C., Croteau, S. & Hardy, P. Histone deacetylase (HDAC) 9: versatile biological functions and emerging roles in human cancer. Cell. Oncol. 44 (5), 997–1017. https://doi.org/10.1007/s13402-021-00626-9 (2021).
Géraldy, M. et al. Selective Inhibition of histone deacetylase 10: hydrogen bonding to the gatekeeper residue is implicated. J. Med. Chem. 62 (9), 4426–4443. https://doi.org/10.1021/acs.jmedchem.8b01936 (2019).
Li, Z. et al. Correction to: Histone deacetylase inhibitor RGFP109 overcomes Temozolomide resistance by blocking NF-κB-Dependent transcription in glioblastoma cell lines. Neurochem Res. 49 (11), 3176–3178. https://doi.org/10.1007/s11064-024-04237-2 (2024).
Li, W., Fu, Y. & Wang, W. A real-world pharmacovigilance study investigating the toxicities of histone deacetylase inhibitors. Ann. Hematol. 103 (8), 3207–3217. https://doi.org/10.1007/s00277-024-05691-2 (2024).
DrugBank Online. Database for Drug and Drug Target Info. (Accessed 26 December 2024). https://go.drugbank.com/drugs/DB02546
ChEMBL Database. (Accessed 20 December 2024). https://www.ebi.ac.uk/chembl/
Hopkins, A. L., Keserü Gm, Leeson, P. D., Rees, D. C. & Reynolds, C. H. The role of ligand efficiency metrics in drug discovery. Nat. Rev. Drug Discov. 13 (2), 105–121. https://doi.org/10.1038/nrd4163 (2014).
Jones, L. & Jones, A. M. Suspected adverse drug reactions of the type 2 antidiabetic drug class dipeptidyl-peptidase IV inhibitors (DPP4i): can polypharmacology help explain? Pharmacol. Res. Perspect. 10 (6), 1029. https://doi.org/10.1002/prp2.1029 (2022).
Zolinza (vorinostat) capsules label. FDA Reference ID: 3043460. (Accessed 27 December 2024). https://www.accessdata.fda.gov/drugsatfda_docs/label/2011/021991s002lbl.pdf (2011).
Romidepsin Injection label. FDA Reference ID: 4575263. (Accessed 27 December 2024). https://www.accessdata.fda.gov/drugsatfda_docs/nda/2020/208574Orig1s000,Orig2s000Approv.pdf (2020).
PHEBURANE® (sodium phenylbutyrate) oral pellets label. FDA Reference ID: 5000442. (Accessed 27 December 2024). https://www.accessdata.fda.gov/drugsatfda_docs/label/2022/216513s000lbl.pdf (2022).
Electronic Medicines Compendium. Home-Electronic Medicines Compendium (EMC). (Accessed 26 December 2024). https://www.medicines.org.uk/emc/
National Library of Medicine. (PubChem, accessed 26 December 2024) https://pubchem.ncbi.nlm.nih.gov/
Gunter, A. S. et al. Cerebrospinal fluid penetration of targeted therapeutics in pediatric brain tumor patients. Acta Neuropathol. Commun. 8 (78), 1–21. https://doi.org/10.1186/s40478-020-00953-2 (2020).
Berg, S. et al. Pharmacokinetics and cerebrospinal fluid penetration of phenylacetate and phenylbutyrate in the nonhuman primate. Cancer Chemother. Pharmacol. 47 (5), 385–390. https://doi.org/10.1007/s002800000256 (2001).
Praz, M. S. et al. Population pharmacokinetics of Panobinostat (LBH589) in patients with advanced solid tumors and hematologic malignancies following intravenous and oral administration. Blood 114 (22), 3780. https://doi.org/10.1182/blood.V114.22.3780.3780 (2009).
Piper, W. L., Waddell, J. A. & Solimmando, D. A. Drug monographs: Belinostat and idelalisib. Hosp. Pharm. 49 (11), 1009–1013. https://doi.org/10.1310/hjp4911-1009 (2014).
Iwamoto, F. M. et al. A phase I/II trial of the histone deacetylase inhibitor Romidepsin for adults with recurrent malignant glioma: North American brain tumor consortium study 03–03. Neuro Oncol. 13 (5), 509–516. https://doi.org/10.1093/neuonc/nor017 (2011).
