
Abstract
Colon cancer, a prevalent malignancy, is subject to intricate immune modulation, which substantially affects both treatment efficacy and prognostic outcomes. Furthermore, colon cancer is highly heterogeneous, and our comprehensive understanding of its immune microenvironment has not yet been fully realized. There is still ample opportunity for in-depth investigation into the composition and interactions of immune cells within colon cancer, as well as their implications for disease prognosis. In this study, we employed single-cell data from colon cancer to distinguish immune cells from non-immune cells through cluster analysis. Furthermore, we conducted an in-depth analysis of myeloid and T cells, which were categorized into 20 distinct cell subpopulations. Functional enrichment analysis revealed T cells’ active involvement in the Fatty Acid Metabolism and Adipogenesis pathways, while immune checkpoint-associated genes (ICGs) were notably upregulated in CD8+ T cells. Subsequent analysis involved calculating gene scores to characterize cell subpopulations, which, when combined with patient survival time analysis, revealed a significant association between the gene characterization score (referred to as “imm-score”) and the survival of colon cancer patients. Specifically, the presence of CD8+-ANXA1hi-T cells was linked to shortened overall survival in the high imm-score subgroup. Subsequently, combined with genomic analysis, patients in the high imm-score subgroup exhibited elevated tumor mutation burden (TMB) and heightened activity in both the epithelial-mesenchymal transition (EMT) and Notch signaling pathway. Finally, according to our new algorithm, scores calculated predicted the effectiveness of immunotherapy for patients. The results revealed that patients with lower scores could achieve better therapeutic outcomes with immunotherapy. This study offers an extensive analysis of the interplay between T cells and myeloid cells within colon cancer tissues, exploring their impact on the survival and prognosis of colon cancer patients. Additionally, it unveils the potential significance of the imm-score in colon cancer, potentially indicating a poor prognosis and providing novel insights into the immune-regulatory mechanisms underlying the disease.
Similar content being viewed by others


Introduction
Colon cancer, ranked among the most prevalent cancers, was estimated to have caused over 1.9 million new cases of colorectal cancer and more than 930,000 related deaths globally in 20201. Less than 20% of patients diagnosed with metastatic colorectal cancer can survive for more than 5 years2. The field of cancer treatment has long grappled with significant challenges, particularly pronounced in the context of colon cancer. While surgery, chemotherapy, and radiotherapy stand as cornerstone treatments in managing cancer progression, their concurrent potent side effects and substantial physiological damage pose an undeniable challenge3. The intricacy of the tumor microenvironment (TME) stands as a primary obstacle in tumor therapy4. The TME encompasses a diverse array of components, including tumor cells, immune cells, blood vessels, and stroma. The intricate interplay among these elements gives rise to a highly dynamic ecosystem. The intricate nature of this microenvironment not only hampers the precise targeting of cancer cells by conventional therapies, but also complicates the prediction of treatment outcomes5.
The tumor microenvironment profoundly influences tumor progression and response to immunotherapy6. Single-cell RNA sequencing (scRNA-seq) offers an unprecedentedly detailed characterization of cellular diversity and the heterogeneous transcriptome, enabling a comprehensive assessment of the complexity of the tumor microenvironment7. Multiple single-cell RNA-seq studies have been conducted in human colorectal cancer (CRC), concentrating on organoid, non-immune, T-cell, myeloid, and multimodal histological analyses8,9,10,11,12. Colorectal cancer, ranking as the third most common cancer globally, imposes a significant burden with its high incidence and mortality rates13. Cumulative genetic alterations in oncogenes and oncogenes (e.g., APC, KRAS, PIK3CA, etc.) drive CRC tumorigenesis14. Additionally, dysfunction of DNA mismatch repair genes (MMR), resulting in microsatellite instability (MSI) and genetic hypermutagenicity, is associated with sporadic CRC tumorigenesis14. Given the complexity of colorectal cancer tumorigenesis and clinical presentations, combinations of surgery, chemotherapy, radiotherapy, and immunotherapy have been employed in the treatment of patients with colorectal cancer. MSI serves as a reliable predictor for CRC patients who derive benefit from PD-1 inhibitors targeting immune checkpoints15. Moreover, higher TMB rates are indicative of improved efficacy of immune checkpoint inhibitors15,16. Despite notable clinical achievements, the response rate to PD-1 blockade among CRC patients remains limited to a minority, with the underlying mechanisms still not fully elucidated. This emphasizes the crucial need for further exploration into the intricacies of the CRC tumor microenvironment.
In this study, we will leverage single-cell sequencing technology to comprehensively analyze the immune cell composition in the tumor microenvironment of colon cancer patients. Through clustering of single-cell data from colon cancer, we delineate immune cells from non-immune cells and subsequently classify them into 20 subpopulations, encompassing myeloid and T cells. The genes characterizing each subpopulation will be scored for each patient using the single-sample gene set enrichment analysis (ssGSEA) algorithm. In the detailed analysis of immune cell subpopulations, we observe that the imm-score is significantly associated with the prognosis of colon cancer patients in multiple independent datasets. Our study aims to conduct an exhaustive functional analysis of the CD8+-ANXA1hi-T cell subpopulation in colon cancer. Through a comprehensive research approach, we seek to elucidate their precise biological characteristics and pivotal role in tumor immunomodulation. Lastly, we will conduct clinical phenotyping to investigate the correlation between the imm-score and clinical outcomes, such as recurrence, in patients with colon cancer. At the same time, we will predict the potential impact of the CD8+-ANXA1hiT cell signature on immunotherapy, aiming to offer more personalized and precise treatment strategies for upcoming colon cancer patients. In this study, we anticipate uncovering novel mechanisms underlying colon cancer immunomodulation, offering fresh perspectives and solutions to address the shortcomings of conventional treatments and improve patient outcomes.
Materials and methods
Data acquisition
Single-cell transcriptome data (GSE132465) and tissue transcriptome data (GSE39582 and GSE17538) related to colon cancer were retrieved from the GEO database. Additionally, tissue transcriptome high-throughput sequencing data from the COAD cohort in the TCGA database were also downloaded. For single-cell data, only 23 tumor samples were included. For tissue transcriptome data, only samples with complete survival information were retained based on clinical records.
