
Abstract
Background: Metabolic associated steatohepatitis (MASH) represents a severe subtype of metabolic associated fatty liver disease (MASLD), with an increased risk of progression to cirrhosis and hepatocellular carcinoma. The nomenclature shift from nonalcoholic steatohepatitis (NASH)/nonalcoholic fatty liver disease (NAFLD) to MASH/MASLD, underscores the pivotal role of metabolic factors in disease progression. Diagnosis of MASH currently hinges on liver biopsy, a procedure whose invasive nature limits its clinical utility. This study aims to identify and validate metabolism-related genes (MRGs) markers for the non-invasive diagnosis of MASH. Methods: This study extracted multiple datasets from the GEO database to identify metabolism-related differentially expressed genes (MRDEGs). Protein-Protein Interaction (PPI) network and machine learning algorithms, including Least Absolute Shrinkage and Selection Operator (LASSO) regression, Support Vector Machine-Recursive Feature Elimination (SVM-RFE), and Random Forest (RF), were applied to screen for signature MRDEGs. The diagnostic performance of these MRDEGs was evaluated using the Receiver Operating Characteristic (ROC) curve and further validated using independent external datasets. Additionally, enrichment analysis was performed to uncover key driver pathways in MASH. The infiltration levels of various immune cell types were assessed using single sample Gene Set Enrichment Analysis (ssGSEA). Finally, Spearman correlation analysis confirmed the association between signature genes and immune cells. Results: We successfully identified seven signature MRDEGs, including CYP7A1, GCK, AKR1B10, HPRT1, GPD1, FADS2, and ENO3, through PPI network analysis and machine learning algorithms. The gene model displayed exceptional diagnostic performance in the training and validation cohorts, as evidenced by the area under ROC curve (AUC) exceeding 0.9. Further enrichment analysis revealed that signature MEDEGs were primarily involved in multiple biological pathways related to glucose and lipid metabolism. Immune infiltration analysis indicated a significant increase in the infiltration levels of activated CD8 T cells, gamma-delta T cells, natural killer cells, and CD56bright NK cells in patients with MASH. Conclusion: This study successfully identified seven signature MRDEGs as significant diagnostic biomarkers for MASH. The findings not only offer novel strategies for non-invasive diagnosis of MASH but also highlight the substantial role of immune cell infiltration in the progression of MASH.
Similar content being viewed by others

Introduction
Metabolic associated liver disease (MASLD), formerly known as nonalcoholic fatty liver disease (NAFLD), encompasses a continuum of liver pathologies from metabolic associated fatty liver (MAFL) to metabolic associated steatohepatitis (MASH) and potentially to cirrhosis and hepatocellular carcinoma1,2. The prevalence of MASLD is increasing and is closely related to the global prevalence of metabolic syndrome, and effective diagnostic and treatment strategies are urgently needed. MASH is an important subtype of MASLD. In China, the prevalence of MASH ranges from 2.4–6.1%3. Compared with MAFL, MASH has a higher risk of progression to cirrhosis or even liver cancer, and a poorer prognosis4. Currently, the diagnosis of MASH relies on liver biopsy. However, it is invasive and has a risk of complications, so its application in clinical practice is limited. Therefore, it is necessary to develop non-invasive diagnostic methods to accurately identify and manage MASH.
The redefinition from nonalcoholic steatohepatitis (NASH) to MASH reflects a more comprehensive understanding of etiology of the disease, emphasizing the central role of metabolic factors5. This shift in nomenclature is intended to reduce stigma and better reflect the multifactorial nature of the disease. Metabolic factors, including genetic predispositions, are now recognized as pivotal in the pathogenesis of MASH. For instance, genetic variants involved in lipid metabolism and insulin resistance have been shown to significantly influence the risk and progression of MASH6,7. These genetic metabolic factors can modulate hepatic steatosis and inflammation, highlighting their potential as diagnostic and therapeutic targets.
Machine learning, a branch of artificial intelligence, has emerged as a powerful tool in the analysis of complex biomedical data8,9. Its ability to discern subtle patterns and correlations in high-dimensional datasets makes it an invaluable tool for stratifying patients. Machine learning has been used to identify key ferroptosis, inflammation, or single metabolic pathway-related genes for the diagnosis of NAFLD/NASH10,11,12. In this study, multiple machine learning algorithms were used to identify metabolic-related genes (MRGs) associated with MASH from 84 KEGG metabolic pathways. The aim is to develop a non-invasive diagnostic model that can accurately identify individuals with MASH, thereby facilitating early intervention and improving patient outcomes. Through this approach, we aim to transform the clinical management of MASH by providing a precision medicine framework grounded in data-driven insights.
Materials and methods
Collection and integration of data
We screened eight datasets containing MASH samples from the Gene Expression Omnibus (GEO) (http://www.ncbi.nlm.nih.gov/) database, including GSE135251, GSE17470, GSE37031, GSE48452, GSE89632, GSE63067, GSE126848, and GSE6126013,14,15,16,17,18,19,20. Detailed information of these datasets was provided in Table 1. For data processing, we initially applied the normalizeBetweenArrays function to perform quantile normalization on the expression matrix of each dataset to ensure data consistency and comparability. Subsequently, we utilized the removeBatchEffect function to eliminate batch effects to enhance the accuracy of our analysis. We combined GSE135251, GSE17470, GSE37031, GSE48452, and GSE89632 to form the training cohort, and merged GSE63067 and GSE126848 as the external validation cohort 1, and designated GSE61260 as the external validation cohort 2. We employed the “ggplot2” R package to generate box plots and PCA plots to intuitively demonstrate the removal effect of the batch effect. Furthermore, we extracted 1731 MRGs in 84 metabolic pathways from the KEGG database using the “KEGGREST” R package (Supplementary Tables 1 and Table 2).