VigiAccess. World Health Organisation. (Accessed 14 October 2024). https://www.vigiaccess.org/
McHugh, M. L. The chi-square test of independence. Biochem Med. 23(2), 143–49. https://doi.org/10.11613/bm.2013.018 (2013).
Berg, S. L. et al. Plasma and cerebrospinal fluid pharmacokinetics of depsipeptide (FR901228) in nonhuman primates. Cancer Chemother. Pharmacol. 54 (1), 85–88. https://doi.org/10.1007/s00280-004-0766-5 (2004).
Frühauf, A. & Meyer-Almes, F. J. Non-Hydroxamate Zinc-Binding groups as warheads for histone deacetylases. Molecules 26 (17), 5151. https://doi.org/10.3390/molecules26175151 (2021).
Citarella, A., Moi, D., Pinzi, L., Bonanni, D. & Rastelli, G. Hydroxamic acid derivatives: from synthetic strategies to medicinal chemistry applications. ACS Omega. 6 (34), 21843–21849. https://doi.org/10.1021/acsomega.1c03628 (2021).
Hayward, D. & Beekman, A. M. Strategies for converting turn-motif and Cyclic peptides to small molecules for targeting protein–protein interactions. RSC Chem. Biol. 5 (3), 198–202. https://doi.org/10.1039/D3CB00222E (2024).
Wilting, R. H. et al. Overlapping functions of Hdac1 and Hdac2 in cell cycle regulation and haematopoiesis. EMBO J. 29 (15), 2586–2597. https://doi.org/10.1038/emboj.2010.136 (2010).
Sandhu, D., Antolin, A. A., Cox, A. R. & Jones, A. M. Identification of different side effects between PARP inhibitors and their polypharmacological multi-target rationale. Br. J. Clin. Pharmacol. 88 (2), 742–752. https://doi.org/10.1111/bcp.15015 (2021).
Mita, M. M. & Mita, A. C. Bromodomain inhibitors a decade later: a promise unfulfilled? Br. J. Cancer. 123 (12), 1713–1714. https://doi.org/10.1038/s41416-020-01079-x (2020).
Hamilton, E. P. et al. First-in-human study of AZD5153, A Small-molecule inhibitor of bromodomain protein 4, in patients with relapsed/refractory malignant solid tumors and lymphoma. Mol. Cancer Ther. 22 (10), 1154–1165. https://doi.org/10.1158/1535-7163.MCT-23-0065 (2023).
Jones, D. K. et al. hERG 1b is critical for human cardiac repolarization. PNAS 111 (50), 18073–18077. https://doi.org/10.1073/pnas.1414945111 (2014).
Hoffmann, P. & Warner, B. Are hERG channel Inhibition and QT interval prolongation all there is in drug-induced torsadogenesis? A review of emerging trends. J. Pharmacol. Toxicol. Methods. 53 (2), 87–105. https://doi.org/10.1016/j.vascn.2005.07.003 (2006).
Hancox, J. C. et al. The hERG potassium channel and hERG screening for drug-induced Torsades de pointes. Pharmacol. Ther. 119 (2), 118–132. https://doi.org/10.1016/j.pharmthera.2008.05.009 (2008).
Pigna, E. et al. HDAC4 preserves skeletal muscle structure following long-term denervation by mediating distinct cellular responses. Skelet. Muscle. 8 (6), 1–16. https://doi.org/10.1186/s13395-018-0153-2 (2018).
Du, G. et al. Histone deacetylase 4 deletion results in abnormal chondrocyte hypertrophy and premature ossification from collagen type 2α1expressing cells. Mol. Med. Rep. 22 (5), 4031–4040. https://doi.org/10.3892/mmr.2020.11465 (2020).
Iannitti, T. & Palmieri, B. Clinical and experimental applications of sodium phenylbutyrate. Drugs R D. 11 (3), 227–249. https://doi.org/10.2165/11591280-000000000-00000 (2012).
Zhou, Y., Dou, X., Zhang, C., He, R. & Ding, Y. Hyperammonemia in a pregnant woman with citrullinemia type I: a case report and literature review. BMC Pregnancy Childbirth. 22 (1), 1–8. https://doi.org/10.1186/s12884-022-05298-3 (2022).
