
Abstract
DNA methylation is a critical epigenetic modification that regulates gene expression and plays a significant role in cancer development. This methylation signature can be detected in cancer-derived DNA from non-invasive samples, such as plasma, urine or Pap smears. However, in early-stage cancers—when detection is most critical—the concentration of cancer DNA is often low, limiting the sensitivity of current detection methods. Traditional DNA methylation detection techniques, which rely on methylation ratio-based measurements, may obscure subtle variations in methylation patterns, further reducing detection sensitivity. In this study, we analyzed cervical scraping specimens and examined whether detecting cancer-specific methylation patterns in cervical cancer could be enhanced using a Highly Methylated Haplotype (HMH) approach. This novel approach captures highly methylated haplotypes at single-molecule resolution using next-generation sequencing, providing greater detail than conventional methods. HMHs in specific DNA regions are a hallmark of cancer and stand out in contrast to sporadic methylation commonly observed in non-cancerous tissues. We applied HMH profiling to a gene panel of four biomarkers (CA10, DPP10, FMN2, and HAS1) previously validated in cervical cancer studies. At pre-specified cutoffs (99th percentile of normals), haplotype-based scoring achieved 89.9% sensitivity for invasive cancer at high specificity (~ 94–98%), outperforming median (78.0%) and single-CpG (71.6%) methods. For clinically relevant endpoints, the combined panel detected 51–52% of CIN2 + and 66–67% of CIN3 + cases, again exceeding the performance of median- and single-CpG-based scoring methods.
These findings demonstrate the potential of HMH to substantially enhance sensitivity in cervical cancer detection, offering a promising approach for non-invasive diagnostics.
Similar content being viewed by others

Introduction
DNA methylation is a critical epigenetic modification that may affect gene expression at key positions in the genome 1,2. During the progression from normal to malignant cells, significant alterations in DNA methylation patterns occur, which are a hallmark of cancer development. These changes provide valuable biomarkers for the early detection of cancer 3. Traditional approaches primarily rely on quantitative comparisons of DNA methylation between normal and cancerous tissues 3,4,5. However, in clinical specimens such as cervical scrapes or self-collected samples, tumor-derived DNA is often mixed with a large background of normal DNA. This dilution effect can mask cancer-specific signals and lead to false negatives, highlighting the need for more sensitive detection methods.
Several studies have already examined DNA methylation markers in cervical scrapes and self-collected vaginal samples. Host-cell methylation markers have been highlighted for managing CIN 6; methylation assays have been examined in self-collected samples 7, and HPV DNA methylation has been analyzed as a marker of disease severity 8. Together, these reports underline the strength of methylation biomarkers as a foundation for new approaches.
Despite these advances, most existing methods rely on averaged methylation levels or cycle threshold (Ct) cutoffs. This approach can miss important details, since it smooths over the complexity of tumor-derived DNA. Detecting cancer-specific methylation is especially difficult in clinical specimens, where tumor DNA is present only in tiny amounts and is heavily diluted by normal DNA. This problem is well known in blood-based liquid biopsies, but it also applies to cervical scrapes and self-collected samples.
In this study, we focus on cervical specimens obtained by scraping during colposcopy, which provide a more direct source of tumor-derived DNA than blood. One way to overcome the challenge of low tumor DNA levels is to target CpG sites that show clear binary differences—fully methylated in cancer cells but unmethylated in normal tissue. This kind of qualitative approach improves specificity and accuracy, as has been demonstrated in hepatocellular carcinoma detection 9,10,11,12,13. However, traditional methods, that calculate average methylation levels across CpGs or regions have limited in a DNA sample or the average methylation of multiple CpGs within a region, have limited sensitivity in samples where tumor DNA is scarce. Averaging methylation levels can be useful for broad comparisons or population-level studies, but it obscures the single-molecule patterns that define heavily methylated haplotypes.