Single-cell data processing
For single-cell data, quality control of tumor samples extracted from GSE132465 was conducted using the Seurat package17. This involved filtering based on sequencing depth, total gene count, and the percentage of mitochondrial genes, with the criteria that each cell had a gene count between 500 and 2,000 and that mitochondria accounted for no more than 10% of the total genes. Subsequently, the quality-controlled data were clustered, and the top 2000 genes exhibiting the highest expression value variability across all samples were selected for normalization. Principal component analysis (PCA) was then performed using the top 20 principal components for spatial dimensionality reduction in principal component clustering, with a resolution set to 0.5. Automatic cell annotation was performed using SingleR and celldex6. Additionally, manual annotation categorized cells into six major groups: B cells, T cells, myeloid cells, endothelial cells, epithelial cells, etc.
Immune checkpoint gene expression analysis
According to Guo et al., a total of 79 ICGs were identified through a literature review, predominantly comprising ligands, receptors, or pivotal molecules in the immune checkpoint pathway (see Supplementary Table S1)18. The relative expression levels of these ICGs in T cells compared to myeloid cells were visualized using pheatmap.
Calculating characterization gene scores using ssGSEA
Based on the classification of immune cell subpopulations in previous studies9, we categorized T cells and myeloid cells into 20 subpopulations. Then, we calculated the differential genes of each subpopulation using the FindAllMarkers function, selecting the top five genes expressed by each cell as the characteristic genes of the respective subpopulation. Additionally, we calculated the TCGA score of each COAD sample using ssGSEA19.
One-way COX as well as survival analysis
Patient samples with complete survival time follow-up records were selected to analyze the relationship between the effect of all cellular scores and the length of patient survival using the R package survival. Settlement results revealed that expression levels with a risk percentage greater than 1 had a negative impact on survival, while those with expression levels less than 1 were protective factors for survival. Additionally, based on the median of cellular characteristic-based scores, the samples were divided into two groups of high and low scores for Kaplan-Meier (KM) survival curves. The log-rank test was used between the two groups, with p-values less than 0.05 indicating a significant difference in survival time between the two groups.
Clinical phenotype evaluation
The clinical information of the corresponding samples was obtained from the TCGA website. Clinical phenotypes with categorical significance and consecutive numerical categories suitable for statistical analysis were organized. The differences among different clinical phenotypes were compared based on the imm-score of each sample or the grouping of the samples. Additionally, recurrence information from GSE17538 and GSE38582 was downloaded from GEO. KM analysis was performed based on the grouping by imm-score.
TMB as well as MSI data processing
Exome sequencing data of COAD samples were obtained from the TCGA website. TMB scores for each sample were calculated using maftools20. Wilcoxon tests were performed between scoring subgroups. Additionally, samples were categorized into two groups of high and low TMB scores based on the median of the TMB scores. The TMB subgroups were then combined with the imm-score for KM survival analysis. MSI information from TCGA-COAD was downloaded, and Wilcoxon tests were performed between scoring subgroups.
Evaluating the efficacy of immunotherapy
The degree of immunotherapy response for each sample was calculated using the IOBR package21 to compare the differences between high and low scoring groups. Additionally, this study utilized gene sets C5 (ontology gene sets), H (hallmark gene sets), and C2 (curated gene sets) from the msigdbr database for GSVA scoring. Finally, model validation was conducted using immunotherapy cohort data from the IMvigor210CoreBiologies package to assess the relationship between score scores and immunotherapy response. KM survival analysis was also performed on immune checkpoint gene expression levels based on score groupings.
Statistical analysis
All statistical analyses were conducted using R software (v3.6.3). Correlation matrices were generated using Pearson or Spearman correlation coefficients. Comparisons between the two groups were assessed using the Wilcoxon test. Survival differences were evaluated using KM curves and log-rank tests. p-values < 0.05 were considered statistically significant.
Result
Deciphering cell diversity and differentiation states in colon cancer
To explore the cell diversity and differentiation status in colon cancer patients in-depth, we conducted a comprehensive single-cell cluster analysis. Firstly, we collected GSE132465 data, retaining only the tumor samples. After quality control of the single-cell data, we obtained 47,285 cells. Subsequently, we selected the cells with the top 2,000 expression variability in the dataset and performed PCA analysis. We retained the top 20 principal components for clustering and subclustering, resulting in the classification of cells into 14 subclusters, as depicted in Supplementary Fig. 1. Subsequently, differential analysis was performed on the 14 subgroups to identify the highly expressed genes in each cell subgroup (Fig. 1A). Based on previous reports9, we annotated the 14 subpopulations into 6 categories using cell-specific marker genes, namely epithelial cells, endothelial cells, myeloid cells, T cells, B cells, and fibroblasts. The expression of the respective marker genes (Fig. 1B). To demonstrate the spatial relationship of the 6 subpopulations more clearly, we calculated the spatial coordinates of each cell using the t-SNE algorithm (Fig. 1C). Subsequently, the marker genes of each subpopulation were displayed based on their expression levels (Fig. 1D).

Resolution of cellular components of colon cancer tumor tissues. (A) Heatmap of the expression of TOP5 genes in the 14 subpopulations clustered by Seurat cells, with darker red color representing higher levels of gene expression. (B) Subpopulation classification based on cell-specific markers. (C) tSNE algorithm clustering analysis. (D) Marker gene staining display for six cell types.