Identification of differentially expressed genes (DEGs)
To identify DEGs between MASH and control groups, we utilized the “limma” R package to analyze the training cohort, GSE135251, GSE17470, GSE37031, GSE48452, and GSE89632. The DEGs of the training cohort were defined as those with p-value less than 0.05 and |logFC| greater than 0.585. The “pheatmap” and “ggplot2” R packages were used to plot heatmaps and volcano plots, respectively, to present the analysis results.
Robust rank aggregation (RRA) analysis and identification of metabolism-related differentially expressed genes (MRDEGs)
To extract consistent DEGs from multiple microarray datasets, we conducted RRA analysis. RRA is an efficient data integration tool that enhances the accuracy and robustness of DEG identification by integrating gene ranking information from multiple independent datasets. Before conducting RRA, we extracted lists of DEGs from each dataset. Subsequently, we utilized the “RobustRankAggreg” R package to evaluate the statistical significance of each gene by calculating the ranking of each gene in the integrated list and combining it with the corresponding P value. In the RRA analysis, the p-value reflects the probability of a gene being ranked at a certain position or higher in the final ranking list, while logFC measures the magnitude of gene expression variation. Genes with a p-value less than 0.05 and |logFC| greater than 0.585 were considered statistically significant. By intersecting the DEGs obtained from the training cohort with those identified through RRA analysis, as well as known MRGs, we ultimately identified the MRDEGs.
Enrichment analysis
To elucidate the biological functions of MRDEGs and their potential mechanisms in MASH progression, we employed the R packages “clusterProfiler” and “org.Hs.eg.db” to conduct a comprehensive functional enrichment analysis, including Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analysis. By setting p-value threshold of less than 0.05, we identified significantly enriched GO terms in biological processes and molecular functions, as well as significantly enriched pathways in the KEGG pathway.
Protein-protein Interaction (PPI) Network and Machine Learning methods
To deeply analyze the potential interactions among MRDEGs, we construct a PPI network using the STRING database (https://string-db.org). By setting a confidence threshold above 0.7, we ensured the high quality of the network, thereby revealing the interconnections of these MRDEGs at the molecular level. The topological features of the network were visualized using “Cytoscape” software (version 3.10.1), where each node represents a gene or protein, and the edges between nodes indicate their interactions.
To screen out MRDEGs with potential biomarker value, we employed a comprehensive analysis using three advanced machine learning algorithms: Least Absolute Shrinkage and Selection Operator (LASSO) regression, Support Vector Machine-Recursive Feature Elimination (SVM-RFE), and Random Forest (RF). The LASSO algorithm was implemented through the “glmnet” package, which optimizes the predictive performance of the model while performing variable selection and regularization. In this study, the LASSO parameters were configured as: family = binomial, type.measure = class, alpha = 1, nfold = 10, to effectively reduce feature dimensions and improve the generalization ability of the model. SVM-RFE, as a supervised learning technique, was implemented through the “e1071” package, which iteratively reduces the number of features to identify the variables that contribute most to the predictive performance of the model. In this study, the SVM-RFE parameters were set as functions = “caretFuncs”, methods = “cv”, to prevent model overfitting and ensure the interpretability of the results. The RF algorithm, implemented through the “randomForest” package, performs classification or regression prediction by constructing multiple decision trees. In this study, the RF algorithm parameters were set as ntree = 500. Genes were selected based on a decreasing accuracy approach (Gini coefficient), and genes with an importance score greater than 2 were ultimately chosen as biomarkers for the diagnosis of MASH.
Identification of signature MRDEGs and diagnostic performance analysis
By integrating the core genes from the PPI network with the results of machine learning algorithms, signature MRDEGs that play a crucial role in the progression of MASH were identified.
To explore the ability of signature MRDEGs to identify MASH patients, we constructed receiver operating characteristic (ROC) curves in the training cohort using the “pROC” R package and calculated the area under the curve (AUC) values to evaluate the diagnostic efficacy of individual genes and the diagnostic model composed of signature MRDEGs. The diagnostic model is a logistic regression model, which has been constructed utilizing the “glmnet” R package, based on the expression of MRDEGs. The model equation: y = β0 + β1*Gene1 + β2*Gene2+⋅⋅⋅+βn*Genen. Furthermore, we further validated the diagnostic efficacy of signature MRDEGs in two external validation cohorts. Additionally, we compared the expression levels of signature MRDEGs between MASH and control groups in both the training and external validation cohorts, with the results presented in the form of violin plots.
According to the expression of signature of MRDEGs, the probability of MASH was predicted using a nomogram constructed by the “rms” R packages. The Calibration curve was further performed to evaluate the forecasting precision of the nomogram.
