
Abstract
Previous research suggests mitochondrial apoptosis alleviates rheumatoid arthritis (RA), but the role of mitochondrial apoptosis-related genes (MARGs) is unclear. Urgent exploration of RA-related mitochondrial apoptosis biomarkers is needed. Gene Expression Ontology (GEO)-derived RA datasets were used to identify differentially expressed genes (DEGs) compared to normal controls, intersected with MARGs to obtain differentially expressed mitochondrial apoptosis-related genes (DE-MARGs). Three ML algorithms screened diagnostic biomarkers. A nomogram was built and validated by receiver operating characteristic (ROC) analysis. Gene Set Enrichment Analysis (GSEA), regulatory network, and drug prediction explored biomarker mechanisms. Finally, key cells analysis included clustering, type annotation, pseudo-temporal study, and interaction, focusing on validated biomarker expression in those cells. A total of 147 DE-MARGs linked to energy & ROS metabolism were identified. Four validated biomarkers (MRPS10, EEF2, HSPA9, TUFM) formed a new RA diagnostic model. Moreover, GSEA linked them to oxidative phosphorylation. YY1 regulates EEF2, HSPA9, MRPS10; FOXO3 regulates EEF2, TUFM. Drugs like Nonoxynol-9, Nedocromil, Gadobutrol target these biomarkers. In addition, biomarkers are expressed in plasmablasts, with CD74 as a key receptor binding multiple ligands. RA biomarkers (MRPS10, EEF2, HSPA9, TUFM) linked to energy & ROS, progression tied to AMPK/mTOR, CD74-MIF crucial. Study advances RA pathogenesis knowledge, supporting clinical diagnosis.
Similar content being viewed by others

Introduction
Rheumatoid arthritis (RA) is an autoimmune disease characterized by swollen joints, pain, and stiffness as its main symptoms, accompanied by synovial hyperplasia and inflammatory cell infiltration within the joint1. As is widely known, RA not only affects the bones and joints but also impacts extra-articular organs such as the heart, eyes, lungs, and blood vessels, shortening the life expectancy of approximately one in five adults worldwide1,2. Although the early treatment could alleviate joint deformities and joint damage, the current treatments for RA mainly focus on symptom relief, deformity correction, and functional rehabilitation exercises, the overall efficacy is far from satisfactory3,4. Therefore, there is a pressing need to explore the key genes associated with RA and discover more useful therapeutic targets for RA.
Mitochondria are the “powerhouses” of eukaryotic cells, producing ATP through the tricarboxylic acid cycle and oxidative phosphorylation5.They are involved in various biological processes, such as signal transduction, reducing oxidation, cell cycle regulation and apoptosis. Apoptosis is a programmed mechanism of cell death, and the apoptosis of mitochondria is associated with a wide range of pathology.Mitochondrial homeostasis imbalance can lead to the development of autoimmune rheumatic diseases6,7. Reactive oxygen species (ROS) play an important role in mitochondrial apoptosis, which can directly damage mitochondrial DNA to induce apoptosis of fine cells8, In response to DNA damage or cellular stress, pro-apoptosis protein activation opens the permeable transition pore (PTP), accumulates ROS, and triggers apoptosis9. Abnormal regulation of mitochondrial apoptosis affects ROS production and release, and indirectly affects synovial inflammation and joint damage10. In recent years, it has been found that the internal environment imbalance caused by mitochondrial damage plays a key role in the pathology of rheumatoid arthritis (RA)11, such as cold air plasma can improve RA through mitochondrial apoptosis pathway and inhibit synovial hyperplasia12 Previous studies have focused on ROS accumulation and its association with synovial inflammation and joint injury, but the mechanisms by which mitochondrial apoptosis affects RA and the role of mitochondrial-associated genes (MARGs) in RA remain unclear. Therefore, this study is no longer limited to the already widely studied aspects of ROS and disease phenotype, extending its focus to the mechanisms by which mitochondrial apoptosis affects rheumatoid arthritis (RA) and the role of mitochondrial-associated genes (MARGs) in RA.
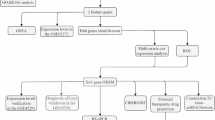
Therefore, this study combined single-cell RNA-seq andbulk transcriptomic data to identify the detailed mechanisms of mitochondrial apoptosis in RA at the cellular and molecular levels.Our aim was to identify key genes associated with mitochondrial apoptosis in RA and to establish an early diagnostic model for the disease, which will provide new references for the clinical diagnosis and treatment of RA.
Results
Functional enrichment analysis of 147 DE-MARGs
A total of 889 DEGs were identified between RA and control samples in the GSE17755, with 491 DEGs were up-regulated and 398 DEGs were down-regulated (Fig. 1a). The 889 DEGs were taken to intersect with MARGs, and a total of 147 DE-MARGs were obtained (Fig. 1b). Next, showed the GO functional enrichment results of the 147 DE-MARGs, among which the most notable ones were ‘mitochondrial inner membrane’, ‘mitochondrial matrix’, ‘response to oxidative stress’, ‘generation ofprecursor metabolites and energy’, ‘mitochondrial protein-containing complex’, ‘organelle outer membrane’, and etc. (Fig. 1c). Then, the KEGG functions of 147 DE-MARGs were mainly distributed in the areas of ‘diabetic cardiomyopathy’, ‘apoptosis’, and ‘necrosis’ (Fig. 1d). Subsequently, a very strong linkage between these 147 DE-MARGs was clearly demonstrated in the PPI network (Fig. 1e), such as the strongest interaction between genes ACTB, HSP90AB1 and GAPDH.

