
Abstract
Delivery of an mRNA formulated with lipid nanoparticles (LNPs) induces robust humoral and cell-mediated branches of the immune response. Depending on the LNP formula, mRNA encoding proteins can be detected in the liver upon intramuscular administration of mRNA/LNP in mice. This study investigated the impact of mRNA/LNP administration on liver-associated macrophages at the transcriptomic and epigenetic levels in a mouse model. An mRNA encoding ovalbumin (OVA) formulated with LNPs, was administered intramuscularly, and a robust OVA-specific antibody was detected in the serum on Day 7. F4/80+ liver-associated macrophages were isolated and subjected to RNA sequencing, which identified 554 genes whose expression levels were altered compared with those in the PBS control group. The expression of genes involved in macrophage inflammatory functions, such as Tnf, Il6 and Marco, were upregulated. Gene ontology enrichment analysis revealed that IL-6/JAK/STAT3 and TNFα/NF-κB hallmarks were significantly enriched, and mRNA/LNP-exposed liver-associated macrophages were characterized as M1-like cells based on the macrophage transcriptomic profiles. Enrichment of the active histone mark H3K4me3 showed that clusters of loci were highly increased in the mRNA/LNP group, indicating an impact of mRNA/LNPs on macrophage epigenetic profiles. The cis-regulatory regions of Tnf, Il6 and Marco showed enrichment of H3K4me3 marks, which correlated well with their increased transcription. Taken together, our data indicated that mRNA/LNP administration via the intramuscular route influences the gene expression and epigenetic profiles of liver-associated macrophages, reflecting its robust ability to induce an immune response.
Similar content being viewed by others

Introduction
Innate immune cells in tissues play important roles in maintaining tissue homeostasis and may exacerbate pathological conditions in disease settings. Tissue-resident macrophages (TRMs) develop from the yolk sac and fetal liver/bone marrow cells and can be found in most organs1. They receive cues from each tissue’s local environment and adopt more tissue-specific roles, reflecting the unique enhancer landscape2. For example, while liver and spleen TRMs are engaged in recycling heme from old red blood cells, alveolar macrophages in the lung play a significant role in controlling surfactant homeostasis. For their effector functions, TRMs phagocytose and eliminate invasive pathogens during infections and may present antigens to T cells3.
The origin and developmental pathways of TRMs have been recently revised, and their roles in maintaining organ functions have become increasingly appreciated. Kupffer cells (KCs) are the major leukocytes in the liver, where they have crucial housekeeping functions and respond to systemic infection4. KCs are of embryonic origin and are capable of self-renewal with limited contributions from circulating monocytes5. It is estimated that KCs make up of approximately 15% of the total liver cell population, or 70-80% of nonparenchymal liver cells6,7. During liver injury, monocyte-derived KCs replenish embryonic KCs and modulate the severity of liver injury and fibrosis8. In recent years, macrophages have been shown to exhibit epigenetic memory that enhances or dampens the response to unrelated stimuli9. Histone modifications by methylation and acetylation of histone subunits influence the cis-regulatory elements of enhancers and promoters that affect the transcription of nearby genes. Active histone marks, including trimethylated lysine 4 of histone subunit 3 (H3K4me3) and acetylated H3K27 (H3K27ac), are crucial for controlling innate immune memory10. Thus, the insults that activate TRMs potentially leave long-term epigenetic marks on these cells and may dictate future responses.
mRNA vaccines have quickly become a vaccine of choice for emerging infectious diseases due to their flexible design and ease of manufacturing. The COVID-19 pandemic has accelerated the emergency use authorization of mRNA vaccines against SARS-CoV-211. mRNA vaccines are expected to be applied to other diseases in the future for both infectious and communicable diseases. The intramuscular administration of mRNA vaccines formulated with lipid nanoparticles (LNPs) results in the expression of encoded protein(s) mainly at the site of injection (such as muscle tissues) and in other organs, mainly the liver and spleen12,13. Depending on the LNP formulation, mRNA/LNPs are localized to different organs, which may influence the local and systemic immune response induced by LNPs in a mouse model14.
In a mouse model, administration of mRNA/LNPs readily resulted in liver expression of the encoded proteins in the liver. In one study, the hepatotropic mRNA-LNP of the reporter gene administered via intravenous injection resulted in detectable reporter signaling in the liver that lasted 3 days15. Interestingly, in this study, the results clearly showed that all hepatocytes were transfected with an mRNA along with a subpopulation of endothelial cells and KCs15. This has led to the development of liver-trophic therapy using mRNA/LNPs that drive the expression of hepatocyte growth factor and epidermal growth factor in the liver16. The effect of liver-associated macrophages on the uptake of mRNA/LNPs in the liver is unknown. Furthermore, how mRNA/LNPs influence the epigenetic profiles of liver-associated macrophages, which may affect the future response of innate immune cells via innate immune memory, is unclear.
Results
Immune response to mRNA/LNP immunization
Mice were intramuscularly immunized with ovalbumin (OVA) encoding mRNA formulated with LNPs (OVA mRNA/LNP) or control PBS. On Day 7 after immunization, blood samples were collected to measure the OVA-specific IgG titer, and livers were harvested to isolate F4/80+ liver-associated macrophages (Fig. 1A). The OVA-specific antibody titer was significantly greater in the OVA mRNA/LNP group than in the PBS control group, indicating that only one dose of OVA mRNA/LNP immunization led to a robust humoral response (Fig. 1B).

Study design and OVA-specific antibody titers after OVA mRNA/LNP immunization. (A) A schematic representation of the study design. (B) Sera obtained from mice (n = 6 per group) immunized with OVA mRNA/LNPs (10 µg per mouse) or PBS control for 7 days were subjected to ELISA. The endpoint titer for OVA-specific IgG was obtained as described in the Methods section. The data are presented as means ± SEM. Statistical analysis was performed using unpaired two-tailed Student’s t test; **** indicates p < 0.0001.