Gene set variation analysis (GSVA)
GSVA, a non-parametric and unsupervised analytical tool, was employed to assess the change in gene set activity, and can analyze gene set activity in a single sample without pre-conditions or groups. By calculating activity scores for the gene set of each sample, GSVA can reveal the enrichment status of the gene set. Subsequently, we grouped the samples based on the median expression values of the MRDEGs and conducted a differential analysis of the activity scores to explore the impact of gene expression changes on biological pathways. In this study, we utilized the “c2.cp.kegg.Hs.symbols.gmt” gene set provided by the MSigDB database to perform GSVA analysis on the training cohorts, aiming to identify the biological pathways involved when signature gene expression changes, so as to gain a deeper understanding of their role in the pathogenesis of the disease.
Assessment of immune cell infiltration
To investigate the differences in the immune microenvironment between MASH patients and control subjects, we employed the single sample gene set enrichment analysis (ssGSEA) to assess the relative abundance of 28 immune cell types in both groups. This analysis generates a composite score for each immune cell type based on the expression levels of specific characteristic genes within the samples. The Wilcoxon test was utilized to compare the differences in immune cell infiltration between the two groups, and the results were depicted using violin plots and heatmaps. Spearman correlation analysis was conducted to examine the correlation between signature MRDEGs and immune cells, with the results presented in a correlation heatmap using the “ggplot” package.
Statistical analysis
Statistical analysis and graphical representation were conducted using R software (version 4.3.0). Visualization of the PPI network was achieved using Cytoscape software (version 3.10.1). Wilcoxon rank-sum test and Student’s t-test were employed to compare differences between groups. Spearman correlation test was utilized to assess the correlation between variables. The diagnostic efficacy of signature genes was evaluated by the ROC curve. A p-value of less than 0.05 was considered statistically significant. Significance levels of p-values less than 0.001, 0.01, and 0.05 are denoted by ***, **, and *, respectively.
Results
Identification of MRDEGs
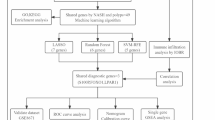
The flow chart of this study was shown in Fig. 1. To correct for individual variations among samples and ensure consistency between datasets, we performed quantile normalization on all included datasets. After removing batch effects, GSE135251, GSE17470, GSE37031, GSE48452, and GSE89632 were merged to construct the training cohort (Fig. 2A-D). Subsequently, the “limma” R package was used to identify DEGs between MASH and control groups in the training cohort. A total of 208 genes were identified, with 99 genes upregulated and 109 genes downregulated (Supplementary Table 3). The top 50 most significantly up- and down-regulated DEGs were presented as a heatmap (Fig. 2E).

The flow chart of this study.

Batch correction analysis and differential expression analysis. (A, B) The boxplots showed five datasets before and after eliminating batch effects. (C, D) PCA plots of five datasets before and after eliminating batch effect. (E) The heatmap showed 100 DEGs between MASH and healthy controls, including 50 upregulated genes and 50 downregulated genes.
RRA methodology compares the rankings of gene expression data across different datasets, rather than the raw expression values, to identify DEGs that consistently rank high across multiple datasets. Prior to RRA analysis, we conducted differential expression analysis on the GSE135251, GSE17470, GSE37031, GSE48452, and GSE89632 datasets, with the results of differential expression shown in Fig. 3A-E. Subsequently, through RRA analysis, we identified 558 significant DEGs (243 downregulated genes, 315 upregulated genes) (Supplementary Table 4). The heatmap displayed the top 25 upregulated and downregulated genes. [Upregulated genes: AKR1B10 (P = 1.98E-11), CYP7A1 (P = 2.43E-11), FNDC5 (P = 3.96E-11), THRSP (P = 2.40E-08), LGALS4 (P = 2.12E-10); Downregulated genes: FOS (P = 1.41E-16), IGFBP1 (P = 2.31E-15), FOSB (P = 1.16E-14), IGFBP2 (P = 3.49E-10), RASD1 (P = 6.62E-12)] (Fig. 3F). To filter for MRDEGs, we intersected the identified DEGs with a set of 1731 MRG, resulting in the identification of 34 MRDEGs (Fig. 4A).

Volcano plots of the five datasets and RRA analysis. (A-E) Volcano plots of GSE135251 (A), GSE17470 (B), GSE37031 (C), GSE48452 (D), GSE89632 (E). (F) Heatmap of the top 20 up- and down-regulated genes in the RRA analysis. Red points represented upregulated genes, and green points represented downregulated genes.

Identification of MRDEGs and enrichment analysis. (A) Venn diagram indicated the overlap of batch-DEG, RRA-DEGs and MRGs to obtain 34 MRDEGs. (B) GO enrichment analysis of 34 MRDEGs. (C) KEGG enrichment analysis of 34 MRDEGs.
Enrichment analysis
To elucidate the biological functions and pathways involved by the 34 MRDEGs, we conducted enrichment analysis. GO analysis encompassed three categories: biological process (BP), cellular component (CC), and molecular function (MF). Fig. 4B illustrated the top ten GO terms under each category. The results indicated that these MRDEGs were predominantly enriched in “sterol biosynthetic process”, “cholesterol metabolic process”, “endoplasmic reticulum lumen” and “response to carbohydrate”. In the KEGG pathway enrichment analysis, the “glycerolipid metabolism”, “PPAR signaling pathway”, and “glycolysis / gluconeogenesis” pathways were significantly enriched by MRDEGs (Fig. 4C). Given that the 34 MRDEGs were a result of filtering for metabolic pathways, the enrichment of metabolic pathways may be anticipated. Consequently, we also conducted enrichment analysis on the 199 DEGs that had not been subjected to filtration by the 1731 MRGs. The findings revealed that several same metabolic pathways, including “endoplasmic reticulum lumen”, “response to carbohydrate”, “glycerolipid metabolism” and “glycolysis / gluconeogenesis”, were significantly enriched (Supplementary Fig. 1). Collectively, these results suggest that the 34 MRDEGs and the enriched metabolic pathways may play a pivotal role in the progression of MASH.