Single-cell data analysis revealed abundant infiltration of immune and non-immune cells in the tumor tissues, with the largest proportion being immune cells, specifically T cells and myeloid cells. To further investigate the role of immune cell heterogeneity in colon carcinogenesis, we subdivided T cells and myeloid cells into subpopulations based on previously reported cellular marker genes. Firstly, T cells were classified as CD8+ T cells based on high expression of CD8A and CD8B, while cells were classified as CD4+ T cells based on high expression of IL7R and CD27. Subsequently, T cells were further subdivided based on the highly expressed genes of the respective subpopulations, resulting in the classification of a total of 20 T cell subpopulations (Fig. 2A). Similarly, according to previous literature, myeloid cells were classified into macrophages, monocytes, mast cells, dendritic cells, and neutrophils based on high or low expression of LYZ, CD68, CD163, CST3, and CSF3R. These myeloid cell populations were further subdivided into a total of 20 subpopulations based on the highly expressed genes of each cell type (Fig. 2B). Next, to analyze the immunosuppression-related status, we examined the expression of 79 immune checkpoint-related genes based on previous literature. Initially, we observed that T cells could be classified into CD4+ and CD8+ T cells based on the expression of immune checkpoint genes. However, we noted exceptions in individual cell subpopulations CD4-C01 and CD8-C16, which exhibited high expression in most cases, except for CD4-C01 and CD4-C13, which displayed partially high and low expression levels in the majority of other CD4+ T cells. This observation suggests that CD8+ T cells are primarily responsible for tumor immunosurveillance within our immune microenvironment. Notably, the CD8-C20 and CD8-C06 subpopulations of CD8+ T cells exhibit prominent expression of specific ligand genes, including HLA, BTNL, and LAG3. Our findings indicate that these highly expressed genes play dual roles in both immunosuppression and immune activation. Meanwhile, CD8-C10 exhibits particularly high expression of the immune checkpoint-associated receptor KIR, which belongs to the class of immunosuppressive receptors. This suggests that our C10 cells may be driving the immunosuppressive function in the microenvironment (Fig. 2C). On the other hand, macrophages and monocytes among myeloid cells exhibited a more activated state compared to other subpopulations. Specifically, the Macro-C5 subpopulation predominantly expressed high levels of immune activation-related receptors such as TBFRSF4, PDCD1, TNFRSF18, KIR2DL4, CTLA4, and CD27. Additionally, the Mono-C3 and Mono-C5 subpopulations of monocytes displayed high expression of immune ligands, including genes from the HLA family and CD86, among others. Notably, these genes play bidirectional roles in immune regulation, suggesting that the C3-C5 subpopulations are primarily involved in immune homeostasis within the microenvironment (Fig. 2D).

Immunoregulatory analysis of T cells and myeloid cells. (A, B) Marker display of T cells and myeloid cells reclustering tsne as well as subpopulations; different colored dots represent different cell subpopulations, and the expression of marker genes for each subpopulation is shown using different colored violin plots. (C, D) expression of immune checkpoint-related genes, on the left, all subpopulations of T cells, blue squares represent CD4+ T cells, yellow squares represent CD8+ T cells; on the right, all subpopulations of myeloid cells including DC cells, macrophages, mast cells, monocytes, and neutrophils; Expression patterns of immune checkpoint genes include IC: expressed on immune cells, TC: expressed on tumor cells, TIC: expressed on immune cells, and TIC: expressed on tumor cells. The expression pattern of immune checkpoint genes includes IC: expressed on immune cells, TC: expressed on tumor cells, and TIC: expressed on both immune cells and tumor cells, including three types: ligand, receptor, and coexpression, which can inhibit and activate the immune function.
Exploring functional diversity and differentiation trajectories of immune cell subpopulations in colon cancer tumor microenvironment
To investigate the functions of individual subpopulations of T cells and myeloid cells in the tumor microenvironment, we initially computed the mean expression levels of genes within each cell subpopulation. Subsequently, we employed the GSVA algorithm to calculate pathway scores for HALLMARK pathways within each cell subpopulation. Our investigation revealed a prevalent state of heightened activity in T cells across pathways associated with Fatty Acid Metabolism, Adipogenesis, Estrogen Response Latency, and Glycolysis. Conversely, CD8-C20, CD4-C08, and CD4-C09 exhibited consistently low activity across all pathways. In contrast, CD4-C01, CD8-C06, and CD4-C07 demonstrated notable levels of activity, particularly CD4-C01 displaying elevated activity in pathways linked to Kras Signaling Upregulation, Early Estrogen Response, and Myogenesis. Additionally, CD8-C06 exhibited activity in pathways related to Unfolded Protein Response, Mtorc1 Signaling, and Apoptosis. CD4-C07 exhibited similar characteristics to C06 and demonstrated activity within the Cholesterol Homeostasis pathway. Myeloid cells exhibited heightened activity in pathways associated with the Apical Surface, Hedgehog Signaling, and Apical Junction, while Neu, Mast-C2, Mono-C6, and Mono-C5 displayed diminished activity levels (Fig. 3A). Conversely, all macrophage subpopulations displayed significant activity in inflammation-related pathways, such as the Inflammatory Response, Reactive Oxygen Species, and TNF-α Signaling via NF (Fig. 3B). These findings strongly suggest their involvement in tumor cell development.
To deepen our comprehension of the interrelationships among cell subpopulations, we conducted the cell trajectory analysis as proposed, aiming to discern potential differentiation trajectory associations between cells. Initially, the algorithm identified five distinct differentiation states within the T cell subpopulations (Fig. 3E). Due to the inability of the computer to ascertain the initial differentiation point of the cells, it differentiated the distance of each cell subpopulation along the differentiation trajectory. Thus, only the distribution of possible differentiation timelines for individual cells is simulated, and the location of differentiation initiation cannot be determined (Fig. 3D). Notably, one end predominantly featured the CD8-C05 and CD8-C06 subpopulations, with CD8-C06 being more prevalent, while the other end primarily comprised CD8-C06 subpopulations (Fig. 3C). In addition to the preceding functional analysis, over-representation of CD4-C01 and CD4-C14 subpopulations was observed at the opposite end (Fig. 3C). The preceding findings suggest that while the differentiation trajectory did not distinctly delineate the 20 identified subpopulations, there exist cells exhibiting diverse differentiation trajectories. To elucidate the functional disparities between cell populations situated at the opposite poles of differentiation, we conducted KEGG and GO database enrichment analysis on the top 100 differentially expressed genes within each subpopulation22,23. Our findings reveal that the CD4-C01 subpopulation primarily engages in pathways associated with protein digestion and inflammatory modulation, such as Regulation of Endopeptidase Activity, Regulation of Inflammatory Response, and the Tumor Necrosis Factor-Mediated Signaling Pathway. The CD4-C14 subgroup predominantly engages in protein folding-related pathways, such as the response to unfolded proteins, correction of topologically incorrect proteins, and various other pathways (Fig. 3F, G). At the opposite end of the differentiation trajectory, CD8-C05 is predominantly associated with pathways related to immunoregulation, such as Lymphocyte-Mediated Immunity, Peptide Antigen Assembly with MHC Class II Protein Complex, and other pathways. Conversely, CD8-C06 is primarily implicated in protein folding pathways, akin to the CD4-C14 subpopulation. This suggests that cells located at various stages of differentiation exhibit similar gene expression patterns (Fig. 3H, I). Simultaneously, we delineated the differentiation trajectories of myeloid cell subpopulations. Utilizing our algorithm, we categorized the 20 cell subpopulations into three distinct states. Notably, we observed significant spatial overlap between the distribution sites of macrophages and monocytes. Furthermore, there was a notable coincidence between the distribution of Mast-C2 and macrophages. Conversely, mast cells and DC cells exhibited consistent distribution patterns at the opposite end of the differentiation trajectory (Supplementary Fig. 2). The aforementioned findings indicate that T cells, along with macrophages and monocytes, constitute the predominant cell types within the tumor immune microenvironment. Moreover, distinct subpopulations of T cells exhibit diverse functions and exert regulatory control over immune checkpoints. Notably, among these subpopulations, CD4-C1, CD4-C14, CD8-C05, and CD8-C06 cells demonstrate heightened activity.

