
Abstract
Breast cancer remains a global health burden, with an increase in deaths related to this particular cancer. Accurately predicting and diagnosing breast cancer is important for treatment development and survival of patients. This study aimed to accurately predict breast cancer using a dataset comprising 1208 observations and 3602 genes. The study employed feature selection techniques to identify the most influential predictive genes for breast cancer using machine learning (ML) models. The study used K-nearest Neighbors (KNN), random forests (RF), and a support vector machine (SVM). Furthermore, the study employed feature- and model-based importance and explainable ML methods, including Shapley values, Partial dependency (PDPS), and Accumulated Local Effects (ALE) plots, to explain the genes’ importance ranking from the ML methods. Shapley values highlighted the significance of some of the genes in predicting cancer presence. Model-based feature ranking techniques, particularly the Leaving-One-Covariate-In (LOCI) method, identified the ten most critical genes for predicting tumor cases. The LOCI rankings from the SVM and RF methods were aligned. Additionally, visualization methods such as PDPS and ALE plots demonstrated how individual feature changes affect predictions and interactions with other genes. By combining feature selection techniques and explainable ML methods, this study has demonstrated the interpretability and reliability of machine learning models for breast cancer prediction, emphasizing the importance of incorporating explainable ML approaches for medical decision-making.
Similar content being viewed by others
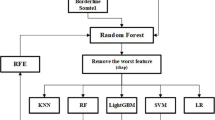


Introduction
Cancer remains a paramount global health challenge, marked by the uncontrolled proliferation and spread of abnormal cells throughout the body1. Its impact is profound, with approximately 19.3 million new cases and nearly 10 million deaths reported worldwide in 2020, spanning 185 countries and encompassing 36 different cancer types2. Among these, breast cancer stands as one of the most prevalent forms among women and ranks as the second leading cause of death in both developed and developing nations3. Recognizing the urgency of the situation, organizations like the World Health Organization and governments globally have strategically prioritized combating breast, cervical, and childhood cancers to alleviate the global cancer burden4. However, comprehensive assessments of this burden are hindered by sparse or unavailable data in certain regions, further compounded by delays in diagnosis and treatment, particularly aggravated by events like the COVID-19 pandemic5,6,7. Enhancing the accuracy of breast cancer screenings, diagnosis, and treatment is paramount in controlling its burden6.
Technological advancements have revolutionized gene expression analysis, making it easier to study the expression of a large set of genes under specific conditions8. RNASeq technology, in particular, has emerged as the preferred method for quantifying gene expression due to its superiority over traditional DNA microarrays9,10,11. Despite its promise, RNASeq gene expression data faces challenges such as small sample sizes and the curse of dimensionality, wherein each sample contains a vast number of genes, many of which may be irrelevant to cancer detection12,13,14. Identifying a concise set of relevant genes can significantly enhance our understanding of breast cancer biology and contribute to its interpretation, biological processes, and pathways15,16.
Machine learning (ML) techniques offer valuable tools for disease prediction and evidence-based decision-making in healthcare17. While ML models have shown promise in cancer studies, their black-box nature limits their interpretability and usability within clinical workflows18. For instance, heat maps have been used to study the resistance of breast cancer to doxorubicin19. While these advancements have improved cancer diagnosis research, the lack of interpretability in the applications restricts physicians’ trust in predicted outcomes, hindering their integration into clinical practice. This paper aims to address these challenges by applying interpretable ML methods to breast cancer classification using genomic data. We outline a comprehensive approach to optimizing gene identification, ML model fitting, and interpretation of model predictions, providing a simple and robust guide that enhances the reproducibility of ML classification and prediction tasks, especially in breast cancer predictions.
Feature selection and machine learning are critical in the analysis of gene expression data, particularly in cancer research. Given the high-dimensional nature of gene expression profiles-where the number of features (genes) greatly exceeds the number of samples-effective feature selection methods are essential for identifying the most relevant genes for accurate cancer diagnosis, prognosis, and treatment prediction20. Numerous feature selection techniques have been developed to tackle this challenge, each with distinct strengths and limitations.
Filter methods, such as t-tests, chi-square tests, and correlation coefficients, assess the relevance of each gene independently of the machine learning model21. These techniques are computationally efficient and widely used in cancer studies, including the identification of genes associated with specific cancer subtypes like breast cancer22. However, they often fail to capture gene interactions, which can be crucial for understanding the underlying biology of cancer.
Wrapper methods address this limitation by using a machine learning model to evaluate the importance of subsets of features23. Recursive Feature Elimination (RFE), for example, recursively removes features based on their impact on model performance, demonstrating success in cancer research applications such as biomarker selection for lung cancer prognosis24. Despite their accuracy, wrapper methods can be computationally intensive, especially with large datasets.
Embedded methods, like LASSO (Least Absolute Shrinkage and Selection Operator), integrate feature selection into the model training process. LASSO is particularly effective in selecting sparse sets of genes for cancer classification, reducing the risk of overfitting-a common issue in high-dimensional datasets25.
Once relevant features are selected, they serve as inputs for various machine learning models that predict cancer outcomes. Support Vector Machines (SVMs) are among the most widely used models for cancer classification tasks due to their ability to handle high-dimensional data. SVMs have been successfully applied to distinguish between malignant and benign tumors based on gene expression profiles26. Random Forests, an ensemble method that aggregates predictions from multiple decision trees, have also been widely used in cancer research, proving particularly useful in identifying key biomarkers in cancers like melanoma and colorectal cancer27.
In recent years, neural networks, especially deep learning models, have gained traction in gene expression analysis. Convolutional Neural Networks (CNNs) and Recurrent Neural Networks (RNNs) have been applied to more complex cancer prediction tasks, such as multi-class classification in breast cancer subtypes28.
These models offer powerful predictive capabilities, although they often require large datasets and can be difficult to interpret. Ensemble methods like the Super Learner algorithm, which combines predictions from different models, have emerged as a promising approach to improve the robustness and accuracy of cancer prognostic models29.
Despite these advancements, several challenges persist. The high dimensionality of gene expression data continues to pose a risk of overfitting, particularly with complex models like deep learning30. Furthermore, not all selected features (genes) are biologically relevant, leading to potential misinterpretation of results. Ensuring that selected features have biological significance is crucial for translating these findings into clinical practice31. Additionally, data heterogeneity-variations in gene expression profiles across different studies and patient populations-complicates the development of universally applicable models32. The computational complexity of some feature selection methods and advanced machine learning models can also be a barrier to their widespread adoption33.
The prediction of breast cancer outcomes using gene expression datasets has been a dynamic area of research, with substantial advancements made over the years. Early seminal work by34 introduced gene expression profiling as a tool for classifying breast cancer into distinct molecular subtypes, revolutionizing our understanding of tumor biology and treatment strategies. This pioneering study laid the groundwork for utilizing gene expression data to categorize breast cancer into subtypes with distinct clinical outcomes and responses to therapy.
Building on this foundational work35, further refined the classification of breast cancer by identifying additional molecular subtypes through comprehensive gene expression analyses. Their study highlighted the heterogeneity within breast cancer and underscored the potential of gene expression profiles in predicting patient prognosis and tailoring personalized treatments. Similarly36, demonstrated the utility of gene expression profiles in predicting breast cancer metastasis, showcasing the potential of these profiles to inform clinical decisions and improve patient outcomes.
Our manuscript builds upon these extensive contributions by addressing current gaps and expanding the scope of gene expression-based predictions. While previous studies have made significant strides in this area, our work address these challenges by applying interpretable ML methods to breast cancer classification using genomic data. We outline a comprehensive approach to optimizing gene identification, ML model fitting, and interpretation of model predictions, offering a simple and robust guide to enhance the reproducibility of ML classification and prediction tasks, especially in breast cancer predictions. By doing so, we aim to advance the field further and enhance the applicability of predictive models in clinical practice, especially in breast cancer predictions.