Lokman, M. & Fitzgerald, C. The effect of cancer on reproductive health. Obstet. Gynecol. Reprod. Med. 30 (1), 6–10. https://doi.org/10.1016/j.ogrm.2019.10.002 (2020).
Bubna, A. K. Vorinostat- an overview. Indian J. Dermatol. 60 (4), 419. https://doi.org/10.4103/0019-5154.160511 (2015).
FDA: Center for Drug Evaluation and Research. Drug approval package. Clinical Pharmacology and Biopharmaceutics Review(s). (Accessed 31 December 2024). https://www.accessdata.fda.gov/drugsatfda_docs/nda/2015/205353Orig1s000PharmR.pdf (2014).
BELEODAQ (belinostat) for injection label. FDA Reference ID: 3536712. (Accessed 31 December 2024). https://www.accessdata.fda.gov/drugsatfda_docs/label/2014/206256lbl.pdf (2014).
Dai, Y. et al. Upregulation of HDAC9 in hippocampal neurons mediates depression-like behaviours by inhibiting ANXA2 degradation. Cell. Mol. Life Sci. 80 (10), 289. https://doi.org/10.1007/s00018-023-04945-y (2023).
Lu, S., Li, H., Li, K. & Fan, X. D. HDAC9 promotes brain ischemic injury by provoking IκBα/NF-κB and MAPKs signaling pathways. Biochem. Biophys. Res. Commun. 503 (3), 1322–1329. https://doi.org/10.1016/j.bbrc.2018.07.043 (2018).
Zhao, M. et al. Fibroblast growth factor Receptor-1 is required for Long-Term potentiation, memory consolidation, and neurogenesis. Biol. Psychiatry. 62 (5), 381–390. https://doi.org/10.1016/j.biopsych.2006.10.019 (2007).
Turner, C. A., Eren-Koçak, E., Inui, E. G., Watson, S. J. & Akil, H. Dysregulated fibroblast growth factor (FGF) signaling in neurological and psychiatric disorders. Semin Cell. Dev. Biol. 53 (1), 136–143. https://doi.org/10.1016/j.semcdb.2015.10.003 (2016).
Klimaschewski, L. & Claus, P. Fibroblast growth factor signalling in the diseased nervous system. Mol. Neurobiol. 58 (8), 3884–3903. https://doi.org/10.1007/s12035-021-02367-0 (2021).
Brooks, L. R. et al. Fibroblast growth factor deficiencies impact anxiety-like behavior and the serotonergic system. Behav. Brain Res. 264 (1), 74–81. https://doi.org/10.1016/j.bbr.2014.01.053 (2014).
Zhu, X., Akiyama, T., Yokoyama, T. & Matsumoto, Y. Stereoselective formation of β-O-4 structures mimicking softwood lignin biosynthesis: effects of solvent and the structures of Quinone Methide lignin models. J. Agric. Food Chem. 67 (25), 6950–6961. https://doi.org/10.1021/acs.jafc.9b01968 (2019).
Bolton, J. L. Quinone Methide bioactivation pathway: contribution to toxicity and/or cytoprotection?? Curr. Org. Chem. 18 (1), 61–69. https://doi.org/10.2174/138527281801140121123046 (2014).
Thompson, D. C., Thompson, J. A., Sugumaran, M. & Moldéus, P. Biological and toxicological consequences of Quinone Methide formation. Chem. Biol. Interact. 86 (2), 129–162. https://doi.org/10.1016/0009-2797(93)90117-H (1993).
Lacouture, M. E. et al. Dermatologic adverse events associated with selective fibroblast growth factor receptor inhibitors: overview, prevention, and management guidelines. Oncologist 26 (2), 316–326. https://doi.org/10.1002/onco.13552 (2021).
Subbiah, V. & Verstovsek, S. Clinical development and management of adverse events associated with FGFR inhibitors. Cell. Rep. Med. 4 (10), 1–13. https://doi.org/10.1016/j.xcrm.2023.101204 (2023).