Functional enrichment analysis. (A, B) HALLMARK pathway GSVA score, darker red color indicates more active pathway, darker blue color indicates inhibited pathway, T cells on the left and myeloid cells shown on the right. (C) Cell subpopulations clustered at both ends of the differentiation timeline, red indicates CD4 + CTLA4hiT cells, yellow indicates CD4+-FOXP3hiT cells, dark green indicates CD8+-CXCL13hiT cells, light green indicates CD8+-ANXA1hiT cells. (D) Cell differentiation trajectory, which is simulating the time sequence of differentiation. (E) Cell differentiation state classification. Classify 20 types of cells into 5 states indicated by different colors respectively. (F-I) KEGG database annotation analysis.
CD8+-ANXA1hi-T cells are associated with poor prognosis in colon cancer
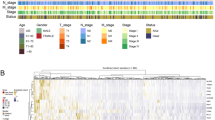
Our previous investigation delved into the distinct roles played by different subpopulations of T cells and myeloid cells within tumor tissues, within the context of the tumor immune microenvironment. Our findings highlighted certain subpopulations intricately linked with cancer progression. Subsequently, to deepen our understanding of how these cells impact the survival outcomes of colon cancer patients, we conducted an analysis of the differential gene expression profiles associated with each of the 40 subpopulations, focusing separately on T cells and myeloid cells. We selected the top 5 highly expressed genes among the differentially expressed genes in each cell type through our calculations to serve as the set of marker genes for respective subpopulations (Fig. 4A, B, Supplementary Fig. 3A, B). The ssGSEA algorithm was subsequently employed to compute the scores of individual cell subpopulations for each sample from TCGA-COAD, GSE17538, and GSE39582 datasets. Subsequently, we categorized the samples into two groups, namely high and low, based on the median scores. Combining this classification with one-way COX analysis of overall survival, we discovered that CD8+-ANXA1hi-T cells, identified by their characteristic score (hereafter referred to as imm-score) (Fig. 4C-E), it is apparent that the observed phenomenon had a notable detrimental effect on the survival outcomes of patients across all three datasets. Concurrently, we conducted survival analysis using Kaplan-Meier methodology for individual cell subpopulations (Supplementary Fig. 4). Consistently, our findings revealed that patients belonging to the high imm-score subgroup exhibited diminished overall survival rates (Fig. 4F-H). The aforementioned findings collectively identify the CD8+-ANXA1hiT cell subpopulation as a significant scoring cell subpopulation in colon cancer. In our investigation, a notable variance in cellular infiltration was observed between the high-scoring and low-scoring groups. Remarkably, the DC-GALhi subpopulation exhibited the highest degree of infiltration in both cohorts, with the low-scoring group exhibiting a notably elevated proportion of this subpopulation, and the proportion of the CD8+-ANXA1hi-T cell subpopulation within the overall CD8+-ANXA1hi-T cell population was relatively low, however, it was evident that the proportion of individuals in the high-scoring group exceeded that in the low-scoring group. Hierarchical cluster analysis was able to partially separate the samples from the high and low score groups, and all of the above results suggest that the imm-score is a poor prognostic feature, and that the survival of patients with a high imm-score is shorter than that of patients with a low imm-score.

Cell subpopulation characterization gene scores influence overall patient survival. (A, B) Heat map of the top five differential genes in T cells. Gene expression levels are higher the darker the red the higher the expression, and conversely, the darker the blue the lower the gene expression level. (C-E) COX univariate analysis of the effect of gene scores characterizing each cell subpopulation on patient survival time. (F-H) Three independent datasets were performed for KM analysis grouped by gene characterization scoring, with red representing the high scoring group and green representing the low scoring group.
Clinical characteristics of the high and low scoring groups
Our prior investigation revealed an association between CD8+-ANXA1hi-T cell subsets and adverse prognosis among patients with colon cancer, particularly evident in those with elevated scores, leading to shortened survival. To provide a comprehensive depiction of the clinical variances between high and low scoring groups, we integrated our analysis with pertinent clinical data sourced from TCGA. In our analysis of cancer clinical staging, we observed a progressive increase in imm-score from stage I to stage IV, with stage I exhibiting the lowest score and stage IV the highest (Fig. 5A). These findings suggest that imm-score may serve as an indicator for determining the clinical staging of patients to some extent. Previously, it was observed that patients exhibiting elevated scores experienced shorter survival durations. Our analysis comparing the survival outcomes between deceased and surviving patients further validated this observation, revealing that deceased patients exhibited higher imm-scores (Fig. 5B). In addition, our findings indicate an increased number of aberrant genomic loci among patients in the high-scoring group (Fig. 5C). This observation implies a potential early cumulative carcinogenic process associated with this group. Additionally, a multitude of clinical features were observed, encompassing cancer cell infiltration into nerves and lymph nodes. Patients exhibiting invasion showed elevated scores compared to those lacking invasion, with notably higher imm-scores evident in individuals with nerve invasion (Fig. 5D). To explore the potential association between high imm-score and shortened patient survival due to cancer recurrence, we analyzed two sets of Bulk RNA-seq sequencing data of colon cancer (GSE17538 and GSE38582). Initially, imm-scores were calculated for individual samples, followed by their categorization into distinct groups. Subsequently, we conducted Kaplan-Meier analysis based on the patients’ tumor recurrence timelines. Our findings revealed a significant correlation between high imm-scores and decreased recurrence-free survival (Fig. 5E, F). We found the same trend of elevated imm-score in samples in the presence of tumor cells invading lymphoid tissue (Fig. 5G, p = 0.081).

