
Abstract
Routine use of genetic data in healthcare is much-discussed, yet little is known about its performance in epidemiological models including traditional risk factors. Using severe COVID-19 as an exemplar, we explore the integration of polygenic risk scores (PRS) into disease models alongside sociodemographic and clinical variables. PRS were optimized for 23 clinical variables and related traits previously-associated with severe COVID-19 in up to 450,449 UK Biobank participants, and tested in 9,560 individuals diagnosed in the pre-vaccination era. Associations were further adjusted for (i) sociodemographic and (ii) clinical variables. Pathway analyses of PRS were performed to improve biological understanding of disease. In univariate analyses, 17 PRS were associated with increased risk of severe COVID-19 and, of these, four remained associated with COVID-19 outcomes following adjustment for sociodemographic/clinical variables: hypertension PRS (OR = 1.1, 95%CI 1.03–1.18), atrial fibrillation PRS (OR = 1.12, 95%CI 1.03–1.22), peripheral vascular disease PRS (OR = 0.9, 95%CI 0.82–0.99), and Alzheimer’s disease PRS (OR = 1.14, 95%CI 1.05–1.25). Pathway analyses revealed enrichment of genetic variants in pathways for cardiac muscle contraction (genes N = 5; beta[SE] = 3.48[0.60]; adjusted-P = 1.86 × 10−5). These findings underscore the potential for integrating genetic data into epidemiological models and highlight the advantages of utilizing multiple trait PRS rather than a single PRS for a specific outcome of interest.
Similar content being viewed by others

Introduction
As genetic testing becomes more cost-effective, interest has grown over its potential utility in epidemiological modelling and ultimately clinical care. Indeed, its use in healthcare has become increasingly common over the past decade, with polygenic risk scores (PRS) developed for several diseases and the first pilot study introducing PRS to clinical practice currently being performed in the UK National Health Service (NHS)1,2,3,4,5. However, limited evidence exists regarding the performance of genetic data in epidemiological models when incorporated alongside common sociodemographic and clinical variables.
There is great potential value in integrating genetic data into epidemiological studies, particularly for complex diseases that are influenced by heritable risk factors. An individual’s germline genotype data could be used to develop a proxy measure of their propensity to develop each of these risk traits. The use of PRS, which summarise an individual’s genetic propensity to a trait6, may reduce the need for time-consuming collection of clinical data and minimize the impact of human bias on disease risk modelling.
Clinical traits and biomarkers may not always be available in electronic health records, and can be difficult to collect consistently across institutions and countries, due to variation in diagnostic criteria and subjective clinical decision making7,8. Predictive models based on historical clinical diagnostic records may also miss a large section of the affected or at-risk population, including those with a high probability of developing future disease but lacking diagnoses (e.g. pre-diabetics8), limiting their efficacy. Therefore, the use of PRS as genetic proxies for both the disease of interest and for related traits where possible could benefit epidemiological studies and ultimately healthcare systems.
Coronavirus disease 2019 (COVID-19), the condition caused by the spread of the highly transmissible severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), had a devastating effect on health and economies worldwide9. However, the rapid scientific response to the COVID-19 pandemic has resulted in a wealth of genetic and clinical data, from which several sociodemographic (e.g. obesity, male sex, older age), clinical (e.g. diabetes, comorbidity count) and genetic risk factors for poor COVID-19 outcomes have been identified2,10,11,12,13,14,15,16,17,18,19. The large volume of data generated during the COVID-19 pandemic, and the strong genetic component observed in COVID-19 outcomes (with heritability estimates of up to 41% for COVID-19 severity)20, make it an ideal case study to investigate the integration of PRS into epidemiological models.
In this study, we tested several trait PRS for associations with hospitalization, critical care admission and death from COVID-19, and sequentially adjust associations for sociodemographic and clinical variables. We highlight the value of integrating genetic data into epidemiological models along with established risk factors, and use in silico pathway analyses of PRS to reveal a shared aetiology of traits which could be leveraged to provide better insights into disease pathogenesis.