Identification of diagnostic signature MRDEGs for MASH
Initially, PPI network of DEGs was performed using the STRING database and the results were visualized using Cytoscape software. The PPI network revealed that 24 out of the 34 MRDEGs interacted with each other (Fig. 5A). Interactions were observed among AKR1B10, GCK, PCK1, ENO3, ALDH3A1, ACSS2, FADS2, FASN, GPAM, GPD1, SQLE, MVD, CH25H, MVK, CYP51A1, LSS, and CYP7A1. Additionally, interactions were identified among PNPLA3, MOGAT2, and LPL. HPRT1 was found to interact with PDE11A, and NAMPT was found to interact with NNMT.

Identification of signature MRDEGs for MASH. (A) 24 MRDEGs were identified through PPI network. (B, C) LASSO regression to extract 15 MRDEGs. (D, E) SVM-RFE algorithm selected 25 MRDEGs. (F) RF algorithm identified 18 MRDEGs. (G) Venn gram to intersect 7 signature MRDEGs.
Subsequently, we conducted three machine learning algorithms to explore signature genes. Through LASSO regression, a total of 15 MRDEGs were identified as diagnostic markers for MASH, such as AKR1B10, GPD1, FADS2, TYMS, CYP7A1 (Fig. 5B and C). For the SVM-RFE algorithm, 25 MRDEGs were selected through 10-fold cross-validation, such as AKR1B10, GPD1, FADS2, FMO1, TYMS, P4HA1, etc. (Fig. 5D and E). The RF algorithm identified 18 MRDEGs with an importance score greater than 2, such as GPD1, FADS2, TYMS, P4HA1, MOGAT2, etc. (Fig. 5F). Ultimately, to obtain the optimal diagnostic signature MRDEGs for MASH, we conducted an intersection analysis of the aforementioned gene subsets, and a total of 7 signature MRDEGs were identified (CYP7A1, GCK, AKR1B10, HPRT1, GPD1, FADS2, ENO3) (Fig. 5G).
To assess the efficacy of the signature MRDEGs in diagnosing MASH, we initially conducted expression level analysis of the seven MRDEGs in the training cohort. The results showed that, compared to the control group, the expression level of the HPRT1 gene was significantly downregulated (p < 0.001) in the MASH group, while the expression levels of CYP7A1, GCK, AKR1B10, GPD1, FADS2, and ENO3 genes were significantly upregulated (p < 0.001) (Fig. 6A). Through ROC curve analysis, we found that except for GCK gene, the AUCs of the other six feature MRDEGs all exceeded 0.7 (Fig. 6B), indicating their high diagnostic potential. Although the AUC for GCK gene was 0.697, it was also very close to 0.7, and this result also indicated that GCK gene had considerable diagnostic value. Notably, the diagnostic model formed by the combination of these MRDEGs exhibited excellent discriminatory performance, with an AUC value reaching 0.915 (Fig. 6C).

The diagnostic performance of signature MRDEGs model. (A) The expression of 7 signature MRDEGs in the training cohort. (B, C) The ROC curves of 7 signature MRDEGs and diagnostic model in the training cohort. (D) The nomogram was used to predict the incidence of MASH. (E) The Calibration curve for evaluated the predictive power of the nomogram model.
We also constructed a nomogram to predict the risk of MASH onset and evaluated predictive power using the calibration curve to predict the probability of MASH (Fig. 6D). The calibration curve indicated a minimal difference between the real and predicted MASH risks, suggesting that the nomograph model of MASH is precise (Fig. 6E).
Validation of the diagnostic efficacy of signature MRDEGs
To further validate the diagnostic value of the MRDEGs, we performed validation in two independent external validation cohorts. The results indicated that in both validation cohorts, the MRDEGs in the MASH group showed significant expression differences compared to the control group (P < 0.05), except for GCK gene in validation cohort 2 (P > 0.05) (Fig. 7A and D). The ROC curve further confirmed the outstanding diagnostic performance of these MRDEGs in the validation cohort 1, with the AUC values all exceeding 0.7 (Fig. 7B), and the AUC value of the diagnostic model reaching 0.979 (Fig. 7C). In the validation cohort 2, except for GCK gene, the AUC values for the other MRDEGs all exceeded 0.6 (Fig. 7E), and the AUC value of the diagnostic model was 0.966 (Fig. 7F). Taken together, these results indicate that signature MRDEGs and the diagnostic model have significant accuracy and reliability in distinguishing MASH patients, potentially providing new molecular markers for the diagnosis of MASH.

Validation of the diagnostic performance of signature MRDEGs model. (A) The expression of 7 signature MRDEGs in the validation cohort 1. (B, C) The ROC curves of 7 signature MRDEGs and diagnostic model in the validation cohort (1) (D) The expression of 7 signature MRDEGs in the validation cohort (2) (E, F) The ROC curves of 7 signature MRDEGs and diagnostic model in the validation cohort 2.