Clinical characteristics differences in performance between high and low imm-score groups. (A) Correlation of patient clinical stage with imm-score, yellow represents clinical stage 1, purple represents clinical stage 2, blue represents clinical stage 3, and orange represents clinical stage 4. (B) Correlation between patient survival status and imm-score, with red representing death and blue representing survival. C Comparison of the number of genomic abnormal loci between high and low imm-score groups. (D) Association of tumor neuroinvasion with imm-score. (E-F) Relationship between imm-score and time to recurrence, with red being the high-score group and green being the low-score group. (G) Difference in imm-score between subgroups with or without lymphatic invasion.
Cancer-associated pathway activation in the high-scoring group
Through our studies, we have observed that patients with high scores frequently exhibit a poor prognosis. Subsequently, to delve into the potential molecular mechanisms underlying this correlation, we aim to investigate the variances in molecular pathways between patients with high and low scores. Initially, the GSVA algorithm was employed to evaluate disparities in pathway scores between the high and low scoring cohorts (Fig. 6A, B). The analysis of KEGG database22,24 annotations unveiled significant activation of glycosaminoglycan synthesis, galactose metabolism, and ECM receptor interaction pathways in the high imm-score group, juxtaposed with notable inhibition in the low imm-score group. Conversely, primary bile acids, selenine, histidine, butyric acid, and butyric acid metabolism, along with bile acid metabolism, exhibited pronounced activation in the low imm-score group. The pathways of histidine metabolism, butyric acid metabolism, and fatty acid metabolism exhibited significant activation and inhibition in the high subgroup, as illustrated in Fig. 6A. Furthermore, we utilized the Gene Ontology (GO) database to annotate the disparities in biological processes between the high and low subgroups, including chromosome attachment to the nuclear envelope, negative regulation of astrocyte activation, phosphatidylinositol synthesis, and fatty acid metabolism, as illustrated in Fig. 6A. Phosphatidylinosito 4 kinase activity and NADH regeneration exhibit heightened activity within the higher subgroup, contrasting with UDP-N-acetylglucosamine transmembrane transport, phosphagen metabolic processes, positive regulation of sister chromatid cohesion maintenance, and pH elevation pathways, which display increased activity within the lower subgroup (Fig. 6B). In addition, we conducted a comprehensive analysis of pathways closely associated with tumorigenesis within the HALLMARK dataset for a thorough investigation of their interrelationships. Our findings reveal a significant positive correlation between the imm-score and the DNA repair, NOTCH signaling, and EMT pathways (Fig. 6C). Genomic instability characterized by alterations in the frequency of microsatellite occurrence is referred to as MSI. Microsatellites exhibit a high mutation rate, and MSI is strongly associated with tumors, including colorectal and gastric cancers. The presence of MSI leads to frameshift mutations, resulting in aberrant protein structures encoded by damaged genes, potentially culminating in tumorigenesis in affected cells. MSI is one of the crucial triggers for colon cancer. By comparing the imm-score between the MSI groups, we found that the immune score of the MSI-H group was significantly lower, indicating a significant negative correlation between the immune score and MSI status. Furthermore, our findings showed that patients in the MSS high immune score subgroup had shorter overall survival, while patients in the MSI-H and low immune score subgroup had significantly prolonged survival (Fig. 6D). Additionally, our analysis of genomic data from the TCGA database revealed that patients with low immune scores had higher tumor mutational burden (TMB), and patients with low TMB and high immune scores exhibited significantly shortened overall survival (Fig. 6E).On the other hand, we observed that patients with low tumor heterogeneity also had low imm-scores (Fig. 6F, G). Finally, among colon cancer patients, the top 10 genes with the highest mutation frequency were APC, TP53, TTN, KRAS, SYNE1, MUC16, PIK3CA, FAT4, RYR2, and CSMD3, and their gene expression was negatively correlated with patients’ imm-scores (Fig. 6H, I). In summary, the imm-score can infer the activity level of tumor-associated signaling pathways in colon cancer patients.

Analysis of functional differences in CD8-C06 score subgroups. (A, B) KEGG versus GO annotated GSVA scores for pathways in the top 10 of differences between high and low score subgroups. (C) Correlation analysis of pathways associated with tumor progression with sample CD8-C06 scores. (D-G) Comparison of differences in MSI, TMB, and tumor heterogeneity scores between high and low scoring groups and KM survival analysis. (H) Correlation of top ten mutated genes with CD8-C06 score. (I) TCGA-COAD mutation analysis of the top 10 mutated genes, with R less than 0 indicating that the mutation frequency was negatively correlated with the imm-score, and R greater than 0 representing a positive correlation between the mutation frequency and the imm-score.
Assessment of immunotherapy efficacy between different imm-score groups
Previously, we observed that cohorts with higher scores exhibited heightened activity in tumor growth and metastatic pathways, correlating with a poorer prognosis. Our aim is to delve deeper into whether the imm-score might serve as a predictive marker for responsiveness to immunotherapy or chemotherapy. Therefore, we calculated the immune phenotype scores for each patient based on the transcriptomic data from TCGA-COAD. Our analysis revealed that in the MHC, CP, AZ, and IPS scores, the high imm-score group exhibited significantly lower IPS scores compared to the low-score group (Fig. 7A-D). This suggests that patients with a low imm-score may achieve better therapeutic efficacy when treated with immune checkpoint inhibitors (ICBs). To validate our findings, we downloaded an immunotherapy cohort from the TIED database and conducted Kaplan-Meier analysis, which showed that patients with high imm-scores had significantly higher relapse rates after ICB therapy compared to those with low imm-scores (Fig. 7E). Additionally, in two other cohorts, we observed that the imm-score of patients could partially predict the effectiveness of ICB therapy (Fig. 7F-G). Finally, we observed that the overall survival time of patients with high scores was significantly shorter after ICB therapy (Fig. 7H, I). In summary, our newly developed algorithm can predict the therapeutic response to ICB therapy by calculating the imm-score of cancer patients.