Methods
Data source
This study was approved by UK Biobank (Application 24559), the population-based cohort that links sources of biological and phenotypic data on > 500,000 individuals in the UK. All methods were performed in accordance with the relevant guidelines and regulations. Self-report questionnaires and baseline biological measurements were recorded from the years 2006–2010, when participants (then aged 40–69 years) were recruited21.
Study population
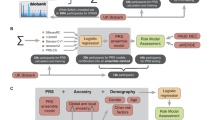
Details of the study population and COVID-19 datasets used in this work may be found in Crossfield et al. (2022)2. To summarize, UK Biobank participants with baseline assessment data, who passed genetic quality control (QC) were included in the study. Individuals included were from assessment centres in England, alive at the start of the study period (1 January 2020) and had not withdrawn consent (Fig. 1). COVID-19 diagnosis was defined as ICD-10 code U071 or U072 from hospital or death certificate data, or a positive laboratory test result. Furthermore, both a transethnic population and a “white European” subpopulation were included in the study. The white European subpopulation was defined as those who lay within the European genetic principal component (PC) cluster, as well as having one of several self-reported “white European” ethnicities in baseline data (n = 404,534/450,577 = 89.78% of entire cohort)2.

Summary of the cohort selection flow used in this study.
Study outcomes
The primary outcome for this study was severe COVID-19, a composite formed from those with a hospital or critical care admission within 28 days of COVID-19 diagnosis (including admissions 1–3 days preceding diagnosis, to account for laboratory testing delays), with a secondary outcome of death within 100 days of COVID-19 diagnosis. Disease controls were defined as those who had a COVID-19 diagnosis but were not hospitalized, had no critical care admission (both within 28 days), and did not die within 100 days following diagnosis. All analyses were performed in both a transethnic cohort (2,109 cases and 5,970 controls for severe COVID-19 and 636 cases and 7,443 controls for COVID-19 mortality) and the white European subset of these (1,833 cases and 5,162 controls for severe COVID-19 and 570 cases and 6,425 controls for COVID-19 mortality).
Variable selection
Clinical variables, including related traits, were selected for PRS modelling based on our previous COVID-19 severity and mortality models2. Additional covariates included previously defined sociodemographic variables (e.g. age and Townsend deprivation index), a previously developed COVID-19 PRS optimised in a white European population (hereafter named “PRSe2” maintaining the nomenclature used in our original publication) and selected clinical variables and related traits based on prior observational evidence (e.g. cardiovascular disease [CVD], angina, and comorbidity count; Supplementary Methods; Supplementary Tables 1–2)2.
Statistical analyses
Statistical analyses were performed in R v3.6.222 to model the risk of severe COVID-19 using logistic regression, and model risk of death (over a period of 100 days post-diagnosis) using Cox proportional hazards regression. Details regarding the modelling of specific variables may be found in Supplementary Methods.
Polygenic risk score associations
PRS were optimized for prediction of the selected clinical variables and related traits in an independent cohort and then tested for association with severe COVID-19 (Fig. 2). Details of QC and PRS optimization are outlined in Supplementary Methods. Briefly, for each PRS, a genome-wide association study (GWAS) was performed in PLINK v1.923, regressing the phenotype on each genetic variant using either linear or logistic regression in the white European subpopulation, including the top 10 PCs from principal component analysis (PCA) as covariates to adjust for population stratification. Samples from the COVID-19 cohort were removed from the UK Biobank cohort prior to trait GWAS analyses, to ensure no overlap between the cohorts at the PRS optimization stage. Summary statistics from each clinical variable GWAS were provided as training datasets to optimize PRS using the clumping and thresholding approach implemented in PRSice v2.3.3, adjusting for the top 10 PCs from PCA as covariates24.

Outline of the analysis steps taken in this study. BMI, body mass index; BMR, basal metabolic rate; WHR, waist-hip ratio; BF, body fat percentage; MI, myocardial infarction; TIA, transient ischaemic attacks; AF, atrial fibrillation; PVD, peripheral vascular disease; HF, heart failure; T1D, type 1 diabetes; T2D, type 2 diabetes; HbA1c, glycated haemoglobin; GWAS, genome-wide association study; FUMA, Functional Mapping and Annotation of Genome-Wide Association Studies; CVD, cardiovascular disease; CRD, chronic respiratory disease; COPD, chronic obstructive pulmonary disease; CKD, chronic kidney disease; CLD, chronic liver disease.