Moreover, while array-based techniques and methylation-specific PCR (MSP) are widely used for rapid methylation assessments, they lack the resolution needed to detect the complex heterogeneity of tumor-derived DNA 14. Next-generation sequencing technologies overcome these limitations by enabling high-resolution methylation profiling at the single-molecule level. Unlike array-based methods that calculate an average methylation level, sequencing provides a more precise and detailed view of individual CpG sites along single DNA molecules 15,16,17. This higher resolution is cruicial for distinguishing cancer-derived DNA from background of normal DNA, ultimately boosting the sensitivity of non-invasive cancer detection.
It is now well-established that cancer-related methylation patterns are not evenly distributed across the genome 18,19. Instead, they manifest as specific combinations of methylated and unmethylated CpG sites, known as haplotypes, on individual DNA strands 17. Studying these haplotypes provides valuable insights into the complexity of methylation in cancer tissue. This approach has become a powerful tool because it reveals details that traditional methods, which rely on averages, often overlook. Using haplotypes improves the resolution of tissue sample analysis, making it easier to distinguish different cell types in mixed samples and to detect disease-associated methylation changes with greater accuracy 20,21,22. Building on this principle, our work specifically demonstrates that haplotype-based methylation profiling can sensitively detect cancer-associated DNA in cervical samples.
In this context, we reanalyzed previously published data on DNA methylation markers in cervical cancer and precancer, specifically the biomarkers CA10, DPP10, FMN2, and HAS1 23, which were validated in independent cohorts 24 but measured using a traditional method. Our goal was to determine whether a haplotype-based profiling strategy, enabled by next-generation sequencing at single-molecule resolution, could improve sensitivity while maintaining high specificity in cervical scraping specimens. We focused on both invasive cancers and precancerous lesions (CIN2 and CIN3), comparing haplotype-based scores to the traditional median-methylation approach across the same genomic regions.
Methods
Study population
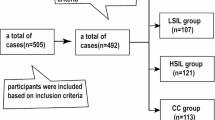
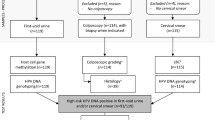
This study analyzed 631 cervical samples from two independent study populations of women referred for colposcopy due to abnormal Pap smear results 24. The Methylation Analysis Revealing Key Epigenetic Regulation (MARKER) study included 100 normal samples, 50 CIN1, 50 CIN2, 50 CIN3, and 8 cancer samples, while the Biomarkers of Cervical Cancer Risk (BCCR) study included 100 normal samples, 57 CIN1, 61 CIN2, 53 CIN3, and 102 cancer samples. A detailed breakdown of participant numbers is provided in Table S1.
DNA collection, processing, and sequencing
The DNA used in this study was originally collected, processed, and sequenced as part of a previous project by our group 24. Briefly, DNA was extracted from cervical samples and treated with the EZ-96 DNA Methylation MagPrep kit (Zymo Research), following the provided instructions for bisulfite conversion. The converted DNA was then amplified through two rounds of PCR targeting the genes CA10, DPP10, FMN2, and HAS1. The purified libraries were sequenced on the Illumina platform using the MiSeq Reagent Nano Kit V2 at the HKG Epitherapeutics Laboratory (Hong Kong), which was blinded to the clinical data. Each amplicon was designed to span a fixed genomic region with a defined number of CpG sites per read. As a result, all sequencing reads covering a given region contained the same number and arrangement of CpG sites, eliminating variability in haplotype length and ensuring consistent haplotype-based methylation scoring across samples. Sequencing data were processed using the Bismark software package for bisulfite mapping and methylation calling. Reads were quality-filtered with Trim Galore (Phred score cutoff: 20) and aligned using Bowtie 2.
Haplotype-based methylation score
We first introduce the notation for methylation profiling, where a methylated CpG site is denoted by 'Z' and an unmethylated site by 'z.' This notation originates from the methylation extraction step performed while using the Bismark software package 25, enabling the representation of methylation haplotypes along individual DNA strands. We applied two haplotype-based scoring approaches for cervical cancer detection:
1. Full Highly Methylated Haplotype (fHMH): Haplotypes in which all CpG sites within the targeted region are fully methylated. For example, a “13Z” haplotype for CA10, DPP10, and FMN2 indicates that all 13 CpG sites in the region are methylated. While a “9Z” haplotype for HAS1 indicates that all 9 CpG sites in the region are methylated.