The rest of the paper is structured as follows: Section 2.1 describes the cancer dataset, sections 2.2 delve into the ML algorithms used in this study while Section 2.3 briefly reviews the Sparse Wrapper Algorithm (SWAG). Finally, Sections 2.4 explore various approaches proposed to assist with explaining and interpreting ML prediction models.
Methods and materials
Data
We utilized the TCGAbiolinks package in R to retrieve the breast cancer data from the Cancer Genome Atlas (TCGA) repository. The BRCA dataset contains 19,948 genes across 1,208 samples. Given the high dimensionality of the gene expression data, filtration and feature selection to reduce the abundance of genes were applied using the TCGAbiolinks package. This process was crucial for removing irrelevant and noisy genes that could potentially hinder the detection of BRCA. However, only the informative genes were returned after implementing normalization, transformation, and filtration processes. Moreover, differentially expressed genes analysis was done to further reduce the high number of genes. Thus, this process retrieved 3602 differentially expressed genes (DEGs) between the tumor and normal samples. Genomic datasets often contain a vast number of features. It is often the case that the number of features is greater than the number of observations. This results in the high-dimensional data challenge37. ML methods may struggle to handle such datasets efficiently due to computational complexity, difficulties in interpreting results, and the risk of overfitting. Therefore, feature selection plays a crucial role in improving the performance of machine learning algorithms by reducing the time required to train the model and improving accuracy during the training process. Methods for feature selection or dimensional reduction for high-dimensional data are often used38.
Methods
Decision trees (CART)
A decision tree algorithm breaks or partitions the input or feature space into regions27,39. The feature space is, therefore, subdivided into non-overlapping regions. It has separate parameters for each region. Unlike linear models, decision trees are known to map non-linear relationships quite well. The partitioning is done through the calculation of some of the data homogeneity measures. Given a set of features, \(\textbf{x}=\left[ x_{1},x_{2},\ldots ,x_{p}\right]\), suppose that they are to be partitioned into \(M\) regions, \(R_{1},R_{2},\ldots ,R_{M}\). A regression tree can be seen as a kind of an additive model:
where \(y_{m}\) are constants, \(\mathbb {I}(.)\) is an indicator function returning 1 if its argument is true and 0 otherwise. \(R_m\) are disjoint partitions or regions of the training data \(D\). To solve a classification task, the learning algorithm is asked to produce a function \(f: \textbf{R}^p \rightarrow \{1,2,\ldots , C\}\). A splitting criteria or node impurity measure is used to build the decision tree. For classification problems there are several impurity measures. In this study, however, we used the entropy measure. The Entropy of a two-class problem is given as:
where \(p\) is the probability of finding a class labeled \(1\). Entropy will have a small value if the given node is pure.

Classification tree algorithm
Random forest
Random Forest is a supervised learning algorithms that works for classification and regression problems27. This is often referred to as an ensemble learning classification method, which creates a series of CART through the bootstrap sampling of the originally supplied data40. Each of the individual trees is built using a random subset of the model features and bagging to create an uncorrelated forest of individual trees working to the same objective. The tree maximizes the classification criteria at each node of the iteration. In many cases, it outperforms many of its parametric equivalents and is less computationally taxing to boot. Below is the working method of the Random Forest algorithm adopted from41.

Random Forest Algorithm
Support vector machine (SVM)
The support vector machine (SVM) is a supervised machine learning algorithm for classification and regression. In addition, SVM has the ability to handle non-linearly separable problems using mapping kernel functions such as the radial basis function (RBF) kernel and polynomial function. The algorithm uses support vectors to find the best hyperplane that separates the classes42. The SVM uses the kernel trick where the original input data is transformed into higher dimensional space and an optimal boundary is found for the possible separation. However, selecting a kernel function can significantly impact the performance of an SVM model42. The SVM model aims to find a hyperplane that separates the two classes in the feature space defined as follows:
and in the dual form can also be written as:
and \(\alpha _i^{'}\)s are the model coefficients (also known as dual coefficients) for each observation in the train set. \(\alpha _i\) is found to be nonzero only for the support vectors in the solution and zero for all the other observations in the train set. Replacing x with the transformation provided by a mapping function \(\phi (x)\), its dot product with the function \(\phi (x_i)\) results into the function called a kernel:
Although there are several kernels for fitting SVM, this study empolyed the radial basis function and the linear Kernels.
K-Nearest neighbours
The K-nearest-neighbor (KNN) idea relies on a distance metric such as the Euclidean distance to assess the similarity between a test sample and the available training samples. This approach is intuitive because it classifies samples by associating them with the same class as their nearest neighbors43. The KNN algorithm has two steps: the first one is to identify the neighbours and the second step is to determine the label (class) using the neighbours. A specific value of k is selected to aid in classifying the unknown label. KNN identifies the class of the unknown data point by considering the class label that is most frequent among the neighbors to the data point to be classified43,44. Below are the basic steps of the KNN algorithm.

k-NN algorithm
Feature selection method
Sparse Wrapper Algorithm (SWAG)
Sparse Wrapper Algorithm (SWAG) is an algorithm that trains a meta-learning procedure that combines screening and wrapper methods to find a set of extremely low-dimensional attribute combinations45. This procedure aims to find a library of extremely low-dimensional attribute combinations that match or improve the predictive performance of any particular method that uses all attributes as input (including sparse learners). Additionally, they provide a low-dimensional network of attributes that are easily interpretable by researchers while also increasing the potential replicability of results due to a diversity of attribute combinations defining strong learners with equivalent predictive power45. The output of the SWAG procedure facilitates the replicability of results46. The SWAG procedure consists of a “greedy” wrapper algorithm that, in the first stage, requires the researcher to specify a model (or learning method), such as classification models including random forest or logistic regression, as well as the maximum number of variables (\(p_{max}\)) to be considered within such a model. For example, the latter choice can be made based on prior knowledge of the problem and interpretability requirements (the smaller this number, the easier the output will be interpreted)45,46. Based on these choices and supposing a total of p features (i.e., genes), the SWAG starts through a first screening step where p models are built, each using a distinct feature. At this stage, the out-of-sample prediction error of each model can be estimated via k-fold cross-validation repeated m times and the best of these models (in terms of lowest prediction error) can be selected thereby providing a list of features that, on their own, appear to be highly predictive for the considered response. The definition of “best” models will be given by the researcher through a parameter \(\alpha\), which represents a proportion (or percentile) and is usually chosen to be considerably small (i.e., between 0.01 and 0.1). With smaller values of \(\alpha\) implying a more strict selection of best models (hence the choice of only the most performing features), the SWAG then uses the features selected in the first step to build higher-dimensional models progressively (i.e., models with an increased number of feature combinations within them) until it reaches the maximum number \(p_{max}\). When building the models for a given dimension, the SWAG takes the best models from the previous step (i.e., the step that built models with one less feature than the current step) and randomly adds a distinct feature from the set of features selected at the first step. Having built m models at each step (where m is also chosen by the user), the final output of the SWAG is a set of “strong” models (i.e., models with high predictive power) where each is based on a combination of 1 to \(p_{max}\)features45,46.