Functional enrichment analysis of de-marg. (a) Volcano plot of differentially expressed genes between RA/CTL samples. Up- and down-regulation of each TOP10 gene is labeled in the figure. (b) Differential MRAGs Gene Wayne Diagram. (c) Bubble map of GO enrichment for genes with differential MRAGs. (d) KEGG pathway enrichment of differential MRAGs genes in Bagua. (e) Network of protein interactions for genes with differential MRAGs.
Determined diagnostic biomarkers for RA
A total of 11 candidate feature genes were screened by performing 10-fold cross-validation in LASSO logistic regression at a lambdamin (lambda value at the lowest cross-validation error rate) of 0.03367 (Fig. 2a).Meanwhile, the SVM-RFE algorithm identified the 20 most important candidate feature genes for predicting RA (Fig. 2b,c). Then the Boruta algorithm then filters out 23 important candidate feature genes (Fig. 2d), and based on the importance rankings, the top10 genes were selected for subsequent analysis. Finally, a total of seven diagnostic biomarkers (MRPS10, CFL1,EEF2, HSPA9, CDC42, TUFM, and HSPA8) were obtained for subsequent analysis by overlapping genes from LASSO, SVM-RFE and Boruta algorithm (Fig. 2e).

Biomarkers for the diagnosis of RA. (a) LASSO regression analysis to screen for characterized genes. On the left is a graph of the gene coefficients, and on the right is a cross-validated error plot. (b,c) Candidate characterization genes were screened using SVM-RFE in GSE17755. (d) Boruta’s algorithm for ranking the importance of genes. Green color represents confirmed important genes, red color is confirmed unimportant genes, and yellow color is uncertain genes. (e) Wayne diagrams for LASSO-Logistic, SVM-RFE, and Boruta algorithms.
The performance of the diagnostic model
The AUC values of these seven diagnostic biomarkers related to mitochondrial apoptosis in the ROC curves were all greater than 0.9, thus indicating that these seven genes have a strong clinical diagnostic value for RA (Fig. 3a). Subsequently, validation in the GSE93272 dataset yielded significant differences in the expression of MRPS10, EEF2, HSPA9, and TUFM and the trend was consistent with the GSE17755 dataset.In these two datasets, except for the upregulation of MRPS10, the other three genes were down - regulated (Fig. 3b,c). Moreover, the RT-qPCR results reveal that EEF2, HSPA9, and TUFM were notably down-regulated in clinical RA sample. However, the results of MRPS10 and the database exhibited a contrasting trend, with no significant difference observed in expression levels between the groups (Fig. 3d). Next, the nomogram model can be used clinically to the diagnose of RA disease (Fig. 3e). The ROC curve of the nomogram model showed an accurate predictive value (AUC > 0.9) (Fig. 3f). In the DCA, the nomogram curve was higher than the gray line as well as the curves of MRPS10, HSPA9, TUFM, and EEF2 (Fig. 3g), which implied that the model has significant discriminative ability and clinical utility in predicting RA at a high risk threshold of 0–1. And the results of the clinical impact curve also proved the significant predictive ability of the nomogram model (Fig. 3h).

The performance of the diagnostic model. (a) GSE17755 Intersecting gene ROC curves. (b) GSE17755 Intersecting Gene Expression Box Plot. (c) GSE93272 Intersecting gene expression box plot. (d) Biomarker expression levels in clinical samples were compared using RT-qPCR.(e) Column line diagram of key genes. (f) Calibration curves for evaluating the predictive power of line graph models. (g) DCA curves for assessing the value of clinical applications of columnar graphical models. (h) Clinical impact curves for nomogram modeling.
GSEA and functional network of biomarkers
The relevant signaled pathways to which these 4 biomarkers were enriched to were demonstrated, and the results showed that they were mainly involved in ‘ribosomes’, ‘oxidative phosphorylation’, ‘neuroactive ligand-receptor interactions’, and etc. (Fig. 4a–d). Secondly, in GeneMANIA results showed that there were various relationships between key genes and other genes such as co-expression, the same location, physical interactions, genetic interactions, sharing protein structural domains, or participating in the same pathway (Fig. 4e). Immediately followed this the NetworkAnalyst database predicted that nine differentially expressed transcription factors have regulatory relationships with key genes. Among them, FOXO3 could regulate EEF2 and TUFMsimultaneously, TRIM28 could regulate both EEF2 and HSPA9, KLF4 could regulate both HSPA9 and MRPS10 at the same time, YY1 could regulate EEF2, HSPA9 and MRPS10 (Fig. 4f). Finally, the targeting relationships between genes and drugs were demonstrated, with each drug being associated with at least 2 biomarkers. For example, Clofarabine and Clocortolone were related to EEF2 and HSPA9, Cholecalciferol and Calcitriol were related to MRPS10 and HSPA9 (Fig. 4g).

GSEA and functional network of biomarkers. (a) MRPS10-KEGG enrichment ridge map. (b) EEF2-KEGG-enriched ridge map. (c) HSPA9-KEGG-enriched ridge map. (d) TUFM-KEGG-enriched ridge map. (e) GeneMANIA key gene co-expression network. (f) DE-TFs-gene regulatory network. (g) DrugBank database of key genes-drugs Sankey map.
Identification of cell subtypes
A total of 2000 genes with high variability in expression among cells were labeled, and the top 10 genes were IGJ, MZB1, S100A9, IGLL5, LYZ, PTGDS, S100A8, IFI27, GNLY, and AL928768.3 (Fig. 5a). The data were then processed by PCA downscaling to divide the core cells into 9 separate cell clusters (Fig. 5b,c). Immediately afterward, these 9 cell clusters were annotated, resulting in 5 cell groups, namely B cells, NK cells, T cells, monocytes and plasmablasts (Fig. 5d,e).

Cell clustering analysis and type annotation. (a) Genes with highly variable intercellular expression. (b,c) Core cell clustering visualization. (d,e) Umap cell clustering distribution map for 9 cell subpopulations.