Prediction of Immunotherapy Effectiveness. (A-D) Differences in immunophenotype scores between the two groups, MHC: represents antigen presentation, CP: represents checkpoint, and IPS represents immunophenotype score. (E) KM analysis of the probability of relapse after application of immune checkpoint inhibitors in two groups of patients. (F-G) Determining whether patients respond to immune checkpoint inhibitors based on scores. (H-I) KM analysis of the probability of patient survival after application of immune checkpoint inhibitors in two groups of patients.
Discussion
The involvement of immune cells within the TME is pivotal in the process of tumorigenesis. Numerous studies have elucidated that immune cells associated with tumors possess the capacity to either suppress tumorigenesis or facilitate tumor progression and metastasis. Surprisingly, immune evasion has emerged as a novel hallmark of cancer, presenting an unprecedented opportunity for a paradigm shift in cancer therapy leveraging immune cells to combat cancerous cells. Thus, comprehending the tumor immune microenvironment (TIME) in colon cancer is pivotal for elucidating the biological characteristics of CRC and for informing cancer therapeutic approaches25. Numerous studies have demonstrated that the composition of the tumor immune microenvironment and tumor heterogeneity are intricately linked to the prognosis of colon cancer patients and the effectiveness of immunotherapy26. Consequently, gaining insights into TIME heterogeneity may enrich our understanding of the biological mechanisms underlying CRC initiation and aid in the formulation of immunotherapeutic strategies. Our study indicates that integrating multidimensional multi-omics sequencing with clinical phenotyping holds promise for precisely delineating risk profiles linked to colon cancer prognosis and dissecting the immune microenvironment of colon cancer. Colorectal cancer elicits heterogeneous anti-tumor immune responses, with tumor-infiltrating T cells being identified as promising prognostic indicators27,28. Nevertheless, immune cells can exert dual effects by not only suppressing tumor growth but also promoting angiogenesis, fostering tumor cell proliferation, inhibiting apoptosis, and facilitating extracellular matrix remodeling and metastatic dissemination through modulation of specific inflammatory pathways29,30, thus, our initial step involved the establishment of the single-cell immune landscape specific to colon cancer. Leveraging insights from prior investigations, we characterized T cells and myeloid cells, delineating 40 distinct cell subpopulations each endowed with unique functionalities, thus elucidating the intricate heterogeneity within the tumor microenvironment. While each cell subpopulation displays distinct pathway activities, it is crucial to recognize the pivotal role of immune checkpoints in modulating these pathways. Immune checkpoints serve as essential immunomodulatory factors, exerting both stimulatory and inhibitory effects, thereby maintaining self-tolerance and finely regulating the nature, intensity, and duration of immune responses1. Tumors often exploit the immune checkpoint pathway to evade immune recognition and dampen immune responses31,32. Informed by prior research, we conducted a comprehensive analysis of the expression profiles of 79 ICGs within T cells and myeloid cells18. These genes predominantly mediate ligand-receptor interactions, exerting diverse effects on immune activity, encompassing inhibition, stimulation, or both. Gene expression of immune checkpoints can effectively classify T cells into distinct subsets, such as CD4+ T cells and CD8+ T cells. Among these subsets, certain genes encoding ligands with both inhibitory and stimulatory immune functions are notably upregulated in CD8+ T cells, notably including MHC molecules, BTN, LAG, among others. On the contrary, the majority of CD4+ T cells exhibited downregulation of immune checkpoint-related genes. This study delves further into the intricacies of immune cell differentiation trajectories. Utilizing single-cell RNA sequencing data, we identified five distinct T cell subpopulations representing various stages of differentiation. Notably, subpopulations 1 and 4 occupied opposite ends of the differentiation spectrum. Subpopulation 1 comprised CD4+-CTLA4hi-T cells and CD4+-FOXP3hi-T cells, while subpopulation 2 encompassed CD8+-CXCL13hi and CD8+-ANXA1hiT cell subsets. Genes characteristic of subpopulation 1 primarily participate in apoptosis and IL17-induced inflammation pathways, whereas those in subpopulation 4 are predominantly involved in antigen processing, presentation, and MHC protein complex assembly.
Gene characterization represents a straightforward and efficient predictive model extensively employed in clinical settings33,34,35. Subsequently, we utilized the TOP5 gene expressions within each cell subpopulation as cellular markers, employing ssGSEA to assign scores to individual samples in bulk RNA data, thereby generating a sample-wise scoring matrix. Utilizing one-way COX analysis, we explored the correlation between representative genes from 40 subpopulations and the prognosis of colon cancer. This investigation was further validated across multiple independent datasets. Our findings revealed that the CD8+-ANXA1hi-T cell marker gene signature is indicative of a poor prognosis, with higher scores correlating to shorter overall survival among patients. This identification holds promise as a potential biomarker for predicting colon cancer prognosis. Subsequently, we employed the CD8+-ANXA1hi-T cell signature score to conduct a comprehensive analysis of the TCGA-COAD dataset. Upon integrating clinical data for comparison, our findings revealed that patients at clinical stage 4 exhibited the highest scores, whereas those at clinical stage 1 demonstrated the lowest scores. Furthermore, patients who succumbed to the disease displayed notably higher scores compared to those who survived. In comparison to patients with low scores, those with high scores exhibited a notable surplus of aberrant genomic loci. Additionally, individuals experiencing peripheral nerve cancer infiltration demonstrated markedly elevated scores in contrast to those without such infiltration. In conclusion, our findings suggest a correlation between scores and tumor recurrence in patients. Specifically, patients with higher scores experienced significantly shorter time to tumor recurrence, potentially contributing to the observed shorter overall survival time in this subgroup. Immune checkpoints play a pivotal role in modulating immune responses by orchestrating stimulatory and inhibitory pathways, thereby crucially maintaining self-tolerance and regulating the nature, extent, and duration of immune reactions1. Tumors often exploit immune checkpoint pathways to evade immune surveillance and dampen immune reactions31,32. Immune checkpoint blockade (ICB) therapy, utilizing antibodies targeting various components of the immune checkpoint pathway, has emerged as a prominent and promising approach in cancer immunotherapy. Among the extensively studied molecules, CTLA-4, PD-1, and PD-L1 stand out36,37. Our analysis revealed a correlation between the expression patterns of ICGs and scores, which were notably elevated in high-scoring patients with TCGA-COAD compared to those with lower scores. The predictive results of immunotherapy indicated a superior response in low-scoring patients compared to their high-scoring counterparts.