Feature interpretability techniques
Partial dependence plot
For a model \(f(\textbf{x})\) that predicts the value of a target variable \(y \in \mathbb {R}\) using a given set of predictor variables \(\textbf{x} \in \mathbb {R}^p\), where \(\textbf{x} = (x_1, x_2, \ldots , x_p)\), visualization is highly valuable in interpreting the relationship between \(y\) and \(\textbf{x}\). However, it is important to note that some challenges exist in creating visualization functions for high-dimension data.
A Partial Dependence Plot (PDP) illustrates the relationship between the outcome and a given set of features of interest47. The plot shows the average partial relationship between the given predictors and the predicted outcome. It illustrates how the predictions partially depend on the values of the input predictors of interest. Due to the fact that human perception is constrained, it is important to keep the size of the set of features of interest small, usually one or two. These features are therefore chosen among the most important features47,48,49.
We normally view the partial dependence of the approximation \(\hat{f}\left( \textbf{x}\right)\) on small subsets of the input variables. By examining the shape of the PDP, we can obtain insight into the relationship between y and some input variable, \(x_j\). The PDP can show whether the relationship between the outcome and the covariate is linear or nonlinear and whether there are any interaction effects between \(x_j\) and other monotonic or more complex predictor variables. This implies that the plot should be a linear relationship if partial dependence plots are applied to a linear regression problem. Furthermore, PDPs can also be used to identify regions of high or low predicted values of the target variable, which can be useful for identifying important subgroups or patterns in the data.
Let \(x_{s}\) be the selected feature subset of size s for which the partial dependence function should be plotted. Consider \(x_{-s}\) as the other features used in the ML model \(\hat{f}\left( \textbf{x}\right)\) such that \(x_{s} \cup x_{-s} = \textbf{x}\). Then, the approximation:
To know the effect of the feature(s) in s on the predicted outcome, we average the predictions over the observed values for the other variables. Then \(\hat{f}\left( \textbf{x}\right)\) can be considered as a function only of the variables in the chosen subset.
For a predictor \(x_j\), the partial dependence of \(f(\textbf{x})\) with respect to the \(j^{th}\) predictor variable \(x_j\) is defined as:
where\(x_{-j}\) is the vector of all predictor variables except for \(x_{j}\), and \(x_{-j}^{(i)}\) is the \(i^{th}\) value of \(\varvec{x}_{-j}\) in the data. The PDP at a specific value \(x_j\) is the average predicted value of the outcome variable when \(x_j\) is fixed at that value, while predictions are averaged over the observed values of all other predictor variables48,49,50. For classification problems where the ML model outputs probabilities, the partial dependence plot displays the probability for a certain class given different values for feature(s)47,51.
Accumulated Local Effects (ALE) plots
Compared to PDPs, ALE plots are useful for detecting non-linear relationships and interactions between features52. The idea behind ALE plots is to calculate the change in the predicted outcome of a model when a single feature is varied over its entire range while averaging over the other features. This gives a function that shows how the feature influences the predicted outcome on average. The ALE plot then accumulates these local effects to show the overall relationship between the feature and the predicted outcome. The algorithm below shows how the ALE plot for the \(j^{th}\) feature is obtained:

Accumulate these local effects by taking the cumulative sum. The resulting function is the ALE function for the \(j^{th}\) feature. The ALE plot shows this function as a step function, where each step corresponds to a change in the local effect of the \(j^{th}\) feature.
Shapely values
Shapley values originated from game theory53. They are used for explaining prediction models for both classification and regression tasks54. Shapley values are advantageous over the other methods of interpretability because they involve all possible subsets of input features. Therefore, interactions and redundancies between features should be taken into account.
For a given feature of interest \(x_j\), the number of iterations \(M\), the data matrix \(\textbf{X}\), and the ML model, \(f\), the Shapley value for \(x_j\) is estimated by averaging the differences between the model’s predictions with and without the feature across all iterations. Specifically, for each iteration \(m = 1, \ldots , M\), the Shapley value for feature \(x_j\) is estimated as:
The detailed algorithm is described within the environment labeled as Algorithm 4.

Shapley values estimation algorithm
The algorithm estimates the Shapley value for a single feature. To obtain Shapley values for all features, the procedure is repeated for each feature index \(j\). This method allows for approximating Shapley values using Monte Carlo sampling techniques, making it computationally feasible, especially when dealing with a large number of features54. The final Shapley value for a feature represents its importance or contribution to a ML prediction model. These values provide insights into understanding the significance of each feature’s contribution to the model’s decision-making. Higher Shapley values suggest the greater importance of a feature in driving predictions process.
Feature importance
In ML, the goal is to predict outcomes based on input features. However, it’s crucial to acknowledge the variability in feature importance rankings across different models trained on the same dataset, necessitating careful consideration when selecting the most suitable feature importance method55. To address this variability, one commonly used technique is permutation importance27, which evaluates the significance of features by randomly shuffling their values, thereby disrupting the relationship between each feature and the outcome. This disruption allows for an assessment of the resulting change in model performance, effectively quantifying the impact of individual features on predictions. A substantial decrease in performance indicates greater importance of the feature to the model’s predictive accuracy.
In addition to permutation importance, another valuable method for feature evaluation is the LOCO (Leave-One-Covariate-Out)56 feature importance method. This approach quantifies feature importance by iteratively removing each feature from the dataset and refitting the machine learning model. By comparing the performance of the original model with that of a refitted model that excludes the feature under evaluation, LOCO provides insights into the contribution of individual features to the model’s predictive power. Moreover, a special case of LOCO known as LOCI (Leaving-One-Covariate-In) focuses specifically on the difference in risk between the optimal prediction and a model that relies solely on the feature being evaluated.
LOCI provides a deeper understanding of each feature’s importance in driving model predictions and offers several advantages over traditional LOCO methods. Firstly, it offers a more direct measure of the contribution of individual features to the model’s predictive performance. By focusing solely on the effect of including or excluding one feature, LOCI provides a clearer understanding of each feature’s unique contribution to the model. Additionally, LOCI can offer insights into feature interactions, aiding in understanding complex relationships between features and improving the model’s overall interpretability. Moreover, LOCI can be computationally more efficient, especially in datasets with a large number of features. Overall, LOCI is a powerful technique for assessing feature importance in ML models, offering a nuanced and direct measure of individual feature contributions while also providing insights into feature interactions.
Data preprocessing
To enhance the predictive performance of the models in our study, we adopted the SWAG method defined in Section 2.3as our feature selection method45,57. While various alternative feature selection techniques exist, they each have their limitations. For instance, filter methods, which rely on statistical measures like correlation or mutual information, may overlook intricate feature interactions20. Similarly, embedded methods such as Lasso regression and decision trees with built-in feature selection may lack the flexibility of SWAG in selecting relevant features58. Lasso regression, while effective at reducing overfitting and selecting features through a penalty on the magnitude of coefficients, is constrained by the strength of its penalty parameter and assumes linear relationships, making it less capable of capturing interactions and complex patterns in the data. Similarly, decision trees select features based on split criteria like impurity or information gain, but they may struggle to capture feature interactions and are prone to instability due to sensitivity to data changes.
In contrast, SWAG is computationally efficient due to its use of stochastic gradient methods, which allow the model to perform feature selection iteratively and in parallel with model fitting. The stochastic gradient updates ensure that only a subset of features is evaluated at each step, significantly reducing the computational burden compared to methods that evaluate all possible combinations of features or require repeated full-batch gradient computations. SWAG constructs multiple models using different feature subsets at each iteration, avoiding the exhaustive search required by more traditional methods. This iterative refinement offers a balance between feature exploration and computational cost, particularly beneficial in high-dimensional datasets like gene expression data. Additionally, SWAG averages weights from multiple points along the optimization trajectory, which reduces the need for retraining models from scratch, leading to faster convergence and lower computational requirements59.