Correlation and enrichment analysis of signature MRDEGs
Understanding the interactions among signature MRDEGs and the biological pathways they participate in is crucial for elucidating the pathogenesis of MASH. Therefore, we initially conducted a correlation analysis of the signature MRDEGs. The results revealed that GPD1 exhibited positive correlations with FADS2, GCK, and ENO3, whereas HPRT1 demonstrated negative correlations with FADS2, GCK, and ENO3 (Fig. 8A). These correlation patterns suggest significant functional similarities and potential synergistic effects among the identified signature genes.

Correlation analysis and enrichment analysis. (A) The correlation between 7 signature MRDEGs. (B) KEGG enrichment analysis of 7 signature MRDEGs. (C-I) GSVA analysis of 7 signature MRDEGs, including CYP7A1 (C), FADS2 (D), GCK (E), GPD1 (F), AKR1B10 (G), ENO3 (H), HPRT1 (I).
Subsequently, we performed KEGG enrichment analysis for signature MRDEGs. We found that they played key roles in multiple biological processes (Fig. 8B), particularly in galactose metabolism (AKR1B10, GCK), glycolysis/gluconeogenesis (ENO3, GCK), PPAR signaling pathway (FADS2, CYP7A1), and carbon metabolism (ENO3, GCK). Furthermore, GSVA analysis confirmed the close association of signature MRDEGs with specific biological processes. The upregulation of CYP7A1 and FADS2 was related to steroid hormone biosynthesis (Fig. 8C and D). The upregulation of GCK was significantly enriched in maturity onset diabetes of the young (Fig. 8E). The upregulation of GPD1 was significantly enriched in beta alanine metabolism and was involved in various metabolic processes including Amino sugar and nucleotide sugar, galactose, and glycolysis (Fig. 8F). The upregulation of AKR1B10 was associated with linoleic acid metabolism, butanoate metabolism, and fructose and mannose metabolism (Fig. 8G). The upregulation of ENO3 was significantly correlated with glycerolipid metabolism (Fig. 8H). In contrast, the downregulation of HPRT1 was primarily associated with the Notch signaling pathway and Fructose and mannose metabolism (Fig. 8I). In summary, our results emphasize the crucial role of signature MRDEGs in regulating key metabolic pathways and provide new insights into their biological functions and potential roles in disease pathogenesis.
Immune cell infiltration
The ssGSEA method was applied to quantitatively assess the relative abundance of 28 immune cell types in the training cohort for both MASH and control groups. The results of the wilcoxon rank-sum test revealed that there were significant differences in the abundance of nine immune cell types between the two groups. Specifically, the infiltration levels of activated CD8 T cells, gamma delta T (γδT) cells, natural killer (NK) cells, and CD56bright NK cells were significantly increased in the MASH group, while the infiltration levels of eosinophils, type 2 T helper (Th2) cells, memory B cells, central memory CD8 T cells, and effector memory CD8 T cells were significantly decreased (Fig. 9A and B). Further correlation analysis demonstrated that the GPD1 exhibited a significant positive correlation with the abundance of activated CD8 T cells and gamma delta T cells, while the GCK showed a significant negative correlation with the abundance of eosinophils and type 2 T helper cells. The HPRT1, on the other hand, displayed a significant positive correlation with the abundance of eosinophils, type 2 T helper cells and effector memory CD8 T cells (Fig. 9C). These findings provide molecular evidence for the specific changes in immune cells within the immunological microenvironment of MASH patients and may significantly impact the understanding of the immunopathological mechanisms in MASH.

Immune cell infiltration analysis. (A) The violin plot indicated the differences in immune infiltrating between MASH and control groups. (B) The heatmap showed the infiltration level of 28 immune cell types in MASH and control groups. (C) The correlation between 28 immune cells and 7 signature MRDEGs.
Discussion
MASH, as a severe subtype of MASLD, is characterized by inflammatory cell infiltration and lipid deposition. Compared to MAFL, MASH is more likely to progress to liver fibrosis, cirrhosis, and hepatocellular carcinoma21. However, to date, the diagnosis of MASH remains a significant challenge. In clinical practice, the diagnosis of MASH relies on liver biopsy, but due to its invasiveness, patients have low acceptance and its application is limited. Therefore, the development of effective non-invasive diagnostic methods is crucial. In 2023, multiple international academic organizations jointly proposed the use of MASH/MASLD to replace NASH/NAFLD, a redefinition that emphasizes the key role of metabolic factors in the pathogenesis of MASH/MASLD5. Identification of MRGs that are critical to MASH may be critical to its diagnosis.
In this study, differential expression of 34 MRGs was observed. These genes play crucial roles in various metabolic pathways in MASH, such as “sterol biosynthetic process”, “cholesterol metabolic process”, “endoplasmic reticulum lumen”, “oxidoreductase activity, acting on paired donors, with incorporation or reduction of molecular oxygen”, and “glycerolipid metabolism”. Notably, these MRDEGs are associated with the etiology of MASH, including dyslipidemia and insulin resistance. To further investigate the significance of MRDEGs in MASH, we processed the 34 MRDEGs and developed a diagnostic model comprising 7 signature MRDEGs through PPI network and machine learning algorithms. The diagnostic model demonstrated an AUC higher than 0.9 for the diagnosis of MASH in the training cohort and two validation cohorts. Additionally, individual genes exhibited AUC values predominantly above 0.7 and 0.6 in the training and validation cohorts, respectively. These results indicate that signature MRDEGs and the diagnostic model possess significant accuracy and reliability in discriminating MASH patients, potentially offering novel molecular biomarkers for the diagnosis of MASH.