Cancer Research, U. K. The immune system and cancer. (Accessed 01 January 2025). https://www.cancerresearchuk.org/about-cancer/what-is-cancer/body-systems-and-cancer/the-immune-system-and-cancer (2023).
Yang, Q. et al. Host HDAC4 regulates the antiviral response by inhibiting the phosphorylation of IRF3. J. Mol. Cell. Biol. 11 (2), 158–169. https://doi.org/10.1093/jmcb/mjy035 (2019).
Lu, Y., Stuart, J. H., Talbot-Cooper, C. & Smith, G. L. Histone deacetylase 4 promotes type I interferon signaling, restricts DNA viruses, and is degraded via vaccinia virus protein C6. PNAS 116 (24), 11997–12006. https://doi.org/10.1073/pnas.1816399116 (2019).
Poralla, L. et al. Histone deacetylase 5 regulates the inflammatory response of macrophages. J. Cell. Mol. Med. 19 (9), 2162–2171. https://doi.org/10.1111/jcmm.12595 (2015).
Ferro, C. J., Solkhan, F., Jalal, Z., Al-Hamid, A. M. & Jones, A. M. Relevance of physicochemical properties and functional Pharmacology data to predict the clinical safety profile of direct oral anticoagulants. Pharmacol. Res. Perspect. 8 (3), 603. https://doi.org/10.1002/prp2.603 (2020).
Stansborough, R. L. & Gibson, R. J. Proteasome inhibitor-induced Gastrointestinal toxicity. Support Palliat. Care. 11 (2), 133–137. https://doi.org/10.1097/SPC.0000000000000266 (2017).
Bandyopadhyay, U., Biswas, K., Bandyopadhyay, D., Ganguly, C. & Banerjee, R. Dexamethasone makes the gastric mucosa susceptible to ulceration by inhibiting prostaglandin synthetase and peroxidase - two important gastroprotective enzymes. Mol. Cell. Biol. 202 (1), 31–36. https://doi.org/10.1023/A:1007018212822 (1999).
Navabi, N. et al. Epithelial histone deacetylase 3 instructs intestinal immunity by coordinating local lymphocyte activation. Cell. Rep. 19 (6), 1165–1175. https://doi.org/10.1016/j.celrep.2017.04.046 (2017).
Reynolds, P. M., MacLauren, R., Mueller, S. W., Fish, D. N. & Kiser, T. H. Management of extravasation injuries: A focused evaluation of noncytotoxic medications. Pharmacother 34 (6), 617–632. https://doi.org/10.1002/phar.1396 (2014).
Grant, C. et al. Romidepsin: a new therapy for cutaneous T-cell lymphoma and a potential therapy for solid tumors. Expert Rev. Anticancer Ther. 10 (7), 997–1008. https://doi.org/10.1586/era.10.88 (2019).
Adil, A., Malik, M., Malik, J. A., Nazir, S. & Ali, I. S. Down regulation in glutathione S-Transferase stimulate oxidative stress and genetic susceptibility of human breast cancer. J. Pharm. Res. Int. 34 (20B), 1–15. https://doi.org/10.9734/jpri/2022/v34i20B35827 (2022).
Wu, X. et al. Clinical application of thioredoxin reductase as a novel biomarker in liver cancer. Sci. Rep. 11 (1), 1–13. https://doi.org/10.1038/s41598-021-85688-3 (2021).
García-Abeijon, P. et al. Factors associated with underreporting of adverse drug reactions by health care professionals: A systematic review update. Drug Saf. 46 (7), 625–636. https://doi.org/10.1007/s40264-023-01302-7 (2023).
Hashiguchi, M. et al. Factors affecting the timing of signal detection of adverse drug reactions. PLoS One. 10 (12), 1–13. https://doi.org/10.1371/journal.pone.0144263 (2015).
Acknowledgements
J.L.P would like to acknowledge support from Cancer Research UK RadNet Birmingham.
Author information
Authors and Affiliations
Contributions
R.B, J.L.P, and A.M.J. wrote the main manuscript text. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Begum, R., Parsons, J.L. & Jones, A.M. Adverse drug reaction profiles of histone deacetylase inhibitors. Sci Rep 15, 35880 (2025). https://doi.org/10.1038/s41598-025-19717-w
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-19717-w