2. Partial Highly Methylated Haplotype (pHMH): Haplotypes in which a minimum number of CpGs within the targeted region are methylated. For example, a “10Z + ” haplotype for CA10, DPP10, and FMN2 indicates that at least 10 of the CpG sites in the region are methylated, while a “6Z + ” haplotype for HAS1 indicates that at least 6 CPG sites are methylated.
In both approaches, the number of reads for each haplotype was normalized to the total number of reads, allowing us to calculate the methylation score for each sample.
Threshold definition for cervical cancer DNA detection
To assess the sensitivity of normalized HMH and median methylation scores in predicting cervical cancer, we first defined thresholds to differentiate between “normal” and cancer samples. These thresholds were established exclusively using samples from ascertained lesion-free individuals to determine a reliable range of normal methylation scores for each gene. The threshold was set at the 99th percentile, assuming that the frequency of cervical cancer DNA among the “normal” population is below 1%. This assumption represents a plausible approximation, as no definitive standard currently exists for identifying the earliest “pre-diagnosed” cervical cancer cases. However, the actual prevalence of individuals with undetected cancerous or precancerous cells may be higher than assumed. We computed thresholds for each gene using four different methylation scoring methods: fHMH Score, pHMH Score, Single CpG Methylation Score, and Median Methylation Scores. This standardized threshold-setting process ensured consistent criteria for evaluating the performance of each method in detecting cervical cancer DNA across varying methylation scores.
Statistical analysis
To evaluate the performance of each scoring method in detecting cervical cancer DNA, we calculated the sensitivity of the four methylation scores (fHMH, pHMH, single CpG methylation, and median methylation) across premalignant stages (CIN1, CIN2, CIN3) and invasive cancer cases. Sensitivity was assessed for both the combined set of genes in the epiCervix panel (CA10, DPP10, FMN2, and HAS1) and individually for each gene in this panel, using the thresholds described above. Additionally, each scoring method was evaluated by calculating the Receiver Operating Characteristic (ROC) area under the curve (AUC).
Ethics statement
All methods were carried out in accordance with relevant guidelines and regulations. All experimental protocols were approved by institutional and/or licensing committees as follows: the MARKER study received ethical approval from the institutional review boards at McGill University and its affiliated hospitals. The BCCR study was approved by McGill’s Institutional Review Board, the Comité d’éthique de la recherche du Centre Hospitalier de l’Université de Montréal, and the Research Ethics Committees of each of the participating hospitals. Participants provided written informed consent for both their participation in the study and the use of their collected cervical specimens in future studies.
Results
Methylation scores, for cancer and normal samples, are shown in Supplemental Fig. 1 for each gene: CA10 (Fig. 1A-D), DPP10 (Fig. 1E-H), FMN2 (Fig. 1I-L), and HAS1 (Fig. 1M-D). For all scoring methods, methylation scores were significantly higher in cancer compared to normal samples (p < 0.0001 for all comparisons.

ROC comparison of scoring methods for each gene in cancer vs normal samples (108 cancers, 169 normals). Panels: (A) CA10, (B) DPP10, (C) FMN2, (D) HAS1. Within each panel, ROC curves for fHMH, pHMH, Median, and Single-CpG scoring are overlaid. The legend in each panel reports the AUC for each method. Axes show Sensitivity versus 1 − Specificity; the 45° gray line indicates chance performance. This figure serves as a method benchmarking comparison (cancer vs normal).
As shown in Table 1, thresholds were defined at the 99th percentile of the normal distribution, which anchors specificity at ~ 94–98% across all methods. At this fixed cutoff, sensitivity provides the most informative comparison. The combined four-gene epiCervix panel achieved the highest performance with haplotype-based methods: fHMH detected 89.9% of cancers, followed by pHMH at 87.2%, both substantially higher than Median (78.0%) or Single CpG (71.6%).