We also found that there was interaction among 7 signature MRDEGs. Furthermore, enrichment analysis indicated that these genes were involved in multiple metabolic processes, including glucose and lipid metabolism. Dysregulation of glycolipid metabolism plays a crucial role in the progression of MASH. Abnormalities in glucose metabolism, particularly the development of insulin resistance, lead to a decreased efficiency of glucose utilization by the liver, thereby promoting the abnormal accumulation of fat in the liver22. Additionally, defects in insulin signaling further impair the regulation of lipid metabolism in the liver, increasing the synthesis and reducing the degradation of fatty acids, resulting in the accumulation of lipids within hepatocytes23. The abnormal accumulation of fatty acids within hepatocytes not only increases the fluidity of the cell membrane but also activates inflammatory signaling pathways, promoting the occurrence of oxidative stress responses24. Chronic oxidative stress and inflammation further impair hepatocyte function and accelerate the progression of MASH. In another study, Wruck et al.25 identified 22 signature genes associated with the progression of NASH based on datasets from du Plessis et al.26, Horvath et al.13, Ahrens et al.17 and Wruck et al.27. Among these genes, only HPRT1 and FADS2 were found to be consistent with our results. This discrepancy may be attributed to the fact that we employed a variety of machine learning approaches to screen for signature genes. Furthermore, they also observed a significant enrichment of pathways related to cholesterol, fatty acid, lipid synthesis, and metabolism. This finding is in full accord with our research outcomes, thereby validating our results and emphasizing the critical role of these biological pathways in the progression of MASH.
AKR1B10, an enzyme belonging to the aldo-keto reductase family, plays a pivotal role in hepatic metabolic pathways including gluconeogenesis, fatty acid metabolism, and bile acid synthesis28. Research has indicated that AKR1B10 facilitates the ubiquitin-dependent degradation of ACCα, thus inhibiting its ubiquitination and proteolysis, promoting the synthesis of fatty acids and lipids, and thus promoting the development of MASH29. Additionally, studies have reported that the combination of serum AKR1B10 and CK18 serves as a potential diagnostic marker for MASH30.
FADS2 (Fatty Acid Desaturase 2) is a crucial enzyme involved in the biosynthesis of polyunsaturated fatty acids (PUFAs), particularly the conversion of linoleic acid to γ-linolenic acid and subsequent metabolic products31. Studies have indicated that increased FADS2 activity in the livers of MASH mice leads to an imbalance between ω-6 fatty acids (pro-inflammatory) and ω-3 fatty acids (anti-inflammatory), with an increased ω-6 to ω-3 ratio, thereby exacerbating lipid accumulation and inflammatory damage within hepatocytes and promoting the progression of MASH18. CYP7A1 (Cholesterol 7α hydroxylase) is the rate-limiting enzyme in the classic bile acid synthesis pathway, which initiates the synthesis of bile acids by catalyzing the conversion of cholesterol to 7α-hydroxycholesterol, and thus can serve as a cholesterol clearance agent32. Enhanced activity of CYP7A1 may expand the pool of toxic bile acids, such as hydrophobic bile acids33. Additionally, Wruck et al.34 discovered that despite the upregulation of CYP7A1 in MASLD, its regulatory level remained lower than cholesterol synthetic enzymes HMGCR and SQLE, leading to the continued accumulation of cholesterol in the live. ENO3 (Enolase 3) is an enzyme involved in glycolysis, but it can also mediate the synthesis of cholesterol esters, increasing the delivery of lipids to the liver and accelerating the accumulation of cholesterol esters in hepatocytes35. Lu et al. reported that ENO3 negatively regulated ferroptosis by upregulating GPX4 expression and lipid accumulation, thereby promoting the progression of MASH36. GCK (Glucokinase), also known as hexokinase IV, is predominantly located in the liver and catalyzes the formation of glucose-6-phosphate37. Studies have found that the regulation of GCK by KLF6 can lead to insulin resistance38. However, the molecular mechanisms by which HPRT1 and GPD1 promote the development of MASH remain unclear. HPRT1 (Hypoxanthine-guanine phosphoribosyltransferase 1) is a key enzyme in the purine nucleotide salvage pathway. A decrease in HPRT1 expression may lead to purine metabolism disorders, subsequently causing elevated levels of uric acid, promoting oxidative stress and inflammatory responses, and exacerbating the pathological progression of MASH39,40. GPD1 (glycerol-3-phosphate dehydrogenase 1) encodes a member of the NAD-dependent glycerol-3-phosphate dehydrogenase family, which plays a critical role in carbohydrate and lipid metabolism and is involved in NADH/NAD+ recycling41. Increased activity of GPD1 may result in excessive accumulation of triglycerides within the liver, thereby promoting steatosis and inflammatory responses42.