PRS were then tested for association with each COVID-19 outcome in univariate analyses (in both the transethnic and white European cohorts) and those PRS with a likelihood ratio (LR) test P-value < 0.05 were combined in a model of severe COVID-19, and another of COVID-19 mortality. To remove highly correlated PRS, a correlation matrix was formed using the regression coefficients from each of these models separately, and one PRS from each pair with a regression coefficient correlation R2 ≥ 0.8 was removed, retaining the most clinically relevant trait guided by a review of the literature. To further refine the model and remove redundant variables, backwards stepwise regression was performed and PRS with a LR test P < 0.05 were retained in the models (henceforth known as “SeverityM1PRS” and “MortalityM1PRS”; Supplementary Methods). PRS odds ratios (ORs) are reported per unit change in standard deviation.
Adjustment for sociodemographic and clinical covariates
PRS in the SeverityM1PRS and MortalityM1PRS models were then adjusted for previously reported socio-demographic variables2, creating “SeverityM2SocioPRS” and “MortalityM2SocioPRS” respectively.
Prior to adjustment of PRS for clinical variables and related traits in our models, univariate analyses were performed to identify clinical traits associated with COVID-19 severity or COVID-19 mortality, and variables with a LR test P-value of < 0.05 were further adjusted for sociodemographic variables. To remove highly correlated clinical variables, a correlation matrix was formed using the regression coefficients of the clinical variables and one variable from each pair with a regression coefficient correlation R2 ≥ 0.8 was removed, guided by a review of the literature. Removal of redundant variables was then performed using backwards stepwise regression, with sociodemographic variables retained in models due to prior evidence of COVID-19 outcome associations, creating “SeverityM3ClinicoDem” and “MortalityM3ClinicoDem” (Supplementary Methods).
Clinico-demographic adjusted PRS associations
Finally, PRS associations in SeverityM2SocioPRS were further adjusted for clinical factors which had a LR test P-value < 0.05 in earlier analyses, creating the “SeverityM4ClinicoDemPRS” model. This was repeated for PRS associated with COVID-19 mortality in MortalityM2SocioPRS, creating “MortalityM4ClinicoDemPRS” (Supplementary Methods).
Comparisons of model fit
To compare the epidemiological models created in this work, model fit was assessed using the Akaike Information Criterion (AIC) and Bayesian Information Criterion (BIC) statistics. This was repeated to compare models for the COVID-19 mortality outcome.
Pathway analysis
Pathway analysis was performed on those PRS associated with COVID-19 outcomes in the final SeverityM4ClinicoDemPRS and MortalityM4ClinicoDemPRS models. This was conducted using the Functional Mapping and Annotation of Genome-Wide Association Studies (FUMA) v1.4.0 tool25, a package that combines multiple in-silico tools (including the Multi-marker Analysis of GenoMic Annotation (MAGMA) gene-based test26) to provide functional interpretation of SNPs in PRS. SNPs analysed by FUMA were restricted to loci found in each PRS, and linkage disequilibrium (LD) thinning was performed using the same parameters as PRSice (R2 < 0.1 in 250 kb blocks) and the 1000 Genomes Phase 3 European panel as reference. More information may be found in the Supplementary Methods.
Results
Polygenic risk score associations
GWAS were performed for 23 UK Biobank clinical variables and related traits, identifying a total of 41,530 independent (LD R2 < 0.6) SNP associations (P < 5 × 10− 8) (Supplementary Table 1). PRS were then optimized using summary statistics produced by these analyses, adjusting for 10 PCs, and associations were found between 17 PRS and COVID-19 outcomes in univariate analyses (Table 1; Supplementary Tables 3–16).
Adjustment for sociodemographic and clinical variables
We then sought to determine whether clinical trait PRS were associated with the COVID-19 outcomes and whether these associations persisted after adjustment for known sociodemographic variables.