The morphology and weight of the livers were observed to investigate the effect of the mRNA/LNPs on the livers. As shown in Fig. 2A,B, no gross changes were observed between the two groups, and the liver weights were similar. After single-cell isolation and magnetic bead column-based sorting of F4/80+ cells, the cells were analyzed for CD45 + CD11b + F4/80+ subsets by flow cytometry (Supplementary Fig. 1). The isolated cells were characterized as CD45 + CD11b + F4/80+ with 90% or greater purity. Compared with that in bone marrow-derived macrophages, the expression of the KC-specific marker Clec4f in isolated cells was detected17. A high level of Clec4f mRNA was detected in sorted F4/80+ liver-associated macrophages, but no difference in the level of Clec4f was found between cells from the PBS control and OVA mRNA/LNP groups (Fig. 2C).

Morphology and weight of livers from OVA mRNA/LNP-immunized mice. (A) The morphology of livers was observed on Day 7 after immunization (n = 6). (B) Liver weight (g) is shown as the mean ± SEM (n = 6). (C) The expression of the Kupffer cell marker Clec4f in isolated liver-associated macrophages (CD45 + CD11b + F4/80+) was examined by qRT‒PCR. Bone marrow-derived macrophages (BMMs) were used as controls. Each bar represents the mean ± SEM (n = 3 per group). Statistical analysis was performed using unpaired two-tailed Student’s t test; **** indicates p < 0.0001, and ns indicates not significant.
Transcriptomic profile of F4/80+ liver-associated macrophages from OVA mRNA/LNP immunized mice
Next, the global transcriptomic changes induced by mRNA/LNP immunization in liver-associated macrophages on Day 7 after immunization were investigated by RNA sequencing. Analysis of the differentially expressed genes (DEGs) in macrophages isolated from OVA mRNA/LNP-immunized mice revealed that 19 genes and 535 genes were upregulated and downregulated, respectively, compared with those in the PBS control group (|log2FC| >1 or <-1, p < 0.05) (Fig. 3A and Supplementary Table 1). Based on the unsupervised clustering, 3 different gene clusters were identified (C1-C3) (Supplementary Fig. 2). Genes in cluster C1 were uniquely upregulated in OVA mRNA/LNP-immunized mice while genes in cluster C3 were uniquely downregulated in OVA mRNA/LNP-immunized mice (Fig. 3B). Genes in cluster C2 did not show such robust differences between OVA and the control group and thus were not subjected to further analysis.

Transcriptomic profiles of liver-associated macrophages from OVA mRNA/LNP-immunized mice. Total RNA from F4/80+ liver-associated macrophages was subjected to RNA sequencing (n = 3 for the control PBS or mRNA/LNP-immunized group). The data were analyzed as described in the Methods section. (A) The volcano plot and (B) the heatmap depicting DEGs between the control and mRNA/LNP-vaccinated groups. DEGs were filtered using |log2FC| > 1 or <-1. The dark blue spots represent genes downregulated in the OVA-treated groups, as depicted in the refined heatmap depicted in (B). The light blue spots represent genes downregulated in the OVA-treated groups from the initial analysis shown in the Supplementary Fig. 2. (C) GO terms associated with DEGs of cluster C1 are shown. (D) GO terms are shown associated with DEGs of cluster C3 are shown (FDR < 0.05). The KEGG database was used for KEGG pathway analysis48,49,50.
Genes associated with macrophage effector functions, such as pro-inflammatory cytokines Il6, Tnf, the chemokines Cxcl13, Cxcl1, Cxcl2 and scavenger receptor Marco, were identified among the upregulated genes. Furthermore, genes involved in antigen presentation, H2-Ab1 and H2-Eb1, were upregulated (Fig. 3B). Among the downregulated genes, genes encoding extracellular matrix (ECM) proteins, including collagen family proteins (Col3a1, Col1a1, Col1a2, Col4a1, Col4a2, Col13a1) were prominent, while macrophage M2a marker Chi3l1 was downregulated (Fig. 3B).
Gene Ontology (GO) enrichment analysis revealed that the C1 cluster contained IL-6/JAK/STAT3 and TNFα signaling/NF-κB hallmarks as the top overrepresented hallmark, which correlated well with the upregulation of both cytokines in OVA mRNA/LNP-immunized mice (Fig. 3C). Among the KEGG pathways enriched in C3, PPAR signaling pathway and TGF-beta signaling pathway were found (Fig. 3D). Taken together, these findings indicate that mRNA/LNP immunization via the intramuscular route induces liver-associated macrophages to adopt proinflammatory phenotypes while decreasing anti-inflammatory function on Day 7 after mRNA/LNP administration.
To understand the activation status of macrophages, MacSpectrum was used to analyze and identify heterogeneous subpopulations and their activation states with condition-specific signature genes18. The results showed that macrophages from the OVA mRNA/LNP group were mainly M1-like cells, whereas macrophages from the PBS group were in a pre-activation state (Fig. 4A). These results are consistent with the GO and GSEA results described above. To corroborate this finding, we compared the expression of several key proinflammatory cytokine genes between the mRNA/LNP and PBS groups using qRT-PCR. The results showed that liver-associated macrophages from the mRNA/LNP group expressed higher levels of Il1b, Il6 and Tnf (Fig. 4B).

Phenotypic characterization of F4/80+ liver-associated macrophages. (A) MacSpectrum characterization was performed using the RNA-seq dataset. The macrophage subsets on the MacSpectrum plot were designated A, “M2-like”; B, “M1-like”; C, “transitional M1-like”; and D, “preactivation”. (B) The expression of inflammatory cytokines was measured by qRT‒PCR. Each bar represents the mean ± SEM (n = 3 per group). Statistical analysis was performed using unpaired two-tailed Student’s t test. * indicates statistical significance at p < 0.05.
H3K4me3 profiles of liver-associated macrophages upon mRNA/LNP immunization
To gain insight into the impact of mRNA/LNPs on the epigenetic profiles of liver-associated macrophages, the CUT&RUN assay and sequencing were performed, focusing on the H3K4me3 profile. The enrichment profiles of this epigenetic mark are summarized in Fig. 5A,B. Compared with those in the PBS group, three clusters of enriched loci in which H3K4me3 was significantly enriched were identified: high, intermediate and low (Clusters 1–3). Loci in Cluster 1 showed enhanced H3K4me3 both upstream and downstream of the TSS (+/- 3 Kbp), which included the coding regions. Cluster 2 had H3K4me3 concentrated around the transcription start sites (TSSs), and Cluster 3 showed little change in H3K4me3 between the groups.