Recent studies have confirmed that the immune system plays a significant role in the initiation and progression of hepatic inflammation in MASH patients43. This study utilized ssGSEA and found that, compared to the control group, the infiltration levels of activated CD8 T cells, γδT cells, NK cells, and CD56bright NK cells were significantly increased in the MASH group. In contrast, the infiltration of Th2 cells, memory B cells, central memory CD8 T cells, effector memory CD8 T cells, and eosinophils was significantly reduced. Various T cell subsets contribute to the immunopathogenesis of MASH. In diet-induced MASH model, activated CD8 T cells promoted inflammation and fibrosis by relying on the expression of myeloid cell MHC I44. γδT cells rapidly respond to inflammation by recognizing danger-associated molecular patterns (DAMPs) and pathogen-associated molecular patterns (PAMPs) through the secretion of immunoregulatory cytokines, including IL-17, IFN-γ, and TGFβ45. In MASH animal models, an increased infiltration of hepatic γδT-cells was observed, mediating IL-17 signaling to accelerate the progression of MASH46. NK cells also rapidly increase in number in MASH patients. In the MASH model, activated hepatic NK cells secreted a large amount of cytokines and induced hepatocyte injury via the JAK/STAT pathway, further promoting the progression of MASH47.
T helper cells serve as pivotal regulators in both pro-inflammatory and anti-inflammatory immune processes, characterized by the expression of CD4 on cytomembrane48. Type 1 T helper (Th1) and Type 17 T helper (Th17) cells are pro-inflammatory subsets that have been reported to increase in peripheral blood of MASH patients49. However, in the current study, no significant differences were observed. Research on the role of Th2 cells in MASH has been limited. Rau et al. reported an increase in peripheral Th2 cells in MASH patients compared to healthy controls with normal body weight; however, there were differences in age among the patients50. It is noteworthy that Th2 cells are considered to possess a high potential for promoting fibrosis, particularly through the action of IL-13, but studies in the context of MASH have yet to explore this aspect in depth. The role of memory CD8 T cells in anti-tumor immunity and inflammatory autoimmune diseases has been well-established51,52. Koda et al. reported the involvement of memory CD8 T cells in the resolution of fibrosis, where these cells significantly accumulated in areas of fibrosis resolution and promoted this process through CCR5-dependent chemotaxis and direct cytotoxic actions against HSCs53. In our study, the infiltration of central memory CD8 T cells and effector memory CD8 T cells in patients with MASH was found to be low, suggesting a diminished protective effect.
However, this study still has some limitations. Firstly, the datasets utilized in our study are sourced from public databases. Secondly, the samples within these datasets originated from various countries, and there is a lack of stratification by gender and region, which may introduce heterogeneity into our results. Consequently, it is imperative that future research employs external cohorts from multiple international centers to validate our findings. Additionally, conducting sensitivity analysis with stratification by gender and region will be essential to ascertain the robustness and generalizability of our results. Thirdly, tissue-based flow cytometry is needed to verify changes in the immune microenvironment.
Conclusion
Our study identified 7 signature MRDEGs that have a good ability to distinguish MASH patients from healthy controls, providing a non-invasive approach for the diagnosis of MASH. Exploration of the metabolic pathways involved in these genes and their association with the immune microenvironment will contribute to our understanding of the mechanisms of MASH progression. Nevertheless, further basic research combined with clinical information is needed to verify our findings.
Data availability
The datasets (GSE135251, GSE17470, GSE37031, GSE48452, GSE89632, GSE63067, GSE126848, and GSE61260) generated and/or analysed during the current study are available in the GEO repository (https://www.ncbi.nlm.nih.gov/geo/).
Abbreviations
- MASLD:
-
Metabolic associated liver disease
- NAFLD:
-
Nonalcoholic fatty liver disease
- MASH:
-
Metabolic associated steatohepatitis
- NASH:
-
Nonalcoholic steatohepatitis
- MAFL:
-
Metabolic associated fatty liver
- MRGs:
-
Metabolism-related genes
- GEO:
-
Gene Expression Omnibus
- DEGs:
-
Differentially expressed genes
- RRA:
-
Robust Rank Aggregation
- MRDEGs:
-
Metabolism-related differentially expressed genes
- GO:
-
Gene Ontology
- KEGG:
-
Kyoto Encyclopedia of Genes and Genomes
- PPI:
-
Protein–Protein Interaction
- LASSO:
-
Least Absolute Shrinkage and Selection Operator
- SVM-RFE:
-
Support Vector Machine-Recursive Feature Elimination
- RF:
-
Random Forest
- ROC:
-
Receiver operating characteristic
- AUC:
-
Calculated the area under the curve
- GSVA:
-
Gene Set Variation Analysis
- ssGSEA:
-
Single sample gene set enrichment analysis
- BP:
-
Biological process
- CC:
-
Cellular component
- MF:
-
Molecular function
- γδT:
-
Gamma delta T
- NK:
-
Natural killer
- Th2:
-
Type 2 T helper
- Th1:
-
Type 1 T helper
- Th17:
-
Type 17 T helper
- DAMPs:
-
Danger-associated molecular patterns
- PAMPs:
-
Pathogen-associated molecular patterns
References
Rinella, M. E. et al. AASLD Practice Guidance on the clinical assessment and management of nonalcoholic fatty liver disease. Hepatology 77 (5), 1797–1835 (2023).
Marjot, T. et al. Nonalcoholic fatty liver disease in adults: Current concepts in etiology, outcomes, and management. Endocr. Rev. 41 (1), 66–117 (2020).
Zou, H. M. et al. Epidemiology and disease burden of non-alcoholic steatohepatitis in greater China: A systematic review. Hep. Intl. 16 (1), 27–37 (2022).
Rafiq, N. et al. Long-term follow-up of patients with nonalcoholic fatty liver. Clin. Gastroenterol. Hepatol. 7 (2), 234–238 (2009).