No PRS were found to be highly correlated (R2 > 0.8). Following removal of redundant PRS using backwards stepwise regression (SeverityM1PRS), and adjustment for sociodemographic variables in the SeverityM2SocioPRS, three PRS remained associated with severe COVID-19 in the transethnic and/or white European models (Supplementary Table 17): BMI PRS (adjusted odds ratio [AOR] = 1.14 95% confidence intervals [CI] 1.07–1.21, P-value [P] = 9.51 × 10− 5 [transethnic]; AOR = 1.15, 95%CI 1.07–1.23, P = 8.00 × 10− 5 [white European]), stroke PRS (AOR = 1.08, 95%CI 1.01–1.15, P = 0.02 [white European]) and hypertension PRS (AOR = 1.11, 95%CI 1.04–1.18, P = 2.63 × 10− 3 [transethnic]; AOR = 1.09, 95%CI 1.02–1.17, P = 0.01 [white European]). More details of correlations, backwards stepwise regression and COVID-19 mortality models (MortalityM1PRS and MortalityM2SocioPRS) may be found in Supplementary Results.
Details of PRS associations with COVID-19 mortality in MortalityM2SocioPRS (Supplementary Table 18) may be found in Supplementary Results. Associated PRS included the AF PRS (AOR = 1.12, 95%CI 1.03–1.22, P = 0.01 [transethnic]; AOR = 1.11, 95%CI 1.02–1.22, P = 0.02 [white European]), the PVD PRS (AOR = 0.9, 95%CI 0.83–0.99, P = 0.03 [white European]), and the Alzheimer’s disease PRS (AOR = 1.14, 95%CI 1.05–1.24, P = 2.50 × 10− 3 [transethnic]; AOR = 1.14, 95%CI 1.04–1.25, P = 4.44 × 10− 3 [white European]). Of note, “PRSe2” was no longer significant in these models.
To select clinical variables/traits for further adjustment of our SeverityM2SocioPRS and MortalityM2SocioPRS models, univariate associations between severe COVID-19 and clinical variables were defined, sociodemographic factors were included in the models (SeverityM3ClinicoDem and MortalityM3ClinicoDem) and highly correlated and residual redundant clinical variables were sequentially removed (Supplementary Tables 19–21).
After the PRS associations were further adjusted for clinical variables in SeverityM4ClinicoDemPRS, one PRS remained associated with severe COVID-19 (Table 2): the hypertension PRS (AOR = 1.1, 95%CI 1.03–1.18, P = 4.83 × 10− 3 [transethnic]). An additional three PRS were associated with COVID-19 mortality in the MortalityM4ClinicoDemPRS, including the Alzheimer’s PRS (AOR = 1.14, 95%CI 1.05–1.25, P = 2.54 × 10− 3 [transethnic] and AOR = 1.14, 95%CI 1.04–1.25, P = 5.22 × 10− 3 [white European]), AF PRS (AOR = 1.12, 95%CI 1.03–1.22, P = 9.98 × 10− 3 [transethnic] AOR = 1.13, 95%CI 1.03–1.23, P = 0.11 [white European]) and PVD PRS in the white European population (AOR = 0.9, 95%CI 0.82–0.99, P = 0.02) (Table 3).
Comparison of model fit
Model fit was compared between epidemiological models in this work, revealing that the addition of sociodemographic variables to the PRS model improved model fit (SeverityM1PRS AIC = 7361.77; SeverityM2SocioPRS AIC = 6332.91 [transethnic]), and the addition of clinical variables to SeverityM2SocioPRS further improved model fit (SeverityM2SocioPRS AIC = 6332.91; SeverityM4ClinicoDemPRS AIC = 6119.35 [transethnic]; Table 4).