Profiles of H3K4me3 in F4/80+ liver-associated macrophages from CUT&RUN analysis. (A) The mapped fragment size distribution from 4 samples subjected to CUT&RUN sequencing. (B) Heatmap showing the genomic regions with enriched H3K4me3 at 3 Kbp flanking the transcriptional start site (TSS) of genes. Gene Ontology (GO) analysis of overrepresented genes from Cluster I is shown. (C) Overrepresentation of pathways in loci of Cluster 1 in the WikiPathway, KEGG, Reactome and Panther databases are shown. The KEGG database was used for KEGG pathway analysis48,49,50.
The loci in Cluster 1 were further analyzed to identify overrepresented loci (Fig. 5B,C). Among the overrepresented sequences were those of genes related to the response to cytokines (biological process), which may reflect the inflammatory nature of mRNA/LNP-stimulated macrophages. Among the enriched pathways, pathways related to the cytokines TNFa and IL-6 were identified, indicating that macrophages in the liver may be epigenetically poised to initiate the transcription of inflammatory cytokines after mRNA/LNP immunization (Fig. 5C). The H3K4me3 profiles of Tnf, Il6 and Marco are shown in Fig. 6. These genes are upregulated in liver-associated macrophages from mRNA/LNP mice according to the RNA-seq data described above. The highly enriched H3K4me3 in the promoter regions provides conducive chromatin structure for active gene expression and may reflect the transcription of those genes.

Genome tracks of H3K4me3 profiles in F4/80+ liver-associated macrophages. Genome tracks showing H3K4me3 enrichment at the promoter regions of Tnf, IL6 and Marco are shown. The experiment was conducted with 2 biological replicates.
Motif identification revealed potential binding sites for GABPA, an ETS transcription factor; Sp2/3, an Sp subfamily transcription factor; and CREB1, a cAMP response element binding protein (Supplementary Fig. 3). This finding suggested that mRNA/LNP immunization may induce conducive conditions for binding transcription factors that regulate gene expression. Taken together, the study results showed that immunization with mRNA/LNPs via the intramuscular route influences liver-associated macrophages to adopt an M1-like phenotype and alters histone modification profiles (active histone marks), which may affect the macrophage response to future stimulation and potentiate adaptive immune functions.
Effect of LNP or mRNA/LNP on macrophage responses to LPS
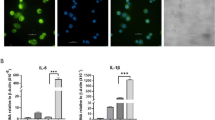
To investigate whether LNP or mRNA/LNP directly trigger responses in macrophages, bone marrow derived macrophages (BMMs) were treated with empty LNP or mRNA/LNP at various doses for 24 h and two inflammatory cytokines (Fig. 7A, TNFα and IL-6 were measured in the culture supernatants. As shown in Fig. 7B, LNP or mRNA/LNP were able to induce TNFα production higher than the untreated control. On the other hand, only mRNA/LNP was able to induce IL-6 production (Fig. 7D). The levels of both cytokines, however, were much lower than LPS stimulation condition. Because mRNA/LNP immunization resulted in active histone mark in liver-associated macrophages, the impact of LNP or mRNA/LNP on innate immune memory was examined using BMM model. After exposure to LNP or mRNA/LNP for 24 h, cells were rested for 48 h before being subjected to LPS stimulation (Fig. 7A). Treatment with β-glucan or LPS was used as the control for trained immune response and tolerance response, respectively19. The amounts of TNFα and IL-6 were used as a readout for innate immune memory response. The results as shown in Fig. 7C,E, exposure to LNP or mRNA/LNP before stimulation with LPS did not change the amounts of both cytokines, whereas treatment with β-glucan further increased LPS-induced cytokine production while treatment with LPS reduced those levels. Taken together, LNP or OVA/LNP exhibited the ability to induce weak but detectable inflammatory response, but either treatment did not affect response after LPS restimulation.

Effect of LNP or mRNA/LNP on Macrophages. (A) Experimental scheme to test the impact of LNP or mRNA/LNP on BMMs. The level of TNFα (B, C) or IL-6 (D, E) were measured by ELISA. Data are representative of four independent experiments. The letter a and b on the bar graph indicated p < 0.001 and p < 0.0001 compared to the unstimulated control, respectively.
Discussion
mRNA technologies provide groundbreaking tools for combating infectious diseases and cancer and for therapeutic purposes20. The delivery of an mRNA for in vivo applications relies mainly on using LNPs, which play crucial roles in intracellular mRNA delivery and as adjuvants in vaccines11. Thus, mRNA/LNP administration can induce tissue responses that alter the homeostasis of affected organs. Our study aimed to investigate the impact of mRNA/LNPs administered via the intramuscular route on macrophages in the liver. mRNAs such as luciferase reporter genes can be detected at the injection site (muscle) and in other tissues, including the liver, spleen, and lymph nodes, with rapid kinetics (within hours) that can last 3–5 days21,22. We designed this study to investigate the post-acute impact of mRNA/LNPs on liver-associated macrophages on Day 7 after one dose of mRNA/LNPs.
The targeted cells in this study were F4/80+ macrophages residing in the liver. Liver macrophages are mainly composed of embryonic-origin KCs and monocyte-derived KCs. The latter is recruited upon depletion of the former, such as in liver injuries23. Our study did not distinguish between embryonic KCs and newly recruited monocyte-derived macrophages. The pools of F4/80+ cells associated with the liver were subjected to transcriptomic analysis to obtain a global view of the gene expression profiles of these cells. On Day 7, we found a total of 554 genes that were considered differentially expressed, where more genes were downregulated. The number of upregulated genes (19 genes) was relatively small, possibly because the time of cell harvesting from the livers was past the acute phase.
GO enrichment analysis identified the enrichment of pro-inflammatory cytokines (IL-6/JAK/STAT3, TNFα/NF-κB pathways) and cytokines related to cell-mediated immune response (interferon gamma response, interferon alpha response) in macrophages from the mRNA/LNP group. This result indicated that intramuscular immunization of mRNA/LNPs triggers inflammatory responses in the livers that favor cell-mediated immune effector functions. The effect observed here may be direct from mRNA/LNPs on macrophages or indirect via inflammatory cytokines such as interferon gamma produced by other immune cells in the liver.