Our findings indicate that MSI-H patients tend to have higher IMM scores. The elevated IMM score in the MSI-H group is consistent with the current understanding of the tumor immune microenvironment in microsatellite instability-high (MSI-H) colorectal cancer. MSI-H tumors are characterized by a higher mutation burden, leading to the production of more neoantigens, which subsequently triggers a stronger immune response. This immune activation may be reflected in the elevated IMM score, making these tumors more sensitive to immune checkpoint inhibitors (ICIs).
In our study, we conducted a thorough examination of the interplay between T cell and macrophage attributes within tumor tissues, illuminating their significance in predicting colon cancer prognosis at the single-cell level. Our findings not only shed light on the underlying biological mechanisms and functions associated with these gene signatures but also offer novel perspectives for further investigation. Our findings contribute to a more nuanced comprehension of the infiltration patterns exhibited by immune cells within the tumor microenvironment of colon cancer, a comprehension pivotal for refining the design and implementation of therapeutic interventions. Moreover, predictive biomarkers and models established through our research hold promise as invaluable tools for guiding immunotherapy and other anti-cancer strategies. Given the escalating utilization of multi-omics data in cancer research, prospective investigations are warranted to delve into the genomic and epigenetic alterations occurring within immune cells within the microenvironment, thus affording a deeper insight into the intricate mechanisms governing intercellular communication among diverse immune cell subpopulations in the context of cancer progression.
In future research, we plan to further explore the specific functions and mechanisms of these cell subsets. To ensure the robustness and reproducibility of our findings, we will use multiple techniques and independent samples for validation. Moreover, we will establish animal models to observe the actual role of these cell subsets in tumor progression and treatment response. Additionally, we aim to perform further clinical data correlation analyses to assess the feasibility and potential of these findings in clinical applications.
Conclusion
This study integrates single-cell transcriptome sequencing, tissue transcriptome sequencing, and genomic analysis to elucidate profound alterations in the dynamics of subpopulations of immune cells within the context of colon cancer progression. Through this comprehensive approach, we identified a distinctive subset of CD8+-ANXA1hi-T cells strongly associated with adverse prognostic outcomes in colon cancer. Furthermore, our investigation delineated the unique biological attributes of CD8+-ANXA1hi-T cells in colon cancer and their correlation with clinical phenotypes among patients. These findings not only offer novel insights into immune regulation mechanisms in colon cancer but also furnish robust evidence supporting the development of future personalized therapeutic strategies.
Data availability
The data that support the findings of this study are openly available in TCGA at https://www.cancer.gov/ccg/research/genome-sequencing/tcga. Single-cell transcriptome data (GSE132465) and tissue transcriptome data (GSE39582 and GSE17538) related to colon cancer were retrieved from the GEO database.
Abbreviations
- ICGs:
-
immune checkpoint-associated genes
- TMB:
-
tumor mutation burden
- EMT:
-
epithelial-mesenchymal transition
- TME:
-
tumor microenvironment
- scRNA-seq:
-
Single-cell RNA sequencing
- CRC:
-
colorectal cancer
- MMR:
-
mismatch repair genes
- ssGSEA:
-
gene set enrichment analysis
- PCA:
-
Principal component analysis
- KM:
-
Kaplan-Meier
- GO:
-
Gene Ontology
- KEGG:
-
Kyoto Encyclopedia of Genes and Genomes
References
Pardoll, D. M. The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer. 12, 252–264. https://doi.org/10.1038/nrc3239 (2012).
Siegel, R. L. et al. Colorectal cancer statistics, CA: a cancer journal for clinicians 73, 233–254, (2023). https://doi.org/10.3322/caac.21772 (2023).
Miller, K. D. et al. Cancer treatment and survivorship statistics, 2022. Cancer J. Clin. 72, 409–436. https://doi.org/10.3322/caac.21731 (2022).
Binnewies, M. et al. Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat. Med. 24, 541–550. https://doi.org/10.1038/s41591-018-0014-x (2018).
Bian, S. et al. Single-cell multiomics sequencing and analyses of human colorectal cancer. Science 362, 1060–1063. https://doi.org/10.1126/science.aao3791 (2018).
Aran, D. et al. Reference-based analysis of lung single-cell sequencing reveals a transitional profibrotic macrophage. Nat. Immunol. 20, 163–172. https://doi.org/10.1038/s41590-018-0276-y (2019).
Papalexi, E. & Satija, R. Single-cell RNA sequencing to explore immune cell heterogeneity. Nat. Rev. Immunol. 18, 35–45. https://doi.org/10.1038/nri.2017.76 (2018).
Roerink, S. F. et al. Intra-tumour diversification in colorectal cancer at the single-cell level. Nature 556, 457–462. https://doi.org/10.1038/s41586-018-0024-3 (2018).
Zhang, L. et al. Lineage tracking reveals dynamic relationships of T cells in colorectal cancer. Nature 564, 268–272. https://doi.org/10.1038/s41586-018-0694-x (2018).
Zhang, L. et al. Single-cell analyses inform mechanisms of myeloid-targeted therapies in Colon cancer. Cell 181, 442–459e429. https://doi.org/10.1016/j.cell.2020.03.048 (2020).
Lee, H. O. et al. Lineage-dependent gene expression programs influence the immune landscape of colorectal cancer. Nat. Genet. 52, 594–603. https://doi.org/10.1038/s41588-020-0636-z (2020).
Li, H. et al. Author correction: reference component analysis of single-cell transcriptomes elucidates cellular heterogeneity in human colorectal tumors. Nat. Genet. 50, 1754. https://doi.org/10.1038/s41588-018-0299-1 (2018).