Pathway analysis
Pathway analysis was performed (using FUMA v1.4.0) on PRS with severe COVID-19 or COVID-19 mortality associations in the SeverityM4ClinicoDemPRS or MortalityM4ClinicoDemPRS models (Supplementary Tables 22–24). This revealed several pathways of potential interest, including enrichment of SNPs in the 994,087 SNP hypertension PRS in the GO ‘voltage gated calcium channel activity involved in cardiac muscle cell action potential’ pathway (N genes = 5; beta[SE] = 3.48 [0.60]; adjusted-P = 1.86 × 10− 5). Other pathways highlighted were the KEGG ‘vascular smooth muscle contraction’ pathway (N genes in gene set = 115; N genes = 24; adjusted-P = 5.18 × 10− 3) and ‘gonadotropin-releasing hormone (GNRH) signalling’ pathway (N genes in gene set = 101; N genes present = 22; adjusted-P = 5.18 × 10− 3) in the Alzheimer’s disease PRS, and the GO ‘membrane repolarization’ pathway (N genes = 43; beta[SE] = 1.25[0.06]; adjusted-P = 4.45 × 10− 13), in the AF PRS. Further details may be found in Supplementary Results.
Discussion
To our knowledge, this study is the first to successfully highlight associations between clinical trait PRS and poor COVID-19 outcomes even following adjustment for other sociodemographic and clinical variables, demonstrating the potential benefits of integrating genetic data into epidemiological models, alongside other risk factors. This work also shows the importance of investigating PRS of multiple clinical traits, which may exhibit stronger associations in models including sociodemographic and clinical variables, compared to using a single PRS optimized for the clinical outcome of interest. In addition to this, pathway analysis of the PRS retained in the fully-adjusted models revealed shared pathogenic mechanisms between several variables and COVID-19 disease, including ‘GNRH signaling’ and ‘cardiac muscle contraction’.
Univariate associations with COVID-19 severity and/or mortality were found for 17 trait PRS, and these PRS were included in a single model, and further adjusted for sociodemographic factors. The weak correlations found between regression coefficients of PRS in the model suggested that the retained PRS had limited overlap and independently contributed predictive value to the model not conferred by other PRS. Four of these associations remained following adjustment for both sociodemographic and clinical factors: the hypertension PRS, AF PRS, Alzheimer’s disease PRS and the PVD PRS. For three of these four results (hypertension, AF and Alzheimer’s disease), the association between the COVID-19 outcome and the PRS proxy of the trait (e.g. hypertension PRS) was stronger than that between the COVID-19 outcome and the trait itself (e.g. hypertension).
There are several reasons why some PRS might be more effective predictors of COVID-19 outcomes compared with their clinical counterparts in these models. Firstly, this enables the identification of individuals who may have a genetic predisposition to certain traits or diseases, even if they have not developed the disease or received a formal diagnosis. By incorporating this information, we can avoid overlooking individuals who may have been missed when relying solely on clinical data to establish associations. Furthermore, including this “at risk” information in the analysis in the form of a continuous predictor may improve the statistical power to detect associations, particularly when the clinical trait under consideration is traditionally defined as a binary variable. Secondly, inconsistencies between clinical definitions are evident in healthcare and epidemiology7. This can result in variation in disease definitions and therefore classifications of individuals in the study, particularly when collating information from self-reports or different healthcare settings. This may lead to inaccurate estimates of effect sizes when testing for associations with the clinical trait. Contrastingly, PRS are calculated systematically using a single algorithm, reducing the impact of bias or variation on classification of individuals and leading to greater consistencies when testing for associations within epidemiological studies. Thirdly, some variables, such as BMI and BF%, may be measured crudely in small epidemiological cohorts, whereas PRS for these traits may benefit from optimization using data from large, consistently measured datasets, improving their uniformity within the sample of interest. Nevertheless, it is important to acknowledge that clinical risk factors played a significant role in enhancing COVID-19 outcome models in this study. Therefore, it is advisable that PRS be considered as supplementary rather than substitutive components in such models when clinical variables are accessible.