Among upregulated genes, pro-inflammatory cytokines, Tnf, Il6, Il1b were significantly increased in macrophages from OVA mRNA/LNPs. This result was expected because mRNA/LNP immunization is known to induce inflammatory response24. Furthermore, several chemokines were upregulated (Cxcl1, Cxcl2, Cxcl13), indicating the ability of mRNA/LNP to recruit immune cells into the liver such as neutrophils25. Overall profiles of F4/80+ liver associated macrophages categorized them to be M1-like phenotypes at 7 days post immunization. This result indicated that mRNA/LNP immunization via intramuscular route conditioned liver-associated macrophages to be pro-inflammatory phenotypes.
The expression of genes encoding ECM proteins was downregulated in the mRNA/LNP groups, while that of Mmp9 was upregulated. The data suggested that mRNA/LNP administration triggered changes in the tissue environment in the liver that may involve remodeling. KCs are known to play a role in liver fibrosis progression and regression, and during liver injury, ECM remodeling is important for supporting liver progenitor cell proliferation and invasion26. Our data and those from studies on the role of monocyte-derived KCs may suggest that after mRNA/LNP exposure, monocyte-derived KCs may be recruited to the liver instead of embryonic KCs, reflecting reduced ECM production8.
LNP is a key component of mRNA delivery system. It has been shown that LNP acts as adjuvant upon co-administration with mRNA or protein antigens27. This study further elucidated that the intrinsic adjuvant activity of LNP formulation promotes strong B cell response and T follicular helper cells via IL-6 induction. Our in vitro data collaborated with this finding that LNP or mRNA/LNP weakly induced TNFα and IL-6 production in BMMs but the cytokine levels were much lower than that induced by strong inflammatory stimulus LPS.
Innate immune memory is controlled by epigenetic reprogramming and metabolic rewiring9. The epigenetic reprogramming of histone proteins allows cells to be in a complex chromatin state readily responsive to unrelated insults. One of our study aims was to elucidate the epigenetic changes to liver-associated macrophages caused by mRNA/LNP administration. The active histone marker H3K4me3 is associated with transcriptionally active/poised chromatin. This histone mark among others is reported to regulate trained immunity in macrophages28,29. The CUT&RUN sequencing data from our study revealed two main clusters of cis-regulatory loci in which H3K4me3 was enriched at high and intermediate levels by mRNA/LNPs. Some loci in these clusters directly regulated the expression of genes (such as Tnf and Marco), whereas others did not directly link to the upregulated genes. This result may indicate that mRNA/LNPs induce epigenetic changes that prepare cells to respond to stimulation by keeping chromatin in a “poised” state. The study of other histone marks, such as H3K27ac or H3K4me1, may provide a more comprehensive view of how mRNA/LNPs induce innate immune memory. The involvement of mRNA/LNPs in inducing trained immunity may be critical for the future design of mRNA-based vaccines or therapeutic tools.
In an effect to investigate whether exposure to LNP or mRNA/LNP alters response to secondary stimuli, an in vitro innate immune memory assay was performed. Exposure to β-glucan before LPS restimulation heightens inflammatory cytokine production which is the characteristic of trained immunity30. In contrast, successive LPS exposure dampens inflammatory cytokine level which is considered tolerance response30. In this study, the in vitro study of innate immune memory induced by LNP or mRNA/LNP did not show enhance or dampen effect upon LPS restimulation. However, other factors such as the amounts of LNP or mRNA/LNP, cytokine milieus may affect this phenomena and further study in an in vivo model is needed to clarify whether exposure to LNP or mRNA/LNP may epigenetically modify innate immune response that result in trained or tolerance response.
The current study has some limitations that may need to be addressed in future studies. In this work, we did not address the direct or indirect impact of mRNA/LNP on macrophages in the liver. The impact of mRNA/LNP administered via intramuscular injection may influence liver associated macrophages directly by being uptake by such cells. Alternatively, the cytokines produced as a result of mRNA/LNP administration may indirectly influence macrophages in the liver. How embryonic KCs and monocyte-derived KCs respond to mRNA/LNPs and influence the immune response is unknown. How long this epigenetic marker identified in our study was maintained is also an intriguing question. Finally, how the response induced in liver-associated macrophages influences the adaptive immune response needs further detailed investigation.
Methods
Mice
C57BL/6 mice (female aged 8–12 weeks) were purchased from Siam Nomura International (Thailand) and housed at Chulalongkorn University Laboratory Animal Center (CULAC) under animal protocol No. 2,173,040, which was approved by CULAC IACUC. All experiments involving mice were performed according to the guidelines issued by the IACUC. Isoflurane was used for anaesthetizing the mice where indicated. The mice were humanely euthanized via carbon dioxide inhalation. The results obtained in this study were reported in accordance with ARRIVE guidelines.
In vitro transcription for mRNA and lipid nanoparticle (LNP) formulation
An mRNA encoding OVA was prepared using previously published pUC-ccTEV-ovalbumin-A101 (OVA)13. The plasmid was propagated in Escherichia coli (Stbl3) and extracted using a ZymoPureII plasmid midiprep kit (Zymo Research, Irvine, CA, USA). Nucleoside-modified mRNA was produced by in vitro transcription (IVT) by substituting uridine triphosphate (UTP) with N1-methylpseudouridine (m1Ψ) triphosphate (TriLink, Biotechnologies, San Diego, CA, USA). The reaction was carried out with T7 RNA polymerase (MegaScript, Thermo Fisher Scientific, MA, USA) on a linearized plasmid. mRNA capping was performed by the trinucleotide cap1 analog CleanCap (TriLink Biotechnologies). The capped mRNA was purified by cellulose column purification31. IVT mRNA was analyzed on agarose to determine its integrity. Additional quality control was performed as described previously to ensure the absence of double-stranded RNA (dsRNA) and endotoxin contamination before encapsulation into LNPs21. mRNA encapsulation was performed by Genevant Sciences Corporation (Vancouver, British Columbia, Canada). The proprietary lipid and LNP compositions are similar to those previously reported32 and are described in the patent application WO2020097540A1. The LNP-encapsulated mRNA was stored at − 80 °C until use.
Immunization with mRNA/LNPs
Mice were randomly divided into 2groups (6 mice per group): the mRNA immunization group and placebo group (PBS). Mice were immunized via the intramuscular (i.m.) route with mRNA encoding OVA formulated with LNPs (10 µg per mouse) or the PBS control. Serum was collected to monitor the specific antibody response.