Moreover, unlike alternative feature selection techniques, SWAG operates as a “greedy” wrapper algorithm, efficiently exploring various combinations of features to identify robust models. After constructing K models at each step, the SWAG algorithm outputs a collection of “strong” models, each of which is constructed by combining a varying number of features, ranging from 1 to the maximum number of features, p46. This approach not only offers better control over model complexity and prevents overfitting but also ensures that selected features are highly predictive while minimizing redundancy, thus enhancing model interpretability.
SWAG’s resistance to overfitting stems from several key aspects. First, SWAG leverages model averaging by combining weights from multiple points in the optimization process, creating a smoother model landscape and reducing the risk of overfitting. This is similar to the regularization effect seen in ensembling methods, which generally improve model generalization by preventing over-reliance on specific models or features60. Furthermore, SWAG’s stochastic exploration of feature subsets ensures that it captures a variety of relevant features, unlike more rigid methods such as Lasso regression, which may penalize features uniformly based on a penalty parameter25. Lasso can sometimes miss complex interactions among features due to its linear assumptions, whereas SWAG is capable of exploring a wider range of feature interactions and relationships.
Moreover, SWAG provides better control over model complexity by progressively selecting features based on their predictive strength, while minimizing redundancy. This approach is particularly effective for high-dimensional datasets, such as gene expression data, where feature selection is critical for preventing overfitting. The iterative nature of SWAG allows it to refine the feature set and identify the most predictive features without introducing unnecessary complexity. The ability to explore various combinations of features, as well as its stochastic weight averaging, contributes to its robustness in preventing overfitting20.
Studies have shown that SWAG improves generalization and reduces prediction error compared to traditional feature selection methods59. Additionally, by leveraging stochastic updates, SWAG can escape local minima and explore the feature space more thoroughly than deterministic methods like decision trees, which can be sensitive to small changes in data27. This flexibility helps SWAG to identify robust models that perform well on unseen data.
In our study, we applied three learners within the SWAG framework: SVM Linear, SVM Radial, and Random Forest (RF). We split the data into training (60%) and testing (40%) sets and implemented the three learners to determine the optimal model with the minimum number of features. Setting parameters like \(\alpha = 0.2\) and the maximum features \(p_{\max } =10\), we performed 10 iterations with 1000 permutations to optimize model performance and feature selection efficiency. By employing SWAG, we aimed to streamline feature selection and model building processes, leading to more accurate, reproducible and interpretable predictive models for breast cancer diagnosis and treatment.
Results
The breast cancer dataset considered comprises 1208 observations with 3602 genes making it a high dimensional dataset. To correctly predict the outcome of whether the genes are associated with cancer cells or not, feature reduction techniques were employed. A SWAG model was employed to reduce the 3602 genes to the most significant predictive features for predicting the outcome variable. We apply three learners of the SWAG model: SVM Linear, SVM Radial, and Random Forest (RF). The data was split into training (\(60\%\)) and testing (\(40\%\)), and then the three learners were implemented to arrive at the best model for the minimum number of features. We chose \(\alpha = 0.2\) and the number of maximum features \((pmax)=10\). The algorithm was set to perform 10 iterations with 1000 permutations of the features.
The analysis was done using a device with the specification of 2nd Gen Intel(R) Core(TM) i7-1260P @ 2.10 GHz and 32.0 GB RAM. It takes about 5.67 hours to run the SWAG algorithm for the genes or variable selection to be modeled.
The results of the SWAG model analysis for the three learners are displayed in Table 1. The results show that all three learners reached the minimum cross-validation (CV) error at the fifth iteration at 0.0014, resulting in the prediction of 5 features per predicting model. The three SWAG learners generated 6 independent sets of models with 5 features per predicting model. It is observed that the SVM radial learner recorded the highest accuracy of 99.59%, and the random forest was the least performing learner with 98.76%.

Selected Genes by the Radial SVM SWAG Learner.
Figure 1 shows the frequencies of genes. The results reveal distinct patterns across the 22 genes examined. Notably, genes COL10A1, NPR1, and SDPR demonstrated higher frequencies than the remaining genes, followed by RPLPOP2.

Correlation plot among the selected variables of the SVM Radial SWAG learner.
Figure 2 illustrates the pairwise relationships between the SVM Radial SWAG learner selected genes. We observed positive and negative correlations between most genes, indicated by the red and purple colors in the correlation matrix.
Table 2 presents the training results for the jointly selected features for the two SWAG learners for all six independent models comprising the seven features each. The selected genes by the SWAG SVM radial were used to build three machine learning models, including SVM-linear, KNN, and RF. The results show that the SVM-linear model outperformed the KNN and RF models with an F1-score of 0.9846 against 0.9531 and 0.9667, respectively.
The importance plot shown in Figure 3 highlights genes such as COL10A1 as the most influential, as indicated by two methods used (KNN, SVM-linear) in predicting positive cancer cells. Additionally, genes like MMP11, SDPR, FIGF, CD300LG, FXYD1, and CLEC3B consistently emerge among the top influential genes across all three models utilized. However, it is important to note that each model assigns different rankings to these predictors. It is challenging to pinpoint a universal agreement on the most influential genes, especially when using three ML models like this study. The lack of consistency in gene rankings complicates the identification of the major contributors to cancer cell prediction. Moreover, the rankings themselves do not offer insights into the specific impact of these genes on the prediction of cancer cells.

The variable importance.
Table 3 shows the top 10 genes selected by the three ML models. The ranking differs for other genes except for the most influential gene (COL10A1). We obtained seven common genes across the three models. However, the ranks of importance for some models differed, as displayed in the common genes.
Our study presents Shapley values, providing an understanding of the magnitude of each feature’s effect in predicting cancer cells. This approach allows for a more comprehensive understanding of the individual contributions of these genes, shining more light on their specific roles in the predictive models.

The shapley values.
In Figure 4, the Shapley values illustrate the influence of features on the model’s cancer predictions. They quantify the impact of individual features, indicating their influence on the model’s prediction. Larger Shapley values emphasize the increased significance of COL10A1, MMP11, SDPR, FIGF, CD300LG, FXYD1, and CLEC3B in predicting cancer presence, contributing notably to the likelihood of detecting cancer cells. However, there are criticisms to the shapely values, as highlighted in a study by61,62, cautioning against their use. The authors emphasized the need for proper consideration in applying Shapley values.
These studies have indicated that Shapley values assume that features contribute independently to model predictions. However, in reality, features may interact in complex ways, leading to challenges in accurately attributing contributions to individual features. Shapley values rely on the feature independence assumption, which may not hold true for all datasets or models. Violations of this assumption can result in biased or misleading interpretations of feature importance. The order in which features are included or removed when calculating Shapley values can impact the results. Different ordering strategies may yield different interpretations of feature importance, introducing potential variability and inconsistency.
In this study, we extended our investigation by incorporating model-based feature rankings to assess their ability to provide consistent rankings across various machine learning models. Specifically, we opted to present the results of LOCI feature importance, as outlined in the section 2.4, based on findings from a recent study55, which indicated its superior performance compared to alternative rankings methodologies.

LOCI feature importance ranking for SVM and RF.
Interestingly, in Figure 5, the two models agree on ranking the top 10 genes that are ranked highest in the importance plots based on the LOCI feature importance method. The genes selected by LOCI are also similar to the genes the Shapely values provided as highly impactful in predicting positive breast cancer cells. We employed more transparent methods such as PDPS and ALE plots to enhance interpretability. PDPS demonstrate how the predicted outcome changes as a single feature varies, while ALE plots showcase how changes in a single feature impact predictions while considering interactions with other variables.