The PVD PRS was present in an epidemiological model for COVID-19 mortality (MortalityM4ClinicoDemPRS) alongside its clinical counterpart, PVD. Both the PVD trait and the PVD PRS had a LR P-value < 0.05, suggesting that both traits independently contributed to risk of the COVID-19 mortality outcome in this study. These results provide further evidence that PRS may provide risk information above and beyond that of their clinical counterpart alone. However, it is noteworthy that the effect size of PVD and the PVD PRS were in opposing directions in this study. Several explanations may account for this outcome. Firstly, there may be unmeasured confounding influencing the effect of these traits on severe COVID-19. For example, pleiotropic SNPs in the PVD PRS could be influencing COVID-19 outcome risk through an alternate pathway to PVD itself. Likewise, PVD is a complex trait which is likely influenced by numerous genetic factors, each with differing effects on disease risk. The PVD PRS described here may capture just a subset of PVD risk, leading to discrepancies between the PRS effect size and the PVD effect size on severe COVID-19 risk. Finally, collider bias could be influencing this association due to the adjustment of genetic PCs. If genetic information in the PCs are also associated with other severe COVID-19 risk factors (e.g. blood group), the observed association between the PVD PRS and severe COVID-19 could be a type one error masking the true causal risk factor. Future studies may employ Mendelian randomization techniques to test for a causal relationship between PVD and COVID-19 outcomes through this PRS, as well as testing for potential confounding pathways.
As anticipated, the fit of the PRS model improved with the addition of sociodemographic and clinical variables. Interestingly, when comparing epidemiological models formed in this study, we observed that the fit of the sociodemographic & PRS model (SeverityM2SocioPRS) was better than a model containing sociodemographic variables alone. This was also found when comparing models with sociodemographic, clinical and PRS variables (SeverityM4ClinicoDemPRS) with just sociodemographic and clinical factors (SeverityM3ClinicoDem). Together, these results suggest that the addition of PRS could improve the fit of epidemiological models containing classic sociodemographic and/or clinical risk factors alone. Such findings should be further investigated in future epidemiological and risk prediction studies.
A statistically significant association was found between the 6,887 SNP Alzheimer’s disease PRS and COVID-19 mortality in the transethnic and white European MortalityM5ClinicoPRS models. This association had a positive direction of effect, wherein an increase in Alzheimer’s disease PRS was associated with an elevated risk of both Alzheimer’s disease and COVID-19 mortality, even after adjustment for other clinico-demographic variables. This PRS was enriched for SNPs in both the ‘GNRH signaling’ and ‘vascular smooth muscle cell contraction’ gene sets, highlighting a possible shared aetiology of Alzheimer’s disease and severe COVID-19 through the PRS’s effect on these biological pathways. These results highlight another potential benefit of testing for associations between trait PRS and disease outcomes in epidemiological modelling. By performing pathway analyses on genetic variants in the trait PRS, it is possible shed light on the pathogenic mechanisms underpinning predisposition to not only the trait itself, but also the disease outcome studied in the epidemiological model. However, results of such enrichment studies should be interpreted with caution, given that the inclusion of some false positive SNP associations are inherent to PRS methodologies6.
Shortcomings of this work included the limited availability of non-white European samples in the study cohort. Whilst the study attempted to repeat risk analyses in a transethnic population, because of the predominance of white European samples in the UK Biobank cohort27, this was difficult to conduct. PRS were therefore optimized in a white European population (to minimize issues related to population stratification), meaning that PRS may not be as effective at predicting risk of COVID-19 outcomes in non-European populations due to differences in LD structure and genetic architecture. This is representative of a wider problem in the genetics community, with work needed to recruit more diverse populations into cohort studies.
The optimization of PRS and prediction of risk in COVID-19 outcomes is also limited by statistical power in the current study, which is constrained by sample sizes of current datasets and the need for a complete case approach. For example, instances in this work wherein associations were found for risk factors in predicting COVID-19 severity but not COVID-19 mortality (e.g. hypertension PRS), could be in part due a loss of power in the smaller cohort sizes of the COVID-19 mortality outcome. Interestingly, the use of PRS as clinical proxies in future epidemiological studies could mitigate these issues, as this circumvents the issue of missing values for clinical variables.
It is also important to note that whilst associations were identified between PRS and COVID-19 outcomes after adjustment for clinico-demographic factors, the models created here are not risk prediction models. More work is needed before PRS are integrated in a clinical setting, including cross-validation studies1,4,5,28. This may be possible using other population-based cohorts such as 23andMe29 or the upcoming Our Future Health project in the UK30. Improvements in PRS performance will occur over time with increasing cohort sizes, particularly in transethnic populations.