Involvement of plasmablasts in autoimmunity
The biomarkers were all expressed in plasmablasts (Fig. 6a). Monocytes had the highest number of receptor-ligands with plasmablasts (Fig. 6b). Subsequently, the specific receptor-ligands between cell-cells were demonstrated (Fig. 6c), such as the receptor-ligand between plasmablasts and B cells and T cells, which include CD74_COPA, CD74_MIF, and HLA-C_FAM3C; the receptor ligand between plasmablasts and monocytes include CD74_APP, CD74_COPA, CD74_MIF, and HLA-DPB1_TNFSF13B, all of which are receptor ligands associated with plasmablasts (p < 0.05). Finally, plasmoblasts were divided into 2 subclasses, and C1 was more differentiated than C2 in the results of the proposed time-series analysis (Fig. 6d–f).

Cellular communication analysis. (a) Key gene expression violin map. (b) Heatmap of ligand-receptor number of interactions between cell subpopulations. (c) Cell-cell receptor ligands. (d) Plasmoblast cell clustering umap visualization. (e,f) Proposed temporal sequence analysis of plasmacytoblasts.
Discussion
In recent years, several studies have shown that the internal environment imbalance resulting from mitochondrial damage plays a key role in the pathology of RA, and is one of the core aspects of RA pathology11,12. However, the mechanism by which mitochondrial apoptosis plays a role in RA, and the role that MARGs plays in RA have been unclear. In this study, we first screened 147 DE-MARGs of RA, and these genes were associated with mitochondrial transmembrane transport, energy generation, apoptosis, cell death induced by oxidative stress, reactive oxygen species metabolism process, oxidative stress, and etc.
MARGs may affect the production of ROS by regulating the related proteins on the mitochondrial membrane. The human mitochondrial membrane is extremely sensitive to peroxidation, and oxidative damage can cause membrane dysfunction, which in turn triggers apoptosis13. In addition, reactive oxygen species can damage proteins, fats, and DNA in the mitochondria, activate mitochondrial autophagy, and, in severe cases, also lead to apoptosis14. On the one hand, as we all known, mitochondria are fundamental to cellular metabolism and physiology6,15, and mitochondrial apoptosis and mitophagy are evolutionarily conserved cellular process to remove dysfunctional or redundant mitochondria and maintain energy metabolism16,17 On the other hand, apoptosis is required to control synovial hyperplasia in RA. Apoptosis can occur through two distinct pathways (the extrinsic pathway and the internal pathway), in which the internal pathway can be initiated in the mitochondria in response to oxidative stress18. Oxidative stress is a state of imbalance between oxidation and antioxidation in the body, which can lead to the production of high levels of reactive oxygen species (ROS) in cells. ROS can damage mitochondrial DNA (mtDNA), and the accumulation of mtDNA damage can lead to mitochondrial damage and dysfunction, ultimately triggering mitochondrial diseases19. In patients with rheumatoid arthritis (RA), the oxidative stress state is associated with enhanced inflammatory responses, which can accelerate joint destruction and disease progression20, and can also induce exacerbation of immune-inflammatory imbalances and promote cellular and tissue damage21. In RA patients, the levels of oxidative stress markers, such as malondialdehyde (MDA), are often elevated and associated with enhanced inflammatory responses22. Additionally, the generation and accumulation of reactive oxygen species can cause oxidative stress in the mitochondrial inner membrane, leading to damage to the membrane structure and function, which can trigger mitochondrial diseases. Reactive oxygen species can also directly oxidize proteins on the mitochondrial inner membrane, such as components of the respiratory chain complex, altering the structure and function of the complex and affecting electron transfer efficiency.In summary, both the increased energy demand on mitochondrial electron transport and hypoxia could lead to an enhanced production of reactive oxygen species (ROS), creating oxidative stress in synoviocytes18,23. In our results, the related functions of these 147 DE-MARGs of RA were also annotated to the related functions of energy metabolism and ROS metabolism, which were consistent with the mechanism associated with mitochondrial apoptosis in RA.
Based on the above 147 DE-MARGs, we further obtained four mitochondrial apoptosis-related biomarkers of RA, namely MRPS10, HSPA9, TUFM, and EEF2. The expression levels of HSPA9, TUFM,and EEF2 were significantly lower in RA samples compared tonormal control samples, while the expression level of MRPS10was significantly higher in RA samples. It is worth noting that Guoqi Dong et al. also indentified EEF2 as a biomarker of RA and reported significantly low expression levels in RA samples, which aligns with our findings. Eukaryotic elongation factor2 (EEF2) is the central substrate encoding for EEF2kinase, also known as calmodulin-dependent protein kinase III, which regulates protein synthesis24,25. Previous research has indicated that EEF2 could promote apoptosis by upregulating the expression of autophagy-related genes and by inhibiting the mTOR pathway26. Dean Minnock et al. suggested that EEF2 kinase promoted cellular autophagy by activating the MAPK signaling pathway to improve fibroblast differentiation in RA27. In our analysis, the GSEA results showed a significant positive association between EEF2 with oxidative phosphorylation. Therefore, we speculated that EEF2 was a key mitochondrial apoptosis-related gene in response to oxidative stress, and played a crucial role in the progression of RA pathology.