Specific antibody titer
Serum was collected to measure the level of IgG antibodies specific to OVA by ELISA. Ovalbumin (InvivoGen, San Diego, CA, USA) (10 µg/ml; 100 µL/well) was added to Maxisorb 96-well plates. Twofold serial dilutions of mouse serum were added in duplicate. After 2 h of incubation at RT, the plates were vigorously washed with washing buffer (PBS + 0.5% Tween 20, PBST). Goat anti-mouse IgG-HRP (Jackson ImmunoResearch, West Grove, PA, USA) was added, and the plates were incubated for an additional 30 min. The substrate TMB (Sigma Aldrich, St. Louis, MO, USA) was added, and the reaction was stopped with 1 M H2SO4. The absorbance at 450 and 620 nm was measured on a microplate reader (Thermo Fisher Scientific, Waltham, MA, USA). The endpoint titers were determined as the highest dilution of sera that had an OD greater than the cutoff value. The cutoff value was calculated from the OD of preimmunized sera at a dilution of 1:8033.
Isolation of F4/80+ liver-associated macrophages
On Day 7 post immunization, the mice were sacrificed as described above, and liver-resident macrophages were isolated following the protocol described by Lynch et al. with some modifications34. Briefly, mice were placed under deep anesthesia, and liver perfusion through the inferior vena cava was performed using PBS. After perfusion, the livers were harvested and placed in DMEM, finely chopped using a razor blade and digested in 5 ml of enzyme mixture (DMEM containing collagenase type IV (0.85 mg/mL; Sigma Aldrich) and dispase II (1 mg/mL; Sigma Aldrich)) for 25 min at 37 °C in an orbital shaker. Digested livers were filtered through 100-µm cell strainers (Corning, NY, USA), and the filtrates containing cells were serially washed in 40 mL and 30 mL of DMEM. The cells were harvested by centrifugation at 300 × g for 5 min. RBC lysis buffer was added to the cell pellets, and the solutions were incubated for 5 min, followed by resuspension and washing in FACS buffer (PBS with 0.5% BSA and 2 M EDTA). The cell pellets were resuspended and filtered through a 40-µm cell strainer. Single-cell suspensions were incubated with MojoSortTM Mouse F4/80 Nanobeads (BioLegend, San Diego, CA, USA) for 15 min on ice. Positive selection of F4/80+ macrophages was performed using MojoSort™ Magnetic Cell Separation (BioLegend). Livers from at least 3 mice from each treatment group were subjected to isolation of F4/80+ liver-associated macrophages.
Flow cytometry
To evaluate the purity of the F4/80+ cells obtained from magnetic bead sorting, the cells were stained with anti-mouse CD45-APC Cy7, anti-mouse F4/80-AlexaFluor 488 and anti-mouse CD11b-PE antibodies (all antibodies were obtained from BioLegend). Dead/live cells were distinguished with 7-AAD (BioLegend). The cells were incubated for 15 min at 4 °C in the dark and analyzed by a Beckman Coulter CytoFLEX Flow Cytometer (Beckman Coulter, Brea, CA, USA). The acquired data were analyzed by FlowJo™ V10 software (TreeStar BD Biosciences).
RT-qPCR
Total RNA from liver-associated macrophages from mice receiving OVA mRNA/LNPs or PBS was extracted using an RNeasy Mini Kit (Qiagen, Hilden, Germany) and reverse transcribed using RevertAid reverse transcriptase (Thermo Fisher Scientific) following the manufacturer’s protocol. Five hundred nanograms of RNA per sample was converted to cDNA, which was used for quantitative PCR using iQ SYBR Green SuperMix (Bio-Rad, Hercules, CA, USA) according to the manufacturer’s instructions. The primers used in this study are listed in Table 1. The relative expression of all target genes was normalized to GAPDH expression by the 2−∆∆ct method and calculated by comparison with that of bone marrow-derived macrophages (Clec4f) or liver-associated macrophages from mice receiving PBS for other genes35.
RNA sequencing and data analysis
Three samples from each treatment (PBS or mRNA/LNP) were subjected to RNA sequencing. Total RNA was extracted using an RNeasy Mini Kit (Qiagen, USA) following the manufacturer’s protocol as described above. The quality and concentrations of RNA were measured using a NanoDrop™ 2000 spectrophotometer (Thermo Fisher Scientific). cDNA libraries were prepared using a DNBSEQ Eukaryotic Strand-specific mRNA library kit (BGI Genomics, Shenzhen, China). The quantity and quality of the libraries were determined using a 2100 Bioanalyzer. Sequencing was performed on a DNBseq platform (2 × 150 bp paired-end reads). The sequences were subjected to cleaning and quality assessment using FastQC. Subsequently, the sequences were mapped and aligned against Mus musculus reference genome GRCm39 using STAR40. The quantification of the mapped outputs was performed using Kallisto41. The read counts were normalized and analyzed for genes that were differentially expressed (DEGs) using edgeR42 and limma-voom packages43,44. Genes were considered statistically significant (nominal p-value < 0.05) if their log2 fold change was less than − 1 or larger than 1, representing down- or up-regulation, respectively.
The nominal p-value reported in the supplementary figures represents the uncorrected p-value obtained from the limma statistical model. While multiple genes showed nominal p-values below 0.05, only one gene (Gm10337, a predicted protein-coding gene) achieved an FDR-adjusted p-value (i.e., q-value) below 0.05. The remaining genes exhibited FDR values between 0.1 and 0.6, indicating a considerable risk of 10–60% false positives. To address this and ensure the robustness of our findings, we focused on genes demonstrating consistent expression changes across all biological replicates. Visual analysis of the heatmap in Supplementary Fig. 2 revealed that Cluster 2 contained genes predominantly upregulated in a single sample, potentially introducing bias. Consequently, we prioritized genes in Clusters 1 and 3 for further analysis, as shown in Fig. 3B. Importantly, key genes identified in this analysis, such as Il6 and Tnf, were further validated using qRT-PCR (Fig. 4B), providing additional confidence in their significance.