This study identified associations between PRS for clinical traits (e.g. hypertension and AF) and poor COVID-19 outcomes, highlighting the value of including multiple trait PRS over a single PRS optimised for the outcome of interest, and identifying shared biological pathways between these traits. This work demonstrates that genetic data can improve the fit of sociodemographic models for COVID-19 outcomes, and highlights the potential benefits of incorporating PRS in disease modelling. As PRS for complex diseases are further refined, concurrent improvements in disease modelling will be attained.
Data availability
UK Biobank data were provided under a licence that does not permit sharing data. The code-lists used in definitions and the derived results are published in Crossfield et al. (2022)2. UK Biobank data is available online (https://www.ukbiobank.ac.uk/). To request further information, please contact AW Morgan.
References
Slunecka, J. L. et al. Implementation and implications for polygenic risk scores in healthcare. Hum. Genom. 15 (1), 46 (2021).
Crossfield, S. S. R., Chaddock, N. J. M., Iles, M. M., Pujades-Rodriguez, M. & Morgan, A. W. Interplay between demographic, clinical and polygenic risk factors for severe COVID-19. Int. J. Epidemiol. 51 (5), 1384–1395 (2022).
Mars, N. et al. The role of polygenic risk and susceptibility genes in breast cancer over the course of life. Nat. Commun. 11 (1), 6383 (2020).
Weale, M. E. et al. Validation of an integrated risk tool, including polygenic risk score, for atherosclerotic cardiovascular disease in multiple ethnicities and ancestries. Am. J. Cardiol. 148, 157–164 (2021).
England, H. E. NHS launches new polygenic scores trial for heart disease United Kingdom 2021.https://www.genomicseducation.hee.nhs.uk/blog/nhs-launches-new-polygenic-scores-trial-for-heart-disease/
Choi, S. W., Mak, T. S. & O’Reilly, P. F. Tutorial: A guide to performing polygenic risk score analyses. Nat. Protoc. 15 (9), 2759–2772 (2020).
Hajjaj, F. M., Salek, M. S., Basra, M. K. & Finlay, A. Y. Non-clinical influences on clinical decision-making: A major challenge to evidence-based practice. J. R Soc. Med. 103 (5), 178–187 (2010).
Lee, C. M. Y. et al. Comparing different definitions of prediabetes with subsequent risk of diabetes: An individual participant data meta-analysis involving 76 513 individuals and 8208 cases of incident diabetes. BMJ Open. Diabetes Res. Care 7 (1), e000794 (2019).
Zhou, P. et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 579 (7798), 270–273 (2020).
Docherty, A. B. et al. Features of 20 133 UK patients in hospital with covid-19 using the ISARIC WHO clinical characterisation protocol: Prospective observational cohort study. BMJ 369, m1985 (2020).
Williamson, E. J. et al. Factors associated with COVID-19-related death using OpenSAFELY. Nature 584 (7821), 430–436 (2020).
McKeigue, P. M. et al. Rapid Epidemiological Analysis of Comorbidities and treatments as risk factors for COVID-19 in Scotland (REACT-SCOT): A population-based case-control study. PLoS Med. 17 (10), e1003374 (2020).
Initiative, C-H-G. Mapping the human genetic architecture of COVID-19. Nature 600 (7889), 472–477 (2021).
Pairo-Castineira, E. et al. Genetic mechanisms of critical illness in COVID-19. Nature 591 (7848), 92–98 (2021).
Ellinghaus, D. et al. Genomewide association study of severe Covid-19 with respiratory failure. N. Engl. J. Med. 383 (16), 1522–1534 (2020).
Hu, J., Li, C., Wang, S., Li, T. & Zhang, H. Genetic variants are identified to increase risk of COVID-19 related mortality from UK Biobank data. medRxiv (2020).
Shelton, J. F. et al. Trans-ancestry analysis reveals genetic and nongenetic associations with COVID-19 susceptibility and severity. Nat. Genet. 53 (6), 801–808 (2021).
Horowitz, J. E. et al. Genome-wide analysis provides genetic evidence that ACE2 influences COVID-19 risk and yields risk scores associated with severe disease. Nat. Genet. 54 (4), 382–392 (2022).
Dite, G. S., Murphy, N. M. & Allman, R. An integrated clinical and genetic model for predicting risk of severe COVID-19: A population-based case-control study. PLoS One 16 (2), e0247205 (2021).