At the gene level, haplotype-based scoring also improved cancer detection compared to traditional approaches, although the gain varied. CA10 and HAS1 showed the clearest improvement, while FMN2 and especially DPP10 showed more modest differences. Overall, fHMH and pHMH consistently provided greater sensitivity at fixed high specificity, underscoring their advantage over median and single-CpG scoring.
Performance at clinically relevant screening endpoints (CIN2 + and CIN3 +)
To reflect real-world screening practice, we assessed performance at the CIN2 + and CIN3 + thresholds. For CIN2 + , the negative group included Normal and CIN1 cases (n = 301), and positives were CIN2, CIN3, and Cancer (n = 317). As shown in Table 2, haplotype-based scoring (fHMH and pHMH) identified more CIN2 + cases than Median or Single-CpG approaches, while preserving high specificity. The advantage was most evident in the combined epiCervix panel, which detected about half of CIN2 + cases (51–52%) compared with 43–49% for Median and 42% for Single-CpG.
For CIN3 + , the negative group included Normal, CIN1, and CIN2, and positives were CIN3 and Cancer (n = 209). Results are shown in Table 3. Again, haplotype-based scoring outperformed the other methods. The combined epiCervix panel detected two-thirds of CIN3 + cases (66–67%) with fHMH and pHMH, compared with 62% for Median and 55% for Single-CpG, while maintaining specificity in the high-80% range.
Discussion
While cancer-specific methylation alterations are easily observable in tumor tissues and biopsies, detecting them in clinical specimens remains a major challenge. This is well known from blood-based liquid biopsies, where tumor DNA is scarce and easily masked by DNA from healthy cells. A similar problem exists in cervical scrapes and self-collected vaginal samples. Here too, tumor-derived DNA is mixed with DNA from normal cervical tissue, making the cancer signal hard to distinguish. The challenge is even greater in early-stage disease, when the proportion of tumor DNA is at its lowest—precisely when early detection would have the greatest benefit 26,27.
The site of the tumor also significantly influences the release of ctDNA, with less vascular tumors releasing lower amounts of DNA and hence lowering sensitivity even further, especially for localized or early-stage cancers. Traditional methods, which rely on ratio-based methylation analysis and CpGs averaging in the genomic region, are incapable of detecting rare cancer-specific profiles and hence generate false negatives. They often fail to detect cancer-specific methylation patterns within a small fraction of fragmented cfDNA, particularly in samples with a vast excess of unmethylated DNA.
A prior study by Mirabello et al. 28 used a haplotype-based approach to analyze methylation patterns within HPV16 viral DNA and showed its utility for stratifying cervical lesions. While conceptually related, our study differs in that it applies haplotype profiling to host genomic regions rather than viral DNA, offering a human gene-based epigenetic signature for cancer detection.
Several systematic reviews, including a recent meta-analysis by Salta et al. 29, have explored DNA methylation markers for cervical cancer detection, mostly centred around panels like CADM1, MAL, and FAM19A4. Beyond these specific marker panels, broader reviews have also emphasized the potential of methylation biomarkers in cervical screening. Host-cell methylation markers have been discussed for their value in cervical screening and the management of precancerous lesions 6. Other work has emphasized the feasibility of applying methylation testing to self-collected samples, including urine and vaginal swabs 7. HPV DNA methylation has also been systematically reviewed and shown to correlate closely with disease severity 8. Taken together, these reports demonstrate the strength of DNA methylation biomarkers across both host and viral targets, and across a variety of sampling methods.
Most of this work, however, has relied on quantitative methylation-specific PCR (qMSP) or on measuring methylation at individual CpG sites.
While qMSP and bisulfite pyrosequencing panels have demonstrated strong diagnostic performance in clinical validation studies, these approaches rely on averaged methylation values or Ct thresholds. Because they can’t look at methylation patterns along single DNA molecules, these methods may miss the cancer-specific signatures that matter most when DNA is scarce or fragmented. In contrast, haplotype-based analysis keeps that single-molecule detail, offering complementary insights that can improve detection in such difficult samples.