Schrock, A. B. et al. Tumor mutational burden is predictive of response to immune checkpoint inhibitors in MSI-high metastatic colorectal cancer. Ann. Oncol. 30, 1096–1103. https://doi.org/10.1093/annonc/mdz134 (2019).
Bogaert, J. & Prenen, H. Molecular genetics of colorectal cancer. Ann. Gastroenterol. 27, 9–14. https://doi.org/10.1016/j.immuni.2019.06.021 (2014).
Le, D. T. et al. PD-1 blockade in tumors with Mismatch-Repair Deficiency. N Engl. J. Med. 372, 2509–2520. https://doi.org/10.1056/NEJMoa1500596 (2015).
Yarchoan, M., Hopkins, A. & Jaffee, E. M. Tumor mutational burden and response rate to PD-1 inhibition. N Engl. J. Med. 377, 2500–2501. https://doi.org/10.1056/NEJMc1713444 (2017).
Hao, Y. et al. Integrated analysis of multimodal single-cell data. Cell 184, 3573–3587e3529. https://doi.org/10.1016/j.cell.2021.04.048 (2021).
Hu, F. F. et al. Expression profile of immune checkpoint genes and their roles in predicting immunotherapy response. Brief. Bioinform. 22 https://doi.org/10.1093/bib/bbaa176 (2021).
Hänzelmann, S., Castelo, R. & Guinney, J. GSVA: gene set variation analysis for microarray and RNA-seq data. BMC Bioinform. 14 https://doi.org/10.1158/1078-0432.Ccr-14-1860 (2013).
Mayakonda, A. et al. Maftools: efficient and comprehensive analysis of somatic variants in cancer. Genome Res. 28, 1747–1756. https://doi.org/10.1101/gr.239244.118 (2018).
Zeng, D. et al. Multi-omics immuno-oncology biological research to decode tumor microenvironment and signatures. Front. Immunol. 12, 687975. https://doi.org/10.3389/fimmu.2021.687975 (2021).
Kanehisa, M. & Goto, S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 28, 27–30. https://doi.org/10.1093/nar/28.1.27 (2000).
Ashburner, M. et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 25, 25–29. https://doi.org/10.1038/75556 (2000).
Kanehisa, M. et al. KEGG for taxonomy-based analysis of pathways and genomes. Nucleic Acids Res. 51, D587–d592. https://doi.org/10.1093/nar/gkac963 (2023).
Lei, X. et al. Immune cells within the tumor microenvironment: Biological functions and roles in cancer immunotherapy. Cancer Lett. 470, 126–133. https://doi.org/10.1016/j.canlet.2019.11.009 (2020).
Ogino, S. et al. Integrative analysis of exogenous, endogenous, tumour and immune factors for precision medicine. Gut 67, 1168–1180. https://doi.org/10.1136/gutjnl-2017-315537 (2018).
Alexander, P. G., McMillan, D. C. & Park, J. H. The local inflammatory response in colorectal cancer - type, location or density? A systematic review and meta-analysis. Cancer Treat. Rev. 83, 101949. https://doi.org/10.1016/j.ctrv.2019.101949 (2020).
Pagès, F. et al. International validation of the consensus immunoscore for the classification of colon cancer: a prognostic and accuracy study. Lancet (London England). 391, 2128–2139. https://doi.org/10.1016/s0140-6736(18)30789-x (2018).
Shalapour, S., Karin, M., Pas de & Deux Control of anti-tumor immunity by Cancer-Associated inflammation. Immunity 51, 15–26. https://doi.org/10.1016/j.immuni.2019.06.021 (2019).
Hanahan, D. & Weinberg, R. A. Hallmarks of cancer: The next generation. Cell 144, 646–674. https://doi.org/10.1016/j.cell.2011.02.013 (2011).
Beatty, G. L. & Gladney, W. L. Immune escape mechanisms as a guide for cancer immunotherapy. Clin. Cancer Res. 21, 687–692. https://doi.org/10.1056/NEJMc1713444 (2015).
Marin-Acevedo, J. A. et al. Next generation of immune checkpoint therapy in cancer: New developments and challenges. J. Hematol. Oncol. 11, 39. https://doi.org/10.1136/gutjnl-2017-315537 (2018).
Paik, S. et al. A multigene assay to predict recurrence of tamoxifen-treated, node-negative breast cancer. N. Engl. J. Med. 351, 2817–2826. https://doi.org/10.1056/NEJMoa041588 (2004).
Väyrynen, J. P. et al. The prognostic role of macrophage polarization in the colorectal cancer microenvironment. Cancer Immunol. Res. 9, 8–19. https://doi.org/10.1158/2326-6066.Cir-20-0527 (2021).
Parker, J. S. et al. Supervised risk predictor of breast cancer based on intrinsic subtypes. J. Clin. Oncology: Official J. Am. Soc. Clin. Oncol. 27, 1160–1167. https://doi.org/10.1200/jco.2008.18.1370 (2009).
Francisco, L. M., Sage, P. T. & Sharpe, A. H. The PD-1 pathway in tolerance and autoimmunity. Immunol. Rev. 236, 219–242. https://doi.org/10.1038/s41590-018-0276-y (2010).
Romo-Tena, J., Gómez-Martín, D. & Alcocer-Varela, J. CTLA-4 and autoimmunity: New insights into the dual regulator of tolerance. Autoimmun. rev. 12, 1171–1176. https://doi.org/10.1016/j.autrev.2013.07.002 (2013).
Funding
This study was supported by the Tianjin Key Medical Discipline (Specialty) Construction Project (TJYXZDXK-035 A).
Author information
Authors and Affiliations
Contributions
All authors contributed to this article: Jing Guo and Zili Zhang designed the study, Jing Guo acquired the data.Jing Guo analyzed the data and edited the manuscript, Zili Zhang revised the manuscript. All authors read and approved the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Guo, J., Zhang, Z. Integrated multi-omics unveils novel immune signature for predicting prognosis in colon cancer patients. Sci Rep 15, 9788 (2025). https://doi.org/10.1038/s41598-025-85390-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-025-85390-8