Brown, K. L., Ramlall, V., Zietz, M., Gisladottir, U. & Tatonetti, N. P. Estimating the heritability of SARS-CoV-2 susceptibility and COVID-19 severity. Nat. Commun. 15 (1), 367 (2024).
Sudlow, C. et al. UK biobank: An open access resource for identifying the causes of a wide range of complex diseases of middle and old age. PLoS Med. 12 (3), e1001779 (2015).
Team, R. C. R: A language and environment for statistical computing Vienna, Austria: R Foundation for Statistical Computing. https://www.R-project.org/ (2019).
Chang, C. C. et al. Second-generation PLINK: Rising to the challenge of larger and richer datasets. Gigascience 4, 7 (2015).
Choi, S. W. & O’Reilly, P. F. PRSice-2: Polygenic risk score software for biobank-scale data. Gigascience 8 (7) (2019).
Watanabe, K., Taskesen, E., van Bochoven, A. & Posthuma, D. Functional mapping and annotation of genetic associations with FUMA. Nat. Commun. 8 (1), 1826 (2017).
de Leeuw, C. A., Mooij, J. M., Heskes, T. & Posthuma, D. MAGMA: Generalized gene-set analysis of GWAS data. PLoS Comput. Biol. 11 (4), e1004219 (2015).
Bycroft, C. et al. The UK Biobank resource with deep phenotyping and genomic data. Nature 562 (7726), 203–209 (2018).
Sollis, E. et al. The NHGRI-EBI GWAS catalog: Knowledgebase and deposition resource. Nucleic Acids Res. 51 (D1), D977–D85 (2023).
23andMe. 23andMe 2022. https://www.23andme.com/en-gb/
Health NIo. All of Us Research Program (2023).
Acknowledgements
We thank the MRC and NIHR for supporting this work and participants of UK Biobank without whom it would not have been possible to undertake this study. The funders had no role in the study design, in the collection, analysis and interpretation of data, in the writing of the report, and in the decision to submit the paper for publication. The views expressed are those of the author(s) and not necessarily those of the NHS, the NIHR or the Department of Health and Social Care.
Funding
This work was funded by a Medical Research Council (MRC) Confidence in Concept award [MC_PC_19042] and was additionally supported a National Institute for Health Research (NIHR) Senior Investigator award to AWM [NIHR202395] and by the NIHR Leeds Biomedical Research Centre [NIHR203331] and NIHR Leeds MedTech and In Vitro Diagnostic Evaluation Co-operative [MIC-2016-015]. Mark Iles is supported in part by the National Institute for Health and Care Research (NIHR) Leeds Biomedical Research Centre. This work was also supported by a PhD studentship from the MRC Discovery Medicines North Doctoral Training Program.
Author information
Authors and Affiliations
Contributions
Study conception and design AWM, MPR and MI; analysis planning, data collection, verification and data analysis NJMC and SSRC; all authors contributed to data interpretation, drafting and critical revision of the article and approved the final submitted version.
Corresponding author
Ethics declarations
Competing interests
None declared for the current manuscript. AWM has received research grant or educational funding or undertaken consultancy for the following pharmaceutical companies on behalf of the University of Leeds in the last 5 years: AstraZeneca, Kiniska Pharmaceuticals, Regeneron, Roche/Chugai, Sanofi and Vifor. MPR is currently employed by Union Chimique Belge (UCB) Biopharma. None declared for MMI, NJMC, or SSRC.
Ethics approval
The study was approved by UK Biobank (project 24559). UK Biobank has ethical approval from the National Research Ethics Committee (REC reference 11/NW/0382), and obtained informed electronic consent from all participants. There was no patient-public involvement in the study, which used non-identifiable data. All methods were performed in accordance with the relevant guidelines and regulations.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Chaddock, N.J.M., Crossfield, S.S.R., Pujades-Rodriguez, M. et al. Genetic proxies for clinical traits are associated with increased risk of severe COVID-19. Sci Rep 15, 2083 (2025). https://doi.org/10.1038/s41598-025-86260-z
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-86260-z