Hierarchical clustering of DEGs was performed (Euclidean distance and Ward.D2 method) using ComplexHeatmap package45. The up- and down-regulated DEGs from selected clusters were analyzed for their functions using Gene Ontology46,47, KEGG48,49,50, and MSigDB hallmark51 databases using enrichr package52,53,54. In gene functional analysis, a gene set was statistically enriched if its p-value was less than 0.05. The analyses were conducted on triplicate samples. All analyses were carried out using R program version 4.3.2 unless otherwise specified.
MacSpectrum is a two-index platform utilizing macrophage polarization (MPI) and activation-induced macrophage differentiation (AMDI) indices that employ distinct gene signatures to correlate with macrophage activation states18. MPI quantifies the overall inflammatory status of the macrophage population. A higher MPI indicates a more pro-inflammatory (M1-like) phenotype. On the other hand, AMDI measures the extent of macrophage differentiation towards a specific activation state. MacSpectrum compares the gene expression patterns in the input data to the signature genes in its reference database and calculates a score for each signature gene based on its expression level in the samples. MacSpectrum is originally designed to characterize adipose tissue macrophages into M1-like, M2-like, transitional, and pre-activation phenotypes, we adapted it to our context. Briefly, significant genes (nominal p-value < 0.05, absolute log2 fold-change > 1) and their expression levels were formatted for the MacSpectrum web-based software (https://macspectrum.uconn.edu). The resulting outputs were then downloaded and visualized using R to establish the M1-like character of the macrophages in our study.
CUT&RUN and CUT&RUN-qPCR data analysis
The CUT&RUN assay was performed using a CUT&RUN Assay Kit (Cell Signaling Technology, Danvers, MA, USA) according to the manufacturer’s instructions. Briefly, approximately 1 × 106 cells were harvested, washed, and fixed with 37% formaldehyde to a final concentration of 0.1%. The cells were then incubated with activated concanavalin A-coated magnetic beads and permeabilized with digitonin. The complexes of bead-bound cells were incubated with antibodies (rabbit anti-H3K4me3 and normal rabbit IgG as a negative control) at 4 °C overnight with rotation. pAG-MNase was accordingly introduced to the cell: bead suspension and incubated at 4 °C for 1 h before DNA digestion and diffusion. The input DNA was prepared and unfragmented separately via sonication. DNA was purified using a DNA purification kit (Cell Signaling Technology). DNA fragmentation was visualized using gel electrophoresis and a bioanalyzer (Agilent Technology, 2100). Fragmented input DNA and digested DNA were used to amplify rpl30, a housekeeping gene positively related to H3K4me3, using qPCR according to the manufacturer’s protocol. Fold enrichments were normalized and calculated based on the total amount of 10% input and presented as a relative quantification using the 2−∆∆ct method.
CUT&RUN sequencing analyses
Reads were aligned to the Mus musculus reference genome mm10 using Bowtie2. The SAM files were converted and sorted to the BAM format using SAMtools. PCR duplicates were removed using Picard tools. The BAM files were converted to bigwig files by deepTools for visualization and heatmaps. For comparison across samples with a spike-in genome, the bigwig files were normalized to spike-in genome coverage (Saccharomyces cerevisiae R64-1-1). Genome browser tracks were visualized by Integrative Genomics Viewer (IGV) and pyGenomeTracks. Heatmap clustering was generated using deepTools by k-means clustering.
Assay for innate immune memory response in macrophages
BMMs were prepared from bone marrow cells of C57BL/6 mice as described elsewhere55. To investigate the effect of LNP or mRNA/LNP on macrophages, BMMs were treated with various amounts of empty LNP or mRNA/LNP for 24 h, and the culture supernatants were subjected to ELISA for TNFα and IL-6 using ELISA MAX™ Deluxe Set for mouse TNF-α or IL-6 (BioLegend, USA). For β-glucan-induced trained immune response or LPS-induced tolerance response, previous protocol as described by Benjaskulluecha et al. were followed and the scheme was depicted in Fig. 7A19.
Data analysis and statistics
All in vivo experiments were performed using six mice from each treatment. All in vitro experiments were performed in triplicates with two independent experiments. Statistical analyses were performed using GraphPad Prism version 9.0. The data were compared using unpaired two-tailed Student’s t test. *, **, and *** indicate P < 0.05, 0.01, and 0.001, respectively. All the data were represented as means ± SEM.
Data availability
The datasets generated and analyzed during the current study are available in the NCBI’s Gene Expression Omnibus repository. The RNA-seq and CUT&RUN-seq data are accessible through GEO Series accession number GSE267045, and GSE267043, respectively.
References
Ginhoux, F., Schultze, J. L., Murray, P. J., Ochando, J. & Biswas, S. K. New insights into the multidimensional concept of macrophage ontogeny, activation and function. Nat. Immunol. 17, 34–40. https://doi.org/10.1038/ni.3324 (2016).
Lavin, Y. et al. Tissue-resident macrophage enhancer landscapes are shaped by the local microenvironment. Cell 159, 1312–1326. https://doi.org/10.1016/j.cell.2014.11.018 (2014).
Okabe, Y. & Medzhitov, R. Tissue biology perspective on macrophages. Nat. Immunol. 17, 9–17. https://doi.org/10.1038/ni.3320 (2016).
Bennett, H., Troutman, T. D., Sakai, M. & Glass, C. K. Epigenetic regulation of Kupffer cell function in health and disease. Front. Immunol. 11, 609618. https://doi.org/10.3389/fimmu.2020.609618 (2020).
Hoeffel, G. et al. C-Myb(+) erythro-myeloid progenitor-derived fetal monocytes give rise to adult tissue-resident macrophages. Immunity 42, 665–678. https://doi.org/10.1016/j.immuni.2015.03.011 (2015).
Scott, C. L. et al. Bone marrow-derived monocytes give rise to self-renewing and fully differentiated Kupffer cells. Nat. Commun. 7, 10321. https://doi.org/10.1038/ncomms10321 (2016).
MacParland, S. A. et al. Single cell RNA sequencing of human liver reveals distinct intrahepatic macrophage populations. Nat. Commun. 9, 4383. https://doi.org/10.1038/s41467-018-06318-7 (2018).