Unfortunately, none of the three biomarkers were reported in RA. Nonetheless, we found that these genes were significantly associated with mitochondrial apoptosis-related pathways and functions. For example, mitochondrial translation elongation factor Tu (TUFM) has been implicated in protein translation elongation and biosynthesis, oxidative phosphorylation, etc28,29. TUFM has been reported to promote the apoptotic function of the mitochondria. BiRou Zhong et al. suggested that TUFMwas involved in disease pathologies associated with ROS30, and Ruyuan Wei et al. also suggested that TUFM could regulate mitochondrial autophagy and apoptosis via the AMPK/mTOR signaling pathway31, which is consistent with the mechanism of EEF2 in RA that we supposed. Similarly, the down-regulation of heat shock protein family A member 9 (HSPA9) could enhance mitochondrial stress by increasing mitochondrial fragmentation and the production of mitochondrial ROS (mtROS)32. But unlike EEF2 and TUFM, the oxidative stress response of HSPA9 seems to be more relevant with the TP53/Akt signaling pathway32,33,34. Besides, mitochondrial ribosomal small subunit protein 10 (MRPS10) is a subunit encoding mammalian mitochondrial ribosomal proteins that promotes protein engagement in mitochondria35 but its association with mitochondrial apoptosis has not been reported. Together, these studies all implicated oxygen stress in biomarkers associated with mitochondrial apoptosis as a key mechanism affecting RA progression.
Moreover, we performed biomarker mapping of cell subtypes. The distribution of B cell subtypes will change with the progression of RA. Specifically, the circulating total memory B cells and pre-switch antigen experienced memory B cells proportions will be significantly reduced as RA develops, and clinically, increased plasmablasts levels are also one of the pathological hallmarks of RA36,37. In our study, all these biomarkers were expressed in plasmablasts, further enhancing the theoretical basis for clinical detection of RA. In addition, we noted that CD74 was perhaps a critical receptor. CD74 is a membrane receptor whose binding to macrophage migration inhibitory factor (MIF) ligand is critical in the progression and pathogenesis of RA, inducing the expression of inflammatory cytokines and molecules degrading tissues, and promoting the proliferation and survival of fibroblast-like synoviocytes38,39. Seung Ah Yoo et al. further suggested that ligand receptor binding of CD74_MIF would affect the expression of downstream MAPK and RhoA signaling, and lead to fibroblast differentiation in RA40. Interestingly, we also pointed out previously that the EEF2 kinase could promote cellular autophagy by activating the MAPK signaling pathway to improve fibroblast differentiation in RA27. CD74_MIF/EEF2/MAPK perhaps a new regulatory axis involved in the course of RA, which needs further experimental verification.
Immediately afterwards, we made predictions for the upstream TFs of the biomarker. We found that YY1 could regulate EEF2, HSPA9 and MRPS10 simultaneously, FOXO3 could regulate EEF2 and TUFM at the same time, while TRIM28 could regulate EEF2 and HSPA9 concurrently. Additionally, KLF4 could regulate HSPA9 and MRPS10 simultaneously. YY1 has been extensively studied in RA as a key TF, and numerous studies have demonstrated a significant correlation of its transcriptional regulation with apoptosis41,42. Although there are no reports of YY1 associated with these three biomarkers, we noticed that YY1 regulates multiple signaling pathways, including PI3K-Akt-mTOR signaling pathway, production of ROS, and oxidative phosphorylation involved in the progression of RA43,44,45. Similarly, KLF4 was also significantly correlated with the phosphorylation levels in RA46. Notably, JinChen Guo et al. showed that phosphorylation of FOXO3 could enhance its transcriptional activity and activate AMPK, further ameliorating the oxidative stress status in RA patients47. We also pointed out earlier that TUFM and EEF2 could regulate mitochondrial autophagy and apoptosis via the AMPK/mTOR signaling pathway in RA26,27,31. These studies imply that the YY1/FOXO3-TUFM/EEF2-AMPK/mTOR axis perhaps is a key mechanism of mitochondrial apoptosis in RA.
At length, based on these four biomarkers, we constructed a clinical diagnostic model of RA, and further predicted the targeted drugs for the biomarkers. The results showed that clofarabine and clocortolone were related to EEF2 and HSPA9, Cholecalciferol and Calcitriolwere related to MRPS10 and HSPA9. Cholecalciferol and Calcitriol have been used clinically to treat or improve RA48,49,50, but the specific mechanism has yet to be explored. Our study provided a rationale for the clinical application of these drugs.However, it must be acknowledged that this study has certain limitations. Firstly, this study conducted bioinformatics analysis using public databases. Although external datasets and RT-qPCR were used to validate the results, the study was still limited by factors such as sample sources and data standardization. The biological significance of the results still needs to be confirmed through experimental validation. Secondly, there are differences in the RT-qPCR results and data set expression of key genes. On one hand, there are differences in sample sources. Factors such as racial, geographical differences, and sample processing methods may affect the results. On the other hand, the sample size used for RT-qPCR is relatively small. A smaller sample size may not accurately reflect the overall gene expression characteristics, which may lead to results that do not match the actual trend. To address this issue and further verify the reliability of existing research results, we have planned to increase the sample size in subsequent studies. By expanding the sample size, it is expected to reduce the bias caused by small sample size and sample heterogeneity, thereby effectively validating our research conclusions.
Conclusions
In summary, this study identified four mitochondrial apoptosis-related biomarkers of RA, which could affect RA progression by regulating energy metabolism and ROS metabolism. Furthermore, we believe that the progression of the RA is significantly related to AMPK/mTOR signaling pathway, withCD74_MIF playing a key role. Through the study of MARGs, this study deepened the understanding of the pathogenesis of rheumatoid arthritis (RA), provided a theoretical basis for the study of the pathogenesis of RA, and contributed to the clinical diagnosis and improvement of its accuracy. In the future, we will construct cell and animal models to further verify the clinical value of biomarkers, and develop more effective RA therapeutics based on the discovered drug targets, or explore combination drug regimens to improve treatment efficacy.