Traditional techniques such as qMSP and bisulfite pyrosequencing remain attractive because they are simple, low-cost, and compatible with routine diagnostic workflows. However, by design, they cannot capture haplotypes, since they only report average methylation levels at CpGs or across small regions. The critical cancer-specific signal — fully methylated haplotypes — is therefore lost. Next-generation sequencing, in contrast, retains single-molecule information, enabling haplotype profiling that identifies truly cancer-derived DNA molecules against a background of normal DNA. Although sequencing requires greater resources, its ability to detect haplotypes makes it indispensable for this purpose, and the practical gap is narrowing as sequencing costs fall and targeted amplicon assays become more accessible.
One strategy to overcome these limitations and enhance the detection sensitivity is to move away from traditional ratio-based approaches and, instead, focus on haplotype-based methylation profiling, that tracks methylation profiles across groups of more than one CpG site on the same DNA molecule. This approach is only feasible with sequencing technologies capable of single-molecule resolution.
In the current study, we developed the HMH approach, which focuses on identifying highly methylated haplotype profiles characteristic of cancerous tissues, and unlike other haplotype-based methods, it uniquely targets fully and partially methylated sequences to improve sensitivity and specificity. We demonstrated that, for the combined panel of genes, the fHMH method achieved the highest sensitivity (89.91%), closely followed by pHMH (87.16%), both of which outperformed traditional methylation scoring methods. These results apply to invasive cancer versus normal comparisons, and not to CIN2 + or CIN3 + endpoints, which are presented separately in Tables 2 and 3. Analysis of the individual CA10, DPP10, FMN2, and HAS1 genes further confirmed the superior sensitivity of HMH in detecting cancer-specific haplotypes. Notably, pHMH showed superior AUC values compared to fHMH for CA10, DPP10, and FMN2 but not for HAS1, possibly due to the latter having fewer CpG sites.
Several strengths should be acknowledged. Our findings were consistent across two independent study populations, both included in this analysis: the MARKER study and the Biomarkers of Cervical Cancer Risk (BCCR) study. These cohorts, comprising a total of 631 cervical samples from women referred for colposcopy due to abnormal Pap smear results, reinforce the robustness and reproducibility of the HMH method across diverse study populations. Notably, the study design also contributed to its robustness: we increased statistical power to detect methylation differences by oversampling disease cases and undersampling normal samples. In addition to demonstrating improved sensitivity, our approach allowed for the establishment of a robust classification threshold. Since all four genes in the epiCervix biomarker set are consistently fully unmethylated in the normal population, any sample exhibiting a methylation score above the established threshold for one or more of these genes was classified as containing cancer DNA. This classification criterion allowed for the clear distinction between cancerous and non-cancerous samples with high specificity, leveraging the absence of methylation in normal samples as a key discriminating factor. In addition, by focusing on binary methylation differences—sites that are either fully methylated ('Z') or unmethylated ('z')—the HMH approach enhances sensitivity in detecting cancer-specific methylation patterns. As illustrated in Supplemental Fig. 1, when the cancer profile is highly methylated and normal blood shows sporadic methylation at individual CpG sites, haplotype analysis easily identifies the cancer-specific profile. In contrast, the averaging method fails to detect significant differences due to dilution by non-cancerous DNA.
The HMH approach captures precise methylation haplotypes along individual DNA strands, which is particularly valuable for highly diluted cfDNA samples, such as those collected from Pap smears or, even more so, from self-sampling. It has the potential to transform non-invasive cancer diagnostics by allowing precise detection of cancer DNA even when diluted by non-cancerous cfDNA. We also detected a higher number of cancers amongst the premalignant samples, pointing to the promise of more effective early detection than current Pap smear-based diagnosis using HMH-based scores. Such improvements hold promise for early cancer diagnosis and monitoring, ultimately leading to better patient outcomes through timely intervention. Moreover, the principles of HMH profiling could be extended to detect other cancer types, broadening the impact of this advancement across oncology.
Although this study looked specifically at cervical scrapes, the haplotype-based approach can be applied to other types of samples as well. It could also be extended to other types of liquid biopsies. For example: blood, saliva, urine, or even cerebrospinal fluid, where tumor DNA is typically scarce and diluted by DNA from healthy cells. To truly understand how broadly this approach can be used in real-world medicine, larger studies in more diverse groups of people, including long-term follow-ups, will be essential.