Li, W., Yang, Y., Yang, L., Chang, N. & Li, L. Monocyte-derived Kupffer cells dominate in the Kupffer cell pool during liver injury. Cell Rep. 42, 113164. https://doi.org/10.1016/j.celrep.2023.113164 (2023).
Netea, M. G. et al. Defining trained immunity and its role in health and disease. Nat. Rev. Immunol. https://doi.org/10.1038/s41577-020-0285-6 (2020).
Sun, S. & Barreiro, L. B. The epigenetically-encoded memory of the innate immune system. Curr. Opin. Immunol. 65, 7–13. https://doi.org/10.1016/j.coi.2020.02.002 (2020).
Pardi, N., Hogan, M. J., Porter, F. W. & Weissman, D. mRNA vaccines - A new era in vaccinology. Nat. Rev. Drug Discov. 17, 261–279. https://doi.org/10.1038/nrd.2017.243 (2018).
Carrasco, M. J. et al. Ionization and structural properties of mRNA lipid nanoparticles influence expression in intramuscular and intravascular administration. Commun. Biol. 4, 956. https://doi.org/10.1038/s42003-021-02441-2 (2021).
Sittplangkoon, C. et al. mRNA vaccine with unmodified uridine induces robust type I interferon-dependent anti-tumor immunity in a melanoma model. Front. Immunol. 13, 983000. https://doi.org/10.3389/fimmu.2022.983000 (2022).
Kranz, L. M. et al. Systemic RNA delivery to dendritic cells exploits antiviral defence for cancer immunotherapy. Nature 534, 396–401. https://doi.org/10.1038/nature18300 (2016).
Rizvi, F. et al. Murine liver repair via transient activation of regenerative pathways in hepatocytes using lipid nanoparticle-complexed nucleoside-modified mRNA. Nat. Commun. 12, 613. https://doi.org/10.1038/s41467-021-20903-3 (2021).
Smith, A. R. et al. Transient growth factor expression via mRNA in lipid nanoparticles promotes hepatocyte cell therapy to treat murine liver diseases. bioRxiv, https://doi.org/10.1101/2024.01.11.575286 (2024).
Jiang, Y. et al. Kupffer cell receptor CLEC4F is important for the destruction of desialylated platelets in mice. Cell Death Differ. 28, 3009–3021. https://doi.org/10.1038/s41418-021-00797-w (2021).
Li, C. et al. Single cell transcriptomics based-MacSpectrum reveals novel macrophage activation signatures in diseases. JCI Insight 5, https://doi.org/10.1172/jci.insight.126453 (2019).
Benjaskulluecha, S. et al. Screening of compounds to identify novel epigenetic regulatory factors that affect innate immune memory in macrophages. Sci. Rep. 12, 1912. https://doi.org/10.1038/s41598-022-05929-x (2022).
Wang, Y. S. et al. mRNA-based vaccines and therapeutics: An in-depth survey of current and upcoming clinical applications. J. Biomed. Sci. 30, 84. https://doi.org/10.1186/s12929-023-00977-5 (2023).
Pardi, N. et al. Expression kinetics of nucleoside-modified mRNA delivered in lipid nanoparticles to mice by various routes. J. Control Release 217, 345–351. https://doi.org/10.1016/j.jconrel.2015.08.007 (2015).
Trougakos, I. P. et al. Adverse effects of COVID-19 mRNA vaccines: The spike hypothesis. Trends Mol. Med. 28, 542–554. https://doi.org/10.1016/j.molmed.2022.04.007 (2022).
Guilliams, M. & Scott, C. L. Does niche competition determine the origin of tissue-resident macrophages?. Nat. Rev. Immunol. 17, 451–460. https://doi.org/10.1038/nri.2017.42 (2017).
Matsumura, T., Takano, T. & Takahashi, Y. Immune responses related to the immunogenicity and reactogenicity of COVID-19 mRNA vaccines. Int. Immunol. 35, 213–220. https://doi.org/10.1093/intimm/dxac064 (2023).
Zhou, C., Gao, Y., Ding, P., Wu, T. & Ji, G. The role of CXCL family members in different diseases. Cell Death Discov. 9, 212. https://doi.org/10.1038/s41420-023-01524-9 (2023).
Van Hul, N. et al. Kupffer cells influence parenchymal invasion and phenotypic orientation, but not the proliferation, of liver progenitor cells in a murine model of liver injury. Am. J. Pathol. 179, 1839–1850. https://doi.org/10.1016/j.ajpath.2011.06.042 (2011).
Alameh, M. G. et al. Lipid nanoparticles enhance the efficacy of mRNA and protein subunit vaccines by inducing robust T follicular helper cell and humoral responses. Immunity 54, 2877–2892. https://doi.org/10.1016/j.immuni.2021.11.001 (2021).
Foster, S. L., Hargreaves, D. C. & Medzhitov, R. Gene-specific control of inflammation by TLR-induced chromatin modifications. Nature 447, 972–978. https://doi.org/10.1038/nature05836 (2007).
Kleinnijenhuis, J. et al. Bacille Calmette-Guerin induces NOD2-dependent nonspecific protection from reinfection via epigenetic reprogramming of monocytes. Proc. Natl. Acad. Sci. USA 109, 17537–17542. https://doi.org/10.1073/pnas.1202870109 (2012).
Divangahi, M. et al. Trained immunity, tolerance, priming and differentiation: Distinct immunological processes. Nat. Immunol. 22, 2–6. https://doi.org/10.1038/s41590-020-00845-6 (2021).
Baiersdorfer, M. et al. A facile method for the removal of dsRNA contaminant from in vitro-transcribed mRNA. Mol. Ther. Nucleic Acids 15, 26–35. https://doi.org/10.1016/j.omtn.2019.02.018 (2019).
Prompetchara, E. et al. Immunogenicity and protective efficacy of SARS-CoV-2 mRNA vaccine encoding secreted non-stabilized spike in female mice. Nat. Commun. 14, 2309. https://doi.org/10.1038/s41467-023-37795-0 (2023).
Pisuttinusart, N. et al. Immunogenicity of a recombinant plant-produced respiratory syncytial virus F subunit vaccine in mice. Biotechnol Rep (Amst.) 41, e00826. https://doi.org/10.1016/j.btre.2023.e00826 (2024).