Materials and methods
Data source
The RA datasets GSE17755 (training dataset) and GSE159117 (single cell dataset), along with GSE93272 (validation dataset), were retrieved from the Gene Expression Ontology (GEO) database (https://www.ncbi.nlm.nih.gov/geo/). The GSE17755 dataset contained transcriptome sequencing data from 112 peripheral blood samples of RA patients and 45 peripheral blood samples from healthy controls (Supplementary Table S1)51. The GSE159117 single-cell sequencing dataset was derived from RNA-seq of peripheral blood mononuclear cells (PBMC) from 1 RA patient52. The GSE93272 dataset contained peripheral blood samples from 68 RA and 43 controls with transcriptome sequencing data from peripheral blood samples (Supplementary Table S2)53. The search in the MitoMiner database (http://mitominer.mrc-mbu.cam.ac.uk/) identified 1,967 MARGs.
Identify and functional enrichment analysis of differentially expressed mitochondrial apoptosis-related genes (DE-MARGs)
Gene expression data were extracted from RA samples and healthy control samples in the GSE17755 and screened for differentially expressed genes (DEGs) using the ‘limma’ package (version 3.46.0) with |log2FC| > 0.5, p.value < 0.054,55. Then, the intersection of DEGs with MARGs was taken using ‘ggVennDiagram’ package (version 1.2.2) to capture DE-MARGs, which were plotted in a venn diagram for presentation56. Next, the DE-MARGs underwent Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG)57,58,59 enrichment analysis (p. adj < 0.05) using the ‘clusterProfiler’ package (version 4.4.4) and the ‘org.Hs.eg.db’ package (version 3.15.0)60,61.
Construction of protein-protein interaction (PPI) networks and screened of hub genes
The PPI network of DE-MARGs was exhibited using STRING (http://string-db.org), followed by the presentation of the PPI network using Cytoscape (version 3.8.2) (STRING score was set as 0.7)62. Subsequently, the hub genes were screened according to five algorithms, namely, degree, maximum neighborhood component (MNC), maximum clique centrality (MCC), edge percolation component (EPC), and density of maximum neighborhood component (DMNC), using the cytoHubba plug-in in Cytoscape, and the UpSet graph was generated for demonstration using the ‘UpSetR’(version 1.4.0)63.
Identification of diagnostic biomarkers for RA
First, the candidate hub genes were screened based on the optimal coefficients and values of lambda utilising the ‘glmnet’ (version 4.1-6) proceed the least absolute shrinkage and selection 0perator (LASSO) analysis64. Next, support vector machine recursive feature elimination (SVM-RFE) was used to determine the optimal variables to screen by removing the feature vectors generated by SVM. Then, the candidate hub genes were screened using the R package ‘Boruta’ (version 7.0.0), and the top 10 genes were selected for subsequent analysis using the Z-Score as a measure of importance65. Finally, diagnostic biomarkers were identified at the junction of the above methods.
Verification of biomarkers
Genes with consistent expression in the validation and training sets were screened as diagnostic biomarkers for subsequent analyses. The usefulness of biomarkers in diagnosing RA was assessed by generating receiver operating characteristic (ROC) curves using the ‘pROC’ package (version 1.18.0) and calculating the area under the curve (AUC) and 95% confidence intervals66. Then, ‘rms’ (version 6.5-0) was used to create nomograms, and the incidence of RA could be assessed based on the total score67. And was regarded as having diagnostic value when the AUC exceeded 0.7. In addition, decision curve analysis (DCA) was executed to indicate the discriminatory validity of the nomogram for RA.
Potential mechanisms and drug prediction of biomarkers
Single-sample gene set enrichment analysis (ssGSEA) was executed on the biomarkers utilizing the ‘clusterProfiler’ (version 4.4.4) and based on the GSE17755 datasets, with the filtering condition of p. adj < 0.0560. The ssGSEA ridge plot for top10 results (p. adj ordering) was presentedusing GseaVis (version 0.0.5)68. In addition, the co-expression network of hub genes was explored using GeneMANIA (http://genemania.org/). Meanwhile, transcription factors targeting the biomarkers were predicted using the NetworkAnalyst (https://www.networkanalyst.ca/) database and demonstrated by Cytoscape (version 3.8.2)62. Finally, the DrugBank (https://go.drugbank.com/) online database was used to predict the targeted drugs associated with the biomarkers, and the targeting relationship between the biomarkers and drugs was demonstrated using Cytoscape.
Identification of different cell types
The GSE159117 was filtered using the function of the ‘Seurat’ package (version 4.0.5), retaining genes with expression data in at least 3 cells and cells with a number of detected genes above 200 (min.cells = 3, min.features = 200)69. Next, biomarkers were calculated using the ‘PercentageFeatureSet’ function, cells with less than 10% of biomarkers were retained, and violin plots were drawn to characterize the data. Next, the data were normalized using ‘NormalizeData’, and the ‘FindVariableFeatures’ function was used to identify genes with high variability in expression between cells (feature selection) for subsequent identification of cell types (default parameter is ‘vst’). The default parameter was ‘vst’ (2000 highly variable genes were selected by the method). For further examination, data that were chosen before the principal component analysis (PCA) inflection point were used. Then, unsupervised cluster analysis of the cells was done using the ‘Seurat’ package (version 4.0.5) ‘FindNeighbors’ and ‘FindClusters’ functions, with clusters visualized using Uniform Manifold Approximation and Projection (UMAP)69. Subsequently, ‘FindAllMarkers’ was used to set the parameters min.pct = 0.5, only.pos = TRUE, and logfc.threshold = 0.5 to search for positive marker genes in various populations. Finally, the marker genes of each cell subpopulation were used to determine the type of subpopulation of cells by comparing these genes with the marker genes of each cell type in the CellMarker database, and the clusters were visualized by UMAP70.