Accumulated local effects (ALE) Plot for the top four common genes selected by the ranking methods.
In Figure 6, an increasing ALE plot of COL10A1 and MMP11 implies a rising trend. This indicates that as the gene’s expression values increase, their effect on the model’s prediction also increases. The opposite is true for genes SPDR and CD300LG. As their expression values increase, the model’s prediction decreases.

Partial dependence plots (PDP) for the top four common genes selected by the ranking methods.
In Figure 7, the partial dependence plots illustrate an increasing trend for PDPS, COL10A1, and MMP11, indicating that higher values of these genes correspond to increased influence on the model’s predictions. On the other hand, for genes SPDR and CD300LG, the partial dependence plots show a decreasing pattern, suggesting that higher values of these genes are associated with a decrease in the model’s value prediction.
These visualizations aid in understanding a feature’s influence on model predictions. This study, therefore, implies that finding out the most important features and their interpretation is a multifaceted approach. There is no one size fits all, especially when using machine learning models in health studies like breast cancer prediction. One has to use all the available methods to enhance predictions and prevent giving a blanket result with no clear explanation of how we arrived at the prediction.
Discussion
In this study, we used a breast cancer dataset with 1208 observations and 3602 genes, to predict whether the genes contained cancer cells. We applied feature selection techniques such as the SWAG model to narrow down the extensive gene pool to the most influential predictive features. To determine optimal feature set for the prediction task, we employed three learners within the SWAG model, namely; SVM Linear, SVM Radial, and Random Forest (RF). Each SWAG learner produced six independent sets of models. We subsequently ran three predictive models, KNN, RF, and the SVM-linear, to predict breast cancer tumors. Based on the permutation feature importance ranking from the RF, we identified predictors like COL10A1, FIGF, and FYD1 as the most significant in predicting breast cancer tumor. However, this ranking lacked specificity regarding how these features precisely contribute to breast cancer tumor predictions, prompting us to explore explainable ML techniques63.
In our investigation, we employed Shapley values to explicate the variable importance ranking derived from the random forest algorithm. Shapley values revealed the influence of features on cancer predictions, identifying genes such as COL10A1, MMP11, SDPR, FIGF, CD300LG, FXYD1, and CLEC3B as significant predictors of cancer presence. Interestingly, some of the features ranked highest in the importance plot did not exhibit a substantial positive impact based on the Shapley values. This inconsistency complicates the process of pinpointing key genes that reliably contribute to breast cancer predictions and raises questions about the biological relevance of these features. The differing rankings suggest that the impact of specific genes on cancer outcomes might be model-dependent, reflecting the nuances in how various algorithms interpret gene expression data. Consequently, the variability challenges the robustness of the predictive models and highlights the need for a comprehensive approach to validate and understand feature importance.
A major criticism of Shapley values is that they can be computationally intensive, especially in high-dimensional datasets like gene expression profiles, where calculating exact values requires evaluating every possible combination of features. This computational burden can limit the practicality of Shapley values in large-scale analyses, potentially leading to approximations or simplified calculations that may not fully capture the true impact of features55.
Additionally, Shapley values are sensitive to the choice of the baseline or reference model, which can influence the magnitude and direction of the contributions attributed to each feature. If the baseline model is not representative or if it does not adequately capture the complexity of the data, the resulting Shapley values may not accurately reflect the true importance of the features. This sensitivity can lead to inconsistencies in feature rankings and impact the reliability of the results.
Furthermore, Shapley values assume that features are evaluated in isolation or in specific combinations, which might not fully account for interactions between features. In high-dimensional datasets with complex relationships among features, this limitation can lead to an incomplete understanding of how features jointly influence predictions. As a result, features that appear important in a model might not show a substantial positive impact when considering their interactions with other features.
Given these criticisms, we employed additional interpretability techniques, such as the LOCI method and transparent visualizations like PDPS and ALE plots. LOCI focuses on providing detailed, localized interpretations of feature importance by assessing how changes in feature values impact predictions. This method excels in identifying critical features and understanding their specific influence on individual predictions. It is particularly useful when a deep understanding of the role of individual features is required.
In contrast, PDPs offer a broader view of feature effects by illustrating the relationship between a feature and the predicted outcome. PDPs show how variations in a single feature affect predictions while averaging out the effects of other features. This approach is valuable for visualizing general trends and understanding the overall influence of individual features on model outcomes. ALE plots provide a model-agnostic perspective on feature effects and interactions.
Unlike PDPs, ALE plots account for interactions between features, offering a more comprehensive view of how changes in feature values influence predictions. This makes ALE plots particularly useful for understanding the combined effects of features and their interactions.
While each method contributes to the interpretability of our models, they are not equivalent and serve different purposes. LOCI is best suited for detailed analysis of feature importance and localized impact, while PDPs are ideal for visualizing general trends in feature effects. ALE plots are recommended when interactions between features need to be examined in depth. The choice of method should align with the specific goals of the analysis. In this study, the LOCI method successfully identified the 10 most critical genes for predicting breast cancer. This was achieved by evaluating whether the same genes frequently appeared among the top ranks in different iterations of the LOCI method. This consistency was crucial for demonstrating the reliability of the identified genes as significant predictors of breast cancer.
In addition to consistency, we assessed the impact of the identified genes on the performance of our predictive models. By incorporating the genes selected through LOCI into our models, we measured improvements in metrics such as accuracy, precision, recall, and F1-score. A notable enhancement in these performance metrics indicated that the genes identified by LOCI were indeed influential in predicting breast cancer outcomes.
Biological relevance also played a critical role in defining the success of the LOCI method. We cross-referenced the top genes identified by LOCI with existing literature on breast cancer to confirm their relevance. Genes that were previously documented as significant in breast cancer research were particularly valued, reinforcing the validity of the LOCI method’s findings.
The genes COL10A1, MMP11, SDPR, FIGF, CD300LG, FXYD1, and CLEC3B have emerged as significant predictors of breast cancer presence in this study, revealing their crucial roles in the disease’s progression. COL10A1, encoding a component of type X collagen, is primarily associated with endochondral bone formation but has also been implicated in tumor development. In breast cancer, COL10A1 is often overexpressed in the tumor stroma, where it contributes to extracellular matrix remodeling and supports tumor invasion. This gene’s elevated expression has been linked to more aggressive cancer and poorer patient outcomes, highlighting its potential as a target for therapeutic intervention64,65.
MMP11, also known as stromelysin-3, plays a critical role in degrading components of the extracellular matrix, facilitating tumor invasion and metastasis. In breast cancer, high levels of MMP11 are associated with increased tumor aggressiveness and worse clinical outcomes, reflecting its role in remodeling the tumor microenvironment to support cancer progression66. This makes MMP11 a valuable marker for assessing disease severity and progression67.
The gene SDPR, which is involved in regulating cell growth and apoptosis, also shows significant implications in breast cancer. Altered expression of SDPR affects tumor cell proliferation and survival, impacting cancer progression and treatment response. Its role in modulating cellular stress responses and apoptotic pathways underscores its importance in breast cancer biology68.
FIGF, or vascular endothelial growth factor D, is a key player in angiogenesis and lymphangiogenesis. In breast cancer, FIGF promotes the formation of new blood and lymphatic vessels, which are essential for tumor growth and metastasis. High FIGF levels are associated with increased tumor vascularization and a greater likelihood of metastatic spread, highlighting its relevance as a prognostic marker and therapeutic target69,70.
CD300LG, a member of the CD300 receptor family involved in immune regulation, influences tumor immune evasion and progression. In breast cancer, elevated CD300LG expression can affect how tumor cells interact with immune cells, potentially contributing to cancer’s progression by modulating immune responses within the tumor microenvironment71. This suggests that CD300LG might offer new avenues for immunotherapy.