Lynch, R. W. et al. An efficient method to isolate Kupffer cells eliminating endothelial cell contamination and selective bias. J. Leukoc. Biol. 104, 579–586. https://doi.org/10.1002/JLB.1TA0517-169R (2018).
Livak, K. J. & Schmittgen, T. D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 25, 402–408. https://doi.org/10.1006/meth.2001.1262 (2001).
Viceconte, N. et al. Neuromelanin activates proinflammatory microglia through a caspase-8-dependent mechanism. J. Neuroinflammation 12, 5. https://doi.org/10.1186/s12974-014-0228-x (2015).
Lai, C. F. et al. Blockade of cysteine-rich protein 61 attenuates renal inflammation and fibrosis after ischemic kidney injury. Am. J. Physiol. Renal. Physiol. 307, F581-592. https://doi.org/10.1152/ajprenal.00670.2013 (2014).
Minashima, T., Campbell, K. A., Hadley, S. R., Zhang, Y. & Kirsch, T. The role of ANK interactions with MYBBP1a and SPHK1 in catabolic events of articular chondrocytes. Osteoarthritis Cartilage 22, 852–861. https://doi.org/10.1016/j.joca.2014.04.008 (2014).
Sasidharan Nair, V., Toor, S. M., Taha, R. Z., Shaath, H. & Elkord, E. DNA methylation and repressive histones in the promoters of PD-1, CTLA-4, TIM-3, LAG-3, TIGIT, PD-L1, and galectin-9 genes in human colorectal cancer. Clin. Epigenetics 10, 104. https://doi.org/10.1186/s13148-018-0539-3 (2018).
Dobin, A. et al. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. https://doi.org/10.1093/bioinformatics/bts635 (2013).
Bray, N. L., Pimentel, H., Melsted, P. & Pachter, L. Near-optimal probabilistic RNA-seq quantification. Nat. Biotechnol. 34, 525–527. https://doi.org/10.1038/nbt.3519 (2016).
Robinson, M. D., McCarthy, D. J. & Smyth, G. K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140. https://doi.org/10.1093/bioinformatics/btp616 (2010).
Smyth, G. K. Linear models and empirical bayes methods for assessing differential expression in microarray experiments. Stat. Appl. Genet. Mol. Biol. 3, 3. https://doi.org/10.2202/1544-6115.1027 (2004).
Law, C. W., Chen, Y., Shi, W. & Smyth, G. K. voom: Precision weights unlock linear model analysis tools for RNA-seq read counts. Genome Biol. 15, R29. https://doi.org/10.1186/gb-2014-15-2-r29 (2014).
Gu, Z., Eils, R. & Schlesner, M. Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics 32, 2847–2849. https://doi.org/10.1093/bioinformatics/btw313 (2016).
Ashburner, M. et al. Gene ontology: Tool for the unification of biology. The gene ontology consortium. Nat. Genet. 25, 25–29. https://doi.org/10.1038/75556 (2000).
Gene Ontology, C. The gene ontology resource: Enriching a gold mine. Nucleic Acids Res. 49, D325–D334. https://doi.org/10.1093/nar/gkaa1113 (2021).
Kanehisa, M. & Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 28, 27–30. https://doi.org/10.1093/nar/28.1.27 (2000).
Kanehisa, M. Toward understanding the origin and evolution of cellular organisms. Protein Sci. 28, 1947–1951. https://doi.org/10.1002/pro.3715 (2019).
Kanehisa, M., Furumichi, M., Sato, Y., Kawashima, M. & Ishiguro-Watanabe, M. KEGG for taxonomy-based analysis of pathways and genomes. Nucleic Acids Res. 51, D587–D592. https://doi.org/10.1093/nar/gkac963 (2023).
Liberzon, A. et al. The molecular signatures database (MSigDB) hallmark gene set collection. Cell Syst. 1, 417–425. https://doi.org/10.1016/j.cels.2015.12.004 (2015).
Chen, E. Y. et al. Enrichr: Interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinf. 14, 128. https://doi.org/10.1186/1471-2105-14-128 (2013).
Kuleshov, M. V. et al. Enrichr: A comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 44, W90-97. https://doi.org/10.1093/nar/gkw377 (2016).
Xie, Z. et al. Gene set knowledge discovery with Enrichr. Curr. Protoc. 1, e90. https://doi.org/10.1002/cpz1.90 (2021).
Palaga, T. et al. Notch signaling is activated by TLR stimulation and regulates macrophage functions. Eur. J. Immunol. 38, 174–183. https://doi.org/10.1002/eji.200636999 (2008).
Acknowledgements
This research is funded by Thailand Science Research and Innovation Fund Chulalongkorn University (FF67). C.P. is supported by a scholarship from the Science Achievement Scholarship of Thailand (SAST), Faculty of Science, Chulalongkorn University and “The 90th Anniversary Chulalongkorn University Fund (Ratchadaphiseksomphot Endowment Fund)” Chulalongkorn University. A.B. is supported by the Second Century Fund (C2F), Chulalongkorn University. This research (T.P. and T.P.K.) received funding support from the NSRF via the Program Management Unit for Human Resources & Institutional Development, Research Innovation (Grant No. B16F640117). This research is financially supported by the UK International Science Partnerships Fund (ISPF) Institutional Support Grant (ODA) awarded to the University of Sheffield (Prof. Tuck Seng Wong and Dr. Kang Lan Tee) with Chulalongkorn University (Prof. Kiat Ruxrungtham) as the international project partner.
Author information
Authors and Affiliations
Contributions
C.P. and P.K.W. designed and performed all the experiments, analyzed all the data, prepared all the figures and drafted the manuscript. A.B. and P.K.J. analyzed the NGS data. B.W. and T.P.K. performed ELISA and PCR and analyzed the data in some experiments. C.S./M.G. designed the plasmids and prepared mRNA by IVT. J.H. and K. L. formulated LNPs for mRNA. B.S. performed the in vivo experiments. T.P.G. and K.R. supervised the overall project, acquired grant funding, designed all experiments, analyzed the data, and prepared the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Pongma, C., Keawvilai, P., Boonmee, A. et al. Effect of mRNA formulated with lipid nanoparticles on the transcriptomic and epigenetic profiles of F4/80+ liver-associated macrophages. Sci Rep 15, 1146 (2025). https://doi.org/10.1038/s41598-025-85234-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-025-85234-5