Some limitations should be acknowledged. Our study design was enriched for cancer cases compared to a true screening population, which may inflate AUC estimates relative to real-world settings. Performance may therefore be somewhat lower in prospective screening cohorts where cancer prevalence is much lower. In addition, although we defined thresholds using normals and then applied them to CIN1–3 and cancer groups, these estimates should be interpreted cautiously until validated in larger, population-based cohorts.
Data availability
Data were deposited in the NCBI Sequence Read Archive (SRA) under BioProject accession number [PRJNA1254662].
References
Razin, A. & Riggs, A. D. DNA methylation and gene function. Science 210(4470), 604–610 (1980).
Massart, R., Suderman, M., Mongrain, V. & Szyf, M. DNA methylation and transcription onset in the brain. Epigenomics 9(6), 797–809 (2017).
Paluszczak, J. & Baer-Dubowska, W. Epigenetic Diagnostics of Cancer — The Application of DNA Methylation Markers. J. Appl. Genet. 47(4), 365–375 (2006).
Stefanska, B. et al. Definition of the landscape of promoter DNA hypomethylation in liver cancer. Cancer Res. 71(17), 5891–5903 (2011).
Hao, X. et al. DNA Methylation Markers for Diagnosis and Prognosis of Common Cancers. Proc. Natl. Acad. Sci. 114(28), 7414–7419 (2017).
Dick, S., Heideman, D. A. M., Berkhof, J., Steenbergen, R. D. M. & Bleeker, M. C. G. Clinical indications for host-cell DNA methylation markers in cervical screening and management of cervical intraepithelial neoplasia: A review. Tumour Virus Res. 19, 200308 (2025).
Sumiec, E. G., Yim, Z. Y., Mohy-Eldin, H. & Nedjai, B. The current state of DNA methylation biomarkers in self-collected liquid biopsies for the early detection of cervical cancer: A literature review. Infect Agent Cancer. 19(1), 62 (2024).
Bowden, S. J. et al. The use of human papillomavirus DNA methylation in cervical intraepithelial neoplasia: A systematic review and meta-analysis. EBioMedicine 50, 246–259 (2019).
Cheishvili, D. et al. A high-throughput test enables specific detection of hepatocellular carcinoma. Nat Commun. 14(1), 3306 (2023).
Millar, D., Paul, C. L., Molloy, P. L. & Clark, S. J. A Distinct Sequence (ATAAA) Separates Methylated and Unmethylated Domains at the 5′-End of theGSTP1 CpG Island*. J. Biol. Chem. 275(32), 24893–24899 (2000).
Liu, M. C., Oxnard, G. R., Klein, E. A., Swanton, C. & Seiden, M. V. Sensitive and specific multi-cancer detection and localization using methylation signatures in cell-free DNA. Ann Oncol. 31(6), 745–759 (2020).
Cheng, H., Gao, Y. & Lou, G. DNA Methylation of the RIZ1 Tumor Suppressor Gene Plays an Important Role in the Tumorigenesis of Cervical Cancer. Eur. J. Med. Res. 15(1), 20 (2010).
Geng J, Sun J, Lin Q, Gu J, Zhao Y, Zhang H, et al. Methylation Status of NEUROG2 and NID2 Improves the Diagnosis of Stage I NSCLC. Oncology letters. 2012.
Zhang, Z. et al. The DNA methylation haplotype (mHap) format and mHapTools. Bioinformatics 37(24), 4892–4894 (2021).
Kotanidou, E. P. et al. Methylation haplotypes of the insulin gene promoter in children and adolescents with type 1 diabetes: Can a dimensionality reduction approach predict the disease?. Exp. Ther. Med. 26(4), 461 (2023).
Ding, Y. et al. mHapTk: A comprehensive toolkit for the analysis of DNA methylation haplotypes. Bioinformatics 38(22), 5141–5143 (2022).