Identification and correlation analysis of key cells
UMAP was employed to demonstrate the expression of the biomarkers in various cell subpopulations. Secondly, CellPhoneDB was used to perform intercellular ligand-receptor interaction analysis to identify the key cells. Next, unsupervised cluster analysis was done to classify the key cells using the ‘seurat’ package (version 4.0.5) ‘FindNeighbors’ and ‘FindClusters’ functions69. Finally, the differential ‘GeneTest’ function was used to determine which genes were expressed differently among key cell groups, and the proposed time-series analysis was performed using ‘monocle’package (version 2.18.0)71.
Reverse transcription quantitative polymerase chain reaction (RT-qPCR)
To further validate our findings, biomarker expression levels in clinical samples were compared using RT-qPCR. Blood samples from Shanxi Provincial People’s Hospital hospital included 10 control and 10 PR samples (Supplementary Table S3). RNA was extracted and purified from the samples using TRIzol (Ambion, Austin, USA) following the manufacturer’s instructions. Subsequently, the RNA was reverse transcribed into cDNA using the SureScript First-strand cDNA synthesis kit (Servicebio, Wuhan, China). The resulting cDNA was analyzed using 2xUniversal Blue SYBR Green qPCR Master Mix (Servicebio) with specific primer sequences (Supplementary Table S4). RT-qPCR amplification was performed under the following thermal cycling conditions: initial denaturation at 95 °C for 1 min, followed by 40 cycles of denaturation at 95 °C for 20 s, annealing at 55 °C for 20 s, and extension at 72 °C for 30 s. Data analysis was performed using the 2−ΔΔCt method, with GAPDH serving as the internal reference for normalization.
Ethics declarations
This study was performed in line with the principles of the Declaration of Helsinki. Approval was granted by the Ethics Committee of Shanxi Provincial People’s Hospital (No566). Informed consent was obtained from all individual participants included in the study.
Statistical analysis
All analyses were conducted using the R(version 4.2.2) programming language, and the data from different groups were compared by the Wilcoxon test. Unless specified otherwise, pvalueless than 0.05 was considered statistically significant.
Data availability
The datasets analysed during the current study are available in the the [GEO] repository, [https://www.ncbi.nlm.nih.gov/geo/] and the search in the [MitoMiner] repository, [http://mitominer.mrc-mbu.cam.ac.uk/].
References
Smolen, J. S., Aletaha, D. & McInnes, I. B. Rheumatoid arthritis. Lancet (London England). 388(10055), 2023–2038 (2016).
Lee, D. M. & Weinblatt, M. E. Rheumatoid arthritis. Lancet (London England). 358(9285), 903–911 (2001).
Sayah, A. & English, J. C. 3rd Rheumatoid arthritis: A review of the cutaneous manifestations. J. Am. Acad. Dermatol. 53(2), 191–209 (2005). quiz 10 – 2.
Gibofsky, A. Epidemiology, pathophysiology, and diagnosis of rheumatoid arthritis: A synopsis. Am. J. Manag. Care 20(7 Suppl), S128–S135 (2014).
Andrieux, P., Chevillard, C., Cunha-Neto, E. & Nunes, J. Mitochondria as a Cellular hub in infection and inflammation. Int. J. Mol. Sci. 22(21) (2021).
Leishangthem, B. D., Sharma, A. & Bhatnagar, A. Role of altered mitochondria functions in the pathogenesis of systemic lupus erythematosus. Lupus 25(3), 272–281 (2016).
Cui, L. et al. Rheumatoid arthritis and mitochondrial homeostasis: the crossroads of metabolism and immunity. Front. Med. 9(2022).
Xian, S. et al. Inhibition of mitochondrial ROS-mediated necroptosis by Dendrobium Nobile Lindl. Alkaloids in carbon tetrachloride induced acute liver injury. J. Ethnopharmacol. ;330. (2024).
Weindel, C. G. et al. Mitochondrial ROS promotes susceptibility to infection via gasdermin D-mediated necroptosis. Cell 185(17), 3214–31e23 (2022).
Prajapati, P. et al. TRIM32 regulates mitochondrial mediated ROS levels and sensitizes the oxidative stress induced cell death. Cell. Signal. 76, 109777 (2020).
Barrera, M. J. et al. Dysfunctional mitochondria as critical players in the inflammation of autoimmune diseases: Potential role in Sjögren’s syndrome. Autoimmun. rev. 20(8), 102867 (2021).
Ding, C. et al. Cold air plasma improving rheumatoid arthritis via mitochondrial apoptosis pathway. Bioeng. Transl. Med. 8(1), e10366 (2023).
<nfykdxxb-41-202107980.pdf>.
Li, L. et al. A mitochondrial membrane-bridging machinery mediates signal transduction of intramitochondrial oxidation. Nat. Metab.
Lopez, J. & Tait, S. W. Mitochondrial apoptosis: Killing cancer using the enemy within. Br. J. Cancer 112(6), 957–962 (2015).
Modanloo, M. & Shokrzadeh, M. Analyzing mitochondrial dysfunction, oxidative stress, and apoptosis: Potential role of L-carnitine. Iran. J. Kidney Dis. 13(2), 74–86 (2019).
Estaquier, J., Vallette, F., Vayssiere, J. L. & Mignotte, B. The mitochondrial pathways of apoptosis. Adv. Exp. Med. Biol. 942, 157–183 (2012).
Panga, V., Kallor, A. A., Nair, A., Harshan, S. & Raghunathan, S. Mitochondrial dysfunction in rheumatoid arthritis: A comprehensive analysis by integrating gene expression, protein-protein interactions and gene ontology data. PloS One. 14(11), e0224632 (2019).
ATAD3B is a mitophagy receptor mediating clearance of oxidative stress-induced damaged mitochondrial DNA. EMBO J. 40(8) (2021).
<scdxxbyxb-55-2-249.pdf>.
<nfykdxxb-41-202107965.pdf>.
<ZhongNanDaXueXueBaoYiXueBan-47-2-183.pdf>.