FXYD1, a regulator of ion transport across cell membranes, impacts cellular ion homeostasis and pH regulation, which can influence tumor growth and treatment resistance. Elevated FXYD1 expression in breast cancer has been linked to poorer prognosis and resistance to therapies, underscoring its potential as a target for improving treatment strategies72,73.
Lastly, CLEC3B, involved in immune response and inflammation regulation, affects the tumor immune microenvironment and cancer progression. Changes in CLEC3B expression can impact tumor growth and response to immune checkpoint inhibitors, suggesting its role in modulating immune interactions and potentially guiding immune-based therapies in breast cancer74.
Together, these genes provide valuable insights into breast cancer biology and underscore the importance of incorporating both biological and computational analyses to enhance our understanding and treatment of the disease.
In our study, we utilized the TCGA breast cancer dataset, which encompasses 19,948 genes across 1,208 samples. To ensure the relevance of our findings, we have compared our dataset with several widely used datasets in the field. For instance, the METABRIC dataset, known for its extensive coverage, includes data for approximately 50,000 genes, though the number of genes analyzed in individual studies can vary75. Similarly, the PAM50 gene signature dataset, commonly used for breast cancer subtype classification, includes a specific set of genes76. Although our dataset contains fewer genes than some of these larger datasets, it still provides broad coverage that is consistent with current research practices. The TCGA dataset is a well-regarded resource in cancer research, known for its comprehensive integration of gene expression data with clinical information, aligning with contemporary standards. Furthermore, datasets such as the GDC Genomic Data Commons also offer extensive gene expression data and have been used in recent studies to validate findings77. While our dataset may not include as many genes as some other contemporary datasets, it remains a robust and relevant resource for understanding breast cancer gene expression patterns. The breadth and quality of our dataset ensure that our findings are significant and applicable to current research, reinforcing the robustness and applicability of our study within the landscape of breast cancer research.
Moreover, our dataset aligns well with some of the criteria outlined by78, who discuss the importance of using comprehensive and publicly available datasets to mitigate inherent biases in gene expression research. The large number of genes and samples provides a robust foundation for feature selection, supporting diverse and reproducible research outcomes. However, the dataset is not without its challenges78. highlight potential biases such as sample heterogeneity, batch effects, and limited representativeness, which are relevant considerations for TCGA data.
Despite its size, the dataset may still exhibit batch effects due to variations in sample processing, potentially influencing the results of feature selection methods. Additionally, the dataset’s demographic focus may impact its generalizability to broader populations, a concern emphasized by78. To address these issues, careful preprocessing, including normalization and batch effect correction, is essential. The feature selection process itself also requires vigilance to avoid biases towards highly expressed or frequently observed genes. Overall, while the TCGA dataset provides valuable insights for breast cancer research, ongoing attention to these potential biases and adherence to evolving best practices are necessary to ensure the validity and applicability of the findings.
Artificial neural networks (ANNs), including deep learning models, have demonstrated impressive capabilities in handling high-dimensional and complex datasets, such as those in cancer research. They excel at capturing intricate, non-linear relationships between genes and tumor characteristics, which can be crucial for accurate cancer classification. One of the key benefits of ANNs in cancer classification is their ability to automatically learn and extract relevant features from gene expression data. This feature learning process reduces the need for manual feature engineering and allows the model to uncover complex patterns that might not be evident through traditional methods. For instance, deep learning models like Convolutional Neural Networks (CNNs) and Recurrent Neural Networks (RNNs) have shown promise in identifying biomarkers and distinguishing between cancer subtypes with high accuracy78.
However, several reasons influenced our decision not to incorporate ANNs into our analysis. First, the primary focus of our study was on utilizing explainable machine learning methods to enhance the interpretability of our models. While ANNs offer powerful predictive capabilities, their “black-box” nature poses significant challenges in terms of understanding and interpreting the model’s decision-making process. Given our emphasis on transparency and interpretability, methods that provide clearer insights into feature importance and model predictions, such as the SWAG model and Shapley values, were more aligned with our objectives.
Second, the computational complexity of ANNs, particularly deep learning models, was a consideration. Training deep neural networks requires substantial computational resources and time, which may not be feasible given the constraints of our study. The SWAG model and other traditional methods we employed were chosen for their balance between computational efficiency and the ability to provide meaningful insights into feature importance.
Additionally, our dataset, although extensive with 19,948 genes across 1,208 samples, was utilized with methods that are well-suited for handling such dimensionality. The feature selection and interpretability methods applied were chosen to address specific research questions and to ensure that the results could be robustly interpreted and validated. Despite these reasons, we acknowledge that ANNs could provide additional benefits in future research. Their ability to model complex, non-linear interactions and handle large datasets aligns well with the demands of gene expression analysis. Furthermore, ongoing advancements in neural network interpretability could mitigate some of the challenges associated with their use, making them a valuable tool for further studies.
Our study nonetheless has several limitations. One such limitation is using the SWAG (Stochastic Weight Averaging) algorithm, which focuses on selecting the most predictive variables that can inadvertently lead to the exclusion of features that may hold important contextual or explanatory value. While SWAG excels at enhancing predictive performance by identifying the strongest contributors to the model’s outcomes, this feature selection process may overlook variables that are not the most predictive but still provide critical insights into the underlying relationships within the data. This limitation emphasizes the need for a balanced approach in feature selection, where the emphasis on prediction does not come at the expense of interpretability. To address this issue, it is essential to complement SWAG with additional analyses that evaluate the importance of discarded features, ensuring a holistic understanding of the model’s behavior and fostering robust interpretability alongside strong predictive capabilities. Despite this limitation, the benefits of SWAG, such as its ability to escape local minima and capture complex interactions within high-dimensional datasets, contribute significantly to its utility in feature selection, making it a valuable tool in the modeling process. Another limitation of this study is the extended processing time required to run the SWAG algorithm due to the limited processing power of the available computer. The analysis took a considerable amount of time to complete the gene selection process, which may hinder efficiency, especially for larger datasets or more computationally demanding analyses. An additional limitation is the validation of our proposed method on a single dataset without comparison to other recent methods in the literature, which limits the generalizability of our findings. Due to the limited availability of comparable datasets specific to our biological system and the challenges in harmonizing gene expression data, we focused on a single, well-characterized dataset to ensure robustness. Future studies incorporating multiple datasets and benchmarking against other recent methods are encouraged to further evaluate and generalize the performance of our approach.
Conclusion
Our study demonstrated the effectiveness of utilizing a combination of feature selection techniques and explainable ML methods to enhance the reproducibility, interpretability and reliability of machine learning models in predicting breast cancer tumor. By employing advanced feature selection techniques like the SWAG model and explainable ML methods such as Shapley values, partial dependence plots, LOCI, and accumulated local effects, we identified critical predictors while unraveling the underlying mechanisms driving cancer predictions. The transparent visualization techniques, including PDPS and ALE plots, provided invaluable insights into feature impacts are crucial for medical decision-making in breast cancer diagnosis and prognosis. Our findings underscore the importance of incorporating explainable ML frameworks to develop robust, reproducible and interpretable models essential for clinical practice, ultimately paving the way for more accurate and reliable predictive models in breast cancer research and patient care.
Data availability
The data are publicly available in The Cancer Genome Atlas (TCGA) repository.
References
Alharbi, F. & Vakanski, A. Machine learning methods for cancer classification using gene expression data: A review. Bioengineering 10(2), 173 (2023).