Hong, Y. et al. mHapBrowser: A comprehensive database for visualization and analysis of DNA methylation haplotypes. Nucleic Acids Res. 52(D1), D929–D937 (2024).
Esteller M. Cancer epigenomics: DNA methylomes and histone-modification maps. Nat Rev Genet. 2007.
Baylin, S. B. & Jones, P. A. A decade of exploring the cancer epigenome - biological and translational implications. Nat Rev Cancer. 11(10), 726–734 (2011).
Guo, S. et al. Identification of methylation haplotype blocks aids in deconvolution of heterogeneous tissue samples and tumor tissue-of-origin mapping from plasma DNA. Nat Genet. 49(4), 635–642 (2017).
Feng, Y. et al. A DNA methylation haplotype block landscape in human tissues and preimplantation embryos reveals regulatory elements defined by comethylation patterns. Genome Res. 33(12), 2041–2052 (2023).
Unterman I, Avrahami D, Katsman E, Triche TJ, Glaser B, Berman BP. Multi-cell type deconvolution using a probabilistic model for single-molecule DNA methylation haplotypes. bioRxiv. 2023:2023.08.20.554012.
El-Zein, M. et al. Genome-wide DNA methylation profiling identifies two novel genes in cervical neoplasia. Int J Cancer. 147(5), 1264–1274 (2020).
El-Zein M, Cheishvili D, Szyf M, Franco EL. Validation of novel DNA methylation markers in cervical precancer and cancer. Int J Cancer. 2023.
Krueger F, Andrews SR. Bismark: A flexible aligner and methylation caller for Bisulfite-Seq applications. Bioinformatics.27(11):1571–2.
Wang, J. et al. Circulating tumor DNA correlates with microvascular invasion and predicts tumor recurrence of hepatocellular carcinoma. Ann Transl Med. 8(5), 237 (2020).
Zhan, Q. et al. New insights into the correlations between circulating tumor cells and target organ metastasis. Signal Transduct Target Ther. 8(1), 465 (2023).
Mirabello, L. et al. HPV16 methyl-haplotypes determined by a novel next-generation sequencing method are associated with cervical precancer. Int J Cancer. 136(4), E146–E153 (2015).
Salta, S., Lobo, J., Magalhaes, B., Henrique, R. & Jeronimo, C. DNA methylation as a triage marker for colposcopy referral in HPV-based cervical cancer screening: A systematic review and meta-analysis. Clin Epigenetics. 15(1), 125 (2023).
Funding
This work was funded by HKG Epitherapeutics Ltd. and EpiMedTech Global Ltd.
Author information
Authors and Affiliations
Contributions
D.C. (David Cheishvili) conceived the study, developed the methodology, performed the analysis, and wrote the manuscript. M.S. (Moshe Szyf) contributed to the study concept, experimental design, and manuscript review. M.E.Z. (Mariam El-Zein) provided critical feedback on the manuscript and contributed to the interpretation of results. E.F. (Eduardo Franco) supervised the original clinical studies, contributed to data interpretation, and provided critical revision of the manuscript. All authors reviewed and approved the final version of the manuscript. Mariam El-Zein: Contribution to study design and data collection in MARKER study, writing – original draft, writing – review and editing. Eduardo L. Franco: Contribution to study design and supervision in the MARKER and BCCR studies, writing, review and editing. Moshe Szyf: Conceptualization, investigation, methodology, writing, review and editing.
Corresponding author
Ethics declarations
Competing interests
David Cheishvili, Mariam El-Zein, Eduardo L Franco, and Moshe Szyf hold a patent related to the discovery of “DNA methylation markers for early detection of cervical cancer,” registered at the Office of Innovation and Partnerships, McGill University, Montreal, Quebec, Canada (October 2018). Moshe Szyf is a shareholder in MTL Epitherapeutics and EpiMedtech Global.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
David, C., El-Zein, M., Franco, E.L. et al. Enhancing the sensitivity of non-invasive cervical cancer detection using CpG methylation haplotype profiling. Sci Rep 15, 36057 (2025). https://doi.org/10.1038/s41598-025-20050-5
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-20050-5