Ng, C. T. et al. Synovial tissue hypoxia and inflammation in vivo. Ann. Rheum. Dis. 69(7), 1389–1395 (2010).
Ryazanov, A. G., Shestakova, E. A. & Natapov, P. G. Phosphorylation of elongation factor 2 by EF-2 kinase affects rate of translation. Nature 334(6178), 170–173 (1988).
Cheng, Y. et al. eEF-2 kinase dictates cross-talk between autophagy and apoptosis induced by akt inhibition, thereby modulating cytotoxicity of novel akt inhibitor MK-2206. Cancer Res. 71(7), 2654–2663 (2011).
Xie, C. M., Liu, X. Y., Sham, K. W., Lai, J. M. & Cheng, C. H. Silencing of EEF2K (eukaryotic elongation factor-2 kinase) reveals AMPK-ULK1-dependent autophagy in colon cancer cells. Autophagy 10(9), 1495–1508 (2014).
Minnock, D. et al. Effects of acute aerobic, resistance and combined exercises on 24-h glucose variability and skeletal muscle signalling responses in type 1 diabetics. Eur. J. Appl. Physiol. 120(12), 2677–2691 (2020).
Choi, C. Y., Vo, M. T., Nicholas, J. & Choi, Y. B. Autophagy-competent mitochondrial translation elongation factor TUFM inhibits caspase-8-mediated apoptosis. Cell Death Differ. 29(2), 451–464 (2022).
Lei, Y., Wen, H. & Ting, J. P. The NLR protein, NLRX1, and its partner, TUFM, reduce type I interferon, and enhance autophagy. Autophagy 9(3), 432–433 (2013).
Zhong, B. R. et al. TUFM is involved in Alzheimer’s disease-like pathologies that are associated with ROS. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 35(5), e21445 (2021).
Wei, R. et al. Silencing TUFM Inhibits development of monocrotaline-induced pulmonary hypertension by regulating mitochondrial autophagy via AMPK/mTOR signal pathway. Oxid. Med. Cell. Longev. 2022, 4931611 (2022).
Bae, J. E. et al. Enhanced primary ciliogenesis via mitochondrial oxidative stress activates AKT to prevent neurotoxicity in HSPA9/mortalin-depleted SH-SY5Y cells. Mol. Brain. 16(1), 41 (2023).
Liu, T. et al. Knockdown of HSPA9 induces TP53-dependent apoptosis in human hematopoietic progenitor cells. PloS One 12(2), e0170470 (2017).
Butler, C. et al. HSPA9/mortalin inhibition disrupts erythroid maturation through a TP53-dependent mechanism in human CD34+ hematopoietic progenitor cells. Cell. Stress Chaperones 29(2), 300–311 (2024).
Revathi Paramasivam, O., Gopisetty, G., Subramani, J. & Thangarajan, R. Expression and affinity purification of recombinant mammalian mitochondrial ribosomal small subunit (MRPS) proteins and protein-protein interaction analysis indicate putative role in tumourigenic cellular processes. J. BioChem. 169(6), 675–692 (2021).
Kinslow, J. D. et al. Elevated IgA plasmablast levels in subjects at risk of developing rheumatoid arthritis. Arthritis Rheumatol. (Hoboken, NJ) 68(10), 2372–2383 (2016).
Reijm, S. et al. Autoreactive B cells in rheumatoid arthritis include mainly activated CXCR3 + memory B cells and plasmablasts. JCI Insight 8(20) (2023).
Morand, E. F., Leech, M. & Bernhagen, J. MIF: A new cytokine link between rheumatoid arthritis and atherosclerosis. Nat. Rev. Drug Discov. 5(5), 399–410 (2006).
Sánchez-Zuno, G. A. et al. Canonical (CD74/CD44) and non-canonical (CXCR2, 4 and 7) MIF receptors are differentially expressed in rheumatoid arthritis patients evaluated by DAS28-ESR. J. Clin. Med. 11(1) (2021).
Yoo, S. A. et al. MIF allele-dependent regulation of the MIF coreceptor CD44 and role in rheumatoid arthritis. Proc. Natl. Acad. Sci. U.S.A. 113(49), E7917–e26 (2016).
Wang, Y. et al. miR-410-3p Regulates Proliferation and Apoptosis of fibroblast-like Synoviocytes by Targeting YY1 in Rheumatoid Arthritis119109426 (Biomedicine & pharmacotherapy = Biomedecine & pharmacotherapie, 2019).
Xiang, L., Yang, W., Wang, F. & Liu, G. Circ_0083964 knockdown impedes rheumatoid arthritis progression via the mir-204-5p-dependent regulation of YY1. J. Orthop. Surg, Res. 17(1), 558 (2022).
Nierode, G. J. et al. High-throughput identification of factors promoting neuronal differentiation of human neural progenitor cells in microscale 3D cell culture. Biotechnol. Bioeng. 116(1), 168–180 (2019).
Zhan, Y., Yang, Z., Zhan, F., Huang, Y. & Lin, S. SIRT1 is transcriptionally repressed by YY1 and suppresses ferroptosis in rheumatoid arthritis. Adv. Rheumatol. (London, England) 63(1), 9 (2023).
Zheng, Q., Wang, D., Lin, R., Lv, Q. & Wang, W. IFI44 is an immune evasion biomarker for SARS-CoV-2 and Staphylococcus aureus infection in patients with RA. Front. Immunol. 13, 1013322 (2022).
Ye, Q., Luo, F. & Yan, T. Transcription factor KLF4 regulated STAT1 to promote M1 polarization of macrophages in rheumatoid arthritis. Aging 14(14), 5669–5680 (2022).