Sung, H., Ferlay, J., Siegel, R.L., Laversanne, M., Soerjomataram, I., Jemal, A. & Bray, F. Global cancer statistics 2020: Globocan estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA: a cancer journal for clinicians 71(3), 209–249 (2021).
Jiang, Q. & Jin, M. Feature selection for breast cancer classification by integrating somatic mutation and gene expression. Frontiers in genetics 12, 629946 (2021).
Anderson BO, F.E.e.a. Ilbawi AM: The global breast cancer initiative: a strategic collaboration to strengthen health care for non-communicable diseases. Lancet Oncol 22(5), 578–5811010161470204521000711 (2021).
Wild CP, S.B.e. & Weiderpass E. World cancer report: Cancer research for cancer prevention. Lyon, France: International Agency for Research on Cancer, 586 (2020).
Siegel RL, W.N.J.A. Miller KD: Cancer statistics, 2023. CA Cancer J Clin 73(1), 17–481033222176336633525 (2023).
Ghoshal S, C.D.B.R.G.M.H.R.L.K.L.W.S.M & Rigney G. Institutional surgical response and associated volume trends throughout the covid-19 pandemic and postvaccination recovery period. JAMA Netw Open 5(8), 2227443–101001202227443359806369389350 (2022).
Hua, H. B. High-throughput technologies for gene expression analyses: What we have learned for noise-induced cochlear degeneration. Journal of otology 8(1), 25–31 (2013).
Datta, S. & Nettleton, D. Statistical analysis of next generation sequencing data (2014)
Rai, M.F., Tycksen, E.D., Sandell, L.J. & Brophy, R.H. Advantages of rna-seq compared to rna microarrays for transcriptome profiling of anterior cruciate ligament tears. Journal of Orthopaedic Research® 36(1), 484–497 (2018).
Koch, C. M. et al. A beginner’s guide to analysis of rna sequencing data. American journal of respiratory cell and molecular biology 59(2), 145–157 (2018).
Mohammed, M., Mwambi, H., Mboya, I. B., Elbashir, M. K. & Omolo, B. A stacking ensemble deep learning approach to cancer type classification based on tcga data. Scientific reports 11(1), 15626 (2021).
Pyingkodi, M. & Thangarajan, R. Informative gene selection for cancer classification with microarray data using a metaheuristic framework. Asian Pacific journal of cancer prevention: APJCP 19(2), 561 (2018).
Hijazi, H. & Chan, C. A classification framework applied to cancer gene expression profiles. Journal of healthcare engineering 4, 255–283 (2013).
Zhang, Y. et al. Identifying breast cancer-related genes based on a novel computational framework involving kegg pathways and ppi network modularity. Frontiers in Genetics 12, 596794 (2021).
Sun, X. et al. Identification of significant genes and therapeutic agents for breast cancer by integrated genomics. Bioengineered 12(1), 2140–2154 (2021).
Karatza, P., Dalakleidi, K., Athanasiou, M. & Nikita, K.S. Interpretability methods of machine learning algorithms with applications in breast cancer diagnosis. In: 2021 43rd Annual International Conference of the IEEE Engineering in Medicine & Biology Society (EMBC), pp. 2310–2313. IEEE (2021).
Brito-Sarracino, T., dos Santos, M.R., Antunes, E.F., de Andrade Santos, I.B., Kasmanas, J.C. & de Leon Ferreira, A.C.P. Explainable machine learning for breast cancer diagnosis. In: 2019 8th Brazilian Conference on Intelligent Systems (BRACIS), pp. 681–686. IEEE (2019).
Ogunleye, A. Z., Piyawajanusorn, C., Gonçalves, A., Ghislat, G. & Ballester, P. J. Interpretable machine learning models to predict the resistance of breast cancer patients to doxorubicin from their microrna profiles. Advanced Science 9(24), 2201501 (2022).
Guyon, I. & Elisseeff, A. An introduction to variable and feature selection. Journal of machine learning research 3(Mar), 1157–1182 (2003).
Tharwat, A. Classification assessment methods. Applied computing and informatics 17(1), 168–192 (2021).
Wang, Y. et al. Gene-expression profiles to predict distant metastasis of lymph-node-negative primary breast cancer. The Lancet 365(9460), 671–679 (2005).
Kohavi, R. & John, G. H. Wrappers for feature subset selection. Artificial intelligence 97(1–2), 273–324 (1997).
Liu, H., Motoda, H., Setiono, R. & Zhao, Z. Feature selection: An ever evolving frontier in data mining. In: Feature Selection in Data Mining, pp. 4–13. PMLR (2010).
Tibshirani, R. Regression shrinkage and selection via the lasso. Journal of the Royal Statistical Society Series B: Statistical Methodology 58(1), 267–288 (1996).
Guyon, I., Weston, J., Barnhill, S. & Vapnik, V. Gene selection for cancer classification using support vector machines. Machine learning 46, 389–422 (2002).
Breiman, L. Random forests. Machine learning 45, 5–32 (2001).
LeCun, Y., Bengio, Y. & Hinton, G. Deep learning. nature 521(7553), 436–444 (2015).
Polley, E.C. & Van der Laan, M.J. Super learner in prediction (2010).
Domingos, P. A few useful things to know about machine learning. Communications of the ACM 55(10), 78–87 (2012).
Shi, L. et al. The microarray quality control (maqc)-ii study of common practices for the development and validation of microarray-based predictive models. Nature biotechnology 28(8), 827–838 (2010).
Johnson, W. E., Li, C. & Rabinovic, A. Adjusting batch effects in microarray expression data using empirical bayes methods. Biostatistics 8(1), 118–127 (2007).
Hastie, T., Tibshirani, R., Friedman, J. & Franklin, J. The elements of statistical learning: data mining, inference and prediction. The Mathematical Intelligencer 27(2), 83–85 (2005).
Perou, C.M., Sørlie, T., Eisen, M.B., Van De Rijn, M., Jeffrey, S.S., Rees, C.A., Pollack, J.R., Ross, D.T., Johnsen, H. & Akslen, L.A. Molecular portraits of human breast tumours. nature 406(6797), 747–752 (2000).
Sørlie, T. et al. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proceedings of the National Academy of Sciences 98(19), 10869–10874 (2001).
Van’t Veer, L.J., Dai, H., Van De Vijver, M.J., He, Y.D., Hart, A.A., Mao, M., Peterse, H.L., Van Der Kooy, K., Marton, M.J. & Witteveen, A.T. Gene expression profiling predicts clinical outcome of breast cancer. nature 415(6871), 530–536 (2002).
Johnstone, I.M. & Titterington, D.M. Statistical challenges of high-dimensional data. The Royal Society Publishing (2009).
Gnana, D. A. A., Balamurugan, S. A. A. & Leavline, E. J. Literature review on feature selection methods for high-dimensional data. International Journal of Computer Applications 136(1), 9–17 (2016).
Kotsiantis, S. B. Decision trees: a recent overview. Artificial Intelligence Review 39, 261–283 (2013).
Maroco, J. et al. Data mining methods in the prediction of dementia: A real-data comparison of the accuracy, sensitivity and specificity of linear discriminant analysis, logistic regression, neural networks, support vector machines, classification trees and random forests. BMC research notes 4(1), 1–14 (2011).
Yekkala, I. & Dixit, S. Prediction of heart disease using random forest and rough set based feature selection. International Journal of Big Data and Analytics in Healthcare (IJBDAH) 3(1), 1–12 (2018).
Huang, S. et al. Applications of support vector machine (svm) learning in cancer genomics. Cancer genomics & proteomics 15(1), 41–51 (2018).
Cunningham, P. & Delany, S. J. k-nearest neighbour classifiers-a tutorial. ACM computing surveys (CSUR) 54(6), 1–25 (2021).
Taunk, K., De, S., Verma, S. & Swetapadma, A. A brief review of nearest neighbor algorithm for learning and classification. In: 2019 International Conference on Intelligent Computing and Control Systems (ICCS), pp. 1255–1260. IEEE (2019)
Molinari, R., Bakalli, G., Guerrier, S., Miglioli, C., Orso, S., Karemera, M. & Scaillet, O. Swag: A wrapper method for sparse learning. arXiv preprint arXiv:2006.12837 (2020).
Miglioli, C. et al. Evidence of antagonistic predictive effects of mirnas in breast cancer cohorts through data-driven networks. Scientific Reports 12(1), 5166 (2022).
Friedman, J. H. 1999 reitz lecture. Ann. Stat. 29(5), 1189–1232 (2001).
Hastie, T., Tibshirani, R., Friedman, J.H. & Friedman, J.H. The Elements of Statistical Learning: Data Mining, Inference, and Prediction vol. 2. Springer, ??? (2009).
Greenwell, B.M., Boehmke, B.C. & McCarthy, A.J. A simple and effective model-based variable importance measure. arXiv preprint arXiv:1805.04755 (2018).
Strobl, C., Boulesteix, A.-L., Kneib, T., Augustin, T. & Zeileis, A. Conditional variable importance for random forests. BMC bioinformatics 9, 1–11 (2008).
Friedman, J.H. Greedy function approximation: a gradient boosting machine. Annals of statistics, 1189–1232 (2001).
Apley, D.W. & Zhu, J. Visualizing the effects of predictor variables in black box supervised learning models. arXiv preprint arXiv:1612.08468 (2016).
Shapley, L.S., et al. A value for n-person games (1953).
Štrumbelj, E. & Kononenko, I. Explaining prediction models and individual predictions with feature contributions. Knowledge and information systems 41, 647–665 (2014).
Ewald, F.K., Bothmann, L., Wright, M.N., Bischl, B., Casalicchio, G. & König, G. A Guide to Feature Importance Methods for Scientific Inference (2024).
Lei, J., G’Sell, M., Rinaldo, A., Tibshirani, R. J. & Wasserman, L. Distribution-free predictive inference for regression. Journal of the American Statistical Association 113(523), 1094–1111 (2018).
Rafid, A. R. H. et al. An effective ensemble machine learning approach to classify breast cancer based on feature selection and lesion segmentation using preprocessed mammograms. Biology 11(11), 1654 (2022).
Duensing, N. et al. Novel features and considerations for era and regulation of crops produced by genome editing. Frontiers in bioengineering and biotechnology 6, 79 (2018).
Maddox, W.J., Izmailov, P., Garipov, T., Vetrov, D.P. & Wilson, A.G. A simple baseline for bayesian uncertainty in deep learning. Advances in neural information processing systems 32 (2019).
Izmailov, P., Podoprikhin, D., Garipov, T., Vetrov, D. & Wilson, A.G. Averaging weights leads to wider optima and better generalization. arXiv preprint arXiv:1803.05407 (2018).
Fryer, D., Strümke, I. & Nguyen, H. Shapley values for feature selection: The good, the bad, and the axioms. Ieee Access 9, 144352–144360 (2021).
Huang, X. & Marques-Silva, J. On the failings of shapley values for explainability. International Journal of Approximate Reasoning, 109112 (2024).
Holzinger, A., Saranti, A., Molnar, C., Biecek, P., & Samek, W. Explainable ai methods-a brief overview. In: International Workshop on Extending Explainable AI Beyond Deep Models and Classifiers, pp. 13–38 (2020). Springer
Zhou, W., Li, Y., Gu, D., Xu, J., Wang, R., Wang, H. & Liu, C. High expression col10a1 promotes breast cancer progression and predicts poor prognosis. Heliyon 8(10) (2022).
Wang, C. et al. Col10a1 as a prognostic biomarker in association with immune infiltration in prostate cancer. Current Cancer Drug Targets 24(3), 340–353 (2024).
Abdellateif, M.S. Matrix metalloproteinases as a key player in cancer progression (2024).
Ma, B., Ran, R., Liao, H.-Y. & Zhang, H.-H. The paradoxical role of matrix metalloproteinase-11 in cancer. Biomedicine & Pharmacotherapy 141, 111899 (2021).
Zhu, F. & Xu, D. Predicting gene signature in breast cancer patients with multiple machine learning models. Discover Oncology 15(1), 516 (2024).
Bokhari, S. M. Z. & Hamar, P. Vascular endothelial growth factor-d (vegf-d): An angiogenesis bypass in malignant tumors. International Journal of Molecular Sciences 24(17), 13317 (2023).
Janiszewska, M. et al. Subclonal cooperation drives metastasis by modulating local and systemic immune microenvironments. Nature cell biology 21(7), 879–888 (2019).
Bao, Y. et al. Transcriptome profiling revealed multiple genes and ecm-receptor interaction pathways that may be associated with breast cancer. Cellular & molecular biology letters 24, 1–20 (2019).
Zhao, E. et al. The roles of fxyd family members in ovarian cancer: an integrated analysis by mining tcga and geo databases and functional validations. Journal of Cancer Research and Clinical Oncology 149(19), 17269–17284 (2023).
Li, L., Algabri, Y. A. & Liu, Z.-P. Identifying diagnostic biomarkers of breast cancer based on gene expression data and ensemble feature selection. Current Bioinformatics 18(3), 232–246 (2023).
Swami, S., Mughees, M., Mangangcha, I. R., Kauser, S. & Wajid, S. Secretome analysis of breast cancer cells to identify potential target proteins of ipomoea turpethum extract-loaded nanoparticles in the tumor microenvironment. Frontiers in Cell and Developmental Biology 11, 1247632 (2023).
Curtis, C. et al. The genomic and transcriptomic architecture of 2,000 breast tumours reveals novel subgroups. Nature 486(7403), 346–352 (2012).
Parker, J. S. et al. Supervised risk predictor of breast cancer based on intrinsic subtypes. Journal of clinical oncology 27(8), 1160–1167 (2009).
Sparano, J. A. et al. Adjuvant chemotherapy guided by a 21-gene expression assay in breast cancer. New England Journal of Medicine 379(2), 111–121 (2018).
Grisci, B. I., Feltes, B. C., de Faria Poloni, J., Narloch, P. H. & Dorn, M. The use of gene expression datasets in feature selection research: 20 years of inherent bias?. Wiley Interdisciplinary Reviews: Data Mining and Knowledge Discovery 14(2), 1523 (2024).
Acknowledgements
This work was completed as part of the Biostatistics MASAMU group. We would like to thank the MASI Institute for providing an environment conducive to discussing and developing this idea.
Funding
Research reported in this publication was supported by the Fogarty International Center of the National Institutes of Health under Award Number D43TW010547. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Author information
Authors and Affiliations
Contributions
All authors contributed substantially to this work. GKD, MM, and JN computed the features, generated the prediction model, performed experimental comparisons, and drafted the manuscript. All authors participated in the design of the study and helped to draft the manuscript. All authors reviewed the drafts of this manuscript and approved the final version for submission.
Corresponding author
Ethics declarations
Competing interests
The authors declare that there are no competing interests.
Ethics approval
There is no ethical approval required.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Kallah-Dagadu, G., Mohammed, M., Nasejje, J.B. et al. Breast cancer prediction based on gene expression data using interpretable machine learning techniques. Sci Rep 15, 7594 (2025). https://doi.org/10.1038/s41598-025-85323-5
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-85323-5