Guo, J. C. et al. [Effect of Huangqin Qingre Chubi Capsules containing serum on oxidative stress and protein expression of AMPK and FoxO3a in rheumatoid arthritis patients]. Zhongguo Zhong Yao Za Zhi = Zhongguo zhongyao zazhi = China. J. Chin. materia Med. 45(13), 3228–3232 (2020).
Bellan, M. et al. Is cholecalciferol a potential disease-modifying anti-rheumatic drug for the management of rheumatoid arthritis? Clinical and experimental rheumatology. 38(2), 343–349 (2020).
Hee, J. Y., Cai, B. & Thomas, R. Liposomal delivery of self-peptide and calcitriol as tolerogenic immunotherapy in rheumatoid arthritis: An exploration using sensory science. Immunol. Cell Biol. 102(5), 353–357 (2024).
Sonigra, A. et al. Randomized phase I trial of antigen-specific tolerizing immunotherapy with peptide/calcitriol liposomes in ACPA + rheumatoid arthritis. JCI Insight 7(20). (2022).
Lee, H. M., Sugino, H., Aoki, C. & Nishimoto, N. Underexpression of mitochondrial-DNA encoded ATP synthesis-related genes and DNA repair genes in systemic lupus erythematosus. Arthritis Res. Therapy 13(2), R63 (2011).
Li, J. et al. Significance of pyroptosis-related gene in the diagnosis and classification of rheumatoid arthritis. Front. Endocrinol. 14, 1144250 (2023).
Tasaki, S. et al. Multi-omics monitoring of drug response in rheumatoid arthritis in pursuit of molecular remission. Nat. Commun. 9(1), 2755 (2018).
Ritchie, M. E. et al. Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 43(7), e47 (2015).
He, Z., Jiang, Q., Li, F. & Chen, M. Crosstalk between venous thromboembolism and periodontal diseases: A bioinformatics analysis. Dis. Markers 2021, 1776567 (2021).
Gao, C. H., Yu, G. & Cai, P. ggVennDiagram: An intuitive, easy-to-use, and highly customizable R package to generate Venn diagram. Front. Genet. 12, 706907 (2021).
Kanehisa, M. & Goto, S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 28(1), 27–30 (2000).
Kanehisa, M. Toward understanding the origin and evolution of cellular organisms. Protein Sci. 28(11), 1947–1951 (2019).
Kanehisa, M., Furumichi, M., Sato, Y., Kawashima, M. & Ishiguro-Watanabe, M. KEGG for taxonomy-based analysis of pathways and genomes. Nucleic Acids Res. 51(D1), D587–D92 (2023).
Yu, G., Wang, L. G., Han, Y. & He, Q. Y. clusterProfiler: An R package for comparing biological themes among gene clusters. Omics: J. Integr. Biol. 16(5), 284–287 (2012).
Zhang, C. et al. Genome-wide mutation profiling and related risk signature for prognosis of papillary renal cell carcinoma. Ann. Transl. Med. 7(18), 427 (2019).
Shannon, P. et al. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 13(11), 2498–2504 (2003).
Conway, J. R., Lex, A. & Gehlenborg, N. UpSetR: An R package for the visualization of intersecting sets and their properties. Bioinf. (Oxford England). 33(18), 2938–2940 (2017).
Yang, L. et al. Powerful identification of large quantitative trait loci using genome-wide R/glmnet-Based regression. J. Heredity 113(4), 472–478 (2022).
Maurya, N. S., Kushwah, S., Kushwaha, S., Chawade, A. & Mani, A. Prognostic model development for classification of colorectal adenocarcinoma by using machine learning model based on feature selection technique boruta. Sci. Rep. 13(1), 6413 (2023).
Robin, X. et al. pROC: An open-source package for R and S + to analyze and compare ROC curves. BMC Bioinform. 12, 77 (2011).
Wells, B. J. et al. Predicting current glycated hemoglobin values in adults: Development of an algorithm from the electronic health record. JMIR Med. Inf. 6(4), e10780 (2018).
Chen, K., Wang, M., Long, D., Zou, D., Li, X., Wang, R., Wang, Y. & Yang, L. Cerebrospinal fluid proteomic profiles in patients with postherpetic Neuralgia. J. Proteome Res. 22(12), 3879–3892 (2023).
Hao, Y. et al. Integrated analysis of multimodal single-cell data. Cell 184(13), 3573–87e29 (2021).
Zhang, Y. et al. Single-cell analyses of renal cell cancers reveal insights into tumor microenvironment, cell of origin, and therapy response. Proc. Natl. Acad. Sci. U.S.A. 118(24) (2021).
Qiu, X. et al. Single-cell mRNA quantification and differential analysis with census. Nat. Methods. 14(3), 309–315 (2017).
Acknowledgements
We would like to express our sincere gratitude to all individuals and organizations who supported and assisted us throughout this research.
Funding
This work was supported byYouth Project of Natural Science Foundation of Shanxi Province (20191D21517); Natural Science Foundation of Shanxi Province (202203021221267); Research Project of Shanxi Administration of Traditional Chinese Medicine (2024ZYYC037); Shanxi Provincial People’s Hospital Education and Teaching Reform and Innovation Project (2023Z012).
Author information
Authors and Affiliations
Contributions
Conghui Gao analyzed and interpreted the patient data regarding the hematologicaldisease and the transplant. Chengqiang ZhangandLixing We n performed thehistological examination of the kidney, and Lixing Wenwas a major contributor inwriting the manuscript.All authors participated in the protocol design and ideaformulation process of the study and provided valuable comments on the experime ntsand data analysis. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Gao, C., Zhang, C., Wen, L. et al. Regulation of reactive oxygen species and the role of mitochondrial apoptotic-related genes in rheumatoid arthritis. Sci Rep 15, 2165 (2025). https://doi.org/10.1038/s41598-025-85460-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-025-85460-x