Rinella, M. E. et al. A multisociety Delphi consensus statement on new fatty liver disease nomenclature. Hepatology 78 (6), 1966–1986 (2023).
Gabriel-Medina, P. et al. Influence of type 2 diabetes in the association of PNPLA3 rs738409 and TM6SF2 rs58542926 polymorphisms in NASH advanced liver fibrosis. Biomedicines 10 (5), 1015 (2022).
Vilar-Gomez, E. et al. The protection conferred by HSD17B13 rs72613567 on hepatic fibrosis is likely mediated by lowering ballooning and portal inflammation. Clin. Gastroenterol. Hepatol. 21 (11), 2981–2983e3 (2023).
Yuan, T. et al. Machine learning in the identification of prognostic DNA methylation biomarkers among patients with cancer: A systematic review of epigenome-wide studies. Artif. Intell. Med. 143, 102589 (2023).
Danieli, M. G. et al. A machine learning analysis to evaluate the outcome measures in inflammatory myopathies. Autoimmun. Rev. 22 (7), 103353 (2023).
He, Z. et al. Identification of MAP3K4 as a novel regulation factor of hepatic lipid metabolism in non-alcoholic fatty liver disease. J. Transl. Med. 20 (1), 529 (2022).
Dai, L. et al. Machine learning-based integration identifies the ferroptosis hub genes in nonalcoholic steatohepatitis. Lipids Health Dis. 23 (1), 23 (2024).
Yu, R. et al. Analysis of machine learning based integration to identify the crosslink between inflammation and immune response in non-alcoholic fatty liver disease through bioinformatic analysis. Heliyon 10 (14), e32783 (2024).
Horvath, S. et al. Obesity accelerates epigenetic aging of human liver. Proc. Natl. Acad. Sci. U S A. 111 (43), 15538–15543 (2014).
Suppli, M. P. et al. Hepatic transcriptome signatures in patients with varying degrees of nonalcoholic fatty liver disease compared with healthy normal-weight individuals. Am. J. Physiol. Gastrointest. Liver Physiol. 316 (4), G462–g472 (2019).
Frades, I. et al. Integrative genomic signatures of hepatocellular carcinoma derived from nonalcoholic fatty liver disease. PLoS One. 10 (5), e0124544 (2015).
Arendt, B. M. et al. Altered hepatic gene expression in nonalcoholic fatty liver disease is associated with lower hepatic n-3 and n-6 polyunsaturated fatty acids. Hepatology 61 (5), 1565–1578 (2015).
Ahrens, M. et al. DNA methylation analysis in nonalcoholic fatty liver disease suggests distinct disease-specific and remodeling signatures after bariatric surgery. Cell. Metab. 18 (2), 296–302 (2013).
López-Vicario, C. et al. Molecular interplay between ∆5/∆6 desaturases and long-chain fatty acids in the pathogenesis of non-alcoholic steatohepatitis. Gut 63 (2), 344–355 (2014).
Baker, S. S. et al. Role of alcohol metabolism in non-alcoholic steatohepatitis. PLoS One. 5 (3), e9570 (2010).
Govaere, O. et al. Transcriptomic profiling across the nonalcoholic fatty liver disease spectrum reveals gene signatures for steatohepatitis and fibrosis. Sci. Transl. Med., 12(572). (2020).
Hardy, T. et al. Nonalcoholic fatty liver disease: Pathogenesis and disease spectrum. Annu. Rev. Pathol. 11, 451–496 (2016).
Ferdous, S. E. & Ferrell, J. M. Pathophysiological relationship between type 2 diabetes mellitus and metabolic dysfunction-associated steatotic liver disease: Novel therapeutic approaches. Int. J. Mol. Sci., 25(16). (2024).
Feng, X. et al. Mechanism of metabolic dysfunction-associated steatotic liver disease: Important role of lipid metabolism. J. Clin. Transl. Hepatol. 12 (9), 815–826 (2024).
Gancheva, S., Roden, M. & Castera, L. Diabetes as a risk factor for MASH progression. Diabetes Res. Clin. Pract. 217, 111846 (2024).
Wruck, W. et al. Concise review: Current status and future directions on research related to nonalcoholic fatty liver disease. Stem Cells. 35 (1), 89–96 (2017).
du Plessis, J. et al. Association of adipose tissue inflammation with histologic severity of nonalcoholic fatty liver disease. Gastroenterology, 149(3), 635 – 48.e14 (2015).
Wruck, W. et al. Multi-omic profiles of human non-alcoholic fatty liver disease tissue highlight heterogenic phenotypes. Sci. Data. 2, 150068 (2015).
Endo, S., Matsunaga, T. & Nishinaka, T. The role of AKR1B10 in physiology and pathophysiology. Metabolites 11 (6), 332 (2021).
Lin, X. L. et al. Nicotinate-curcumin improves NASH by inhibiting the AKR1B10/ACCα-mediated triglyceride synthesis. Lipids Health Dis. 23(1). (2024).
Choi, S. J. et al. A composite blood biomarker including AKR1B10 and cytokeratin 18 for progressive types of nonalcoholic fatty liver disease. Diabetes Metab. J. 48(4). (2024).
Koletzko, B. et al. FADS1 and FADS2 polymorphisms modulate fatty acid metabolism and dietary Impact on Health. Annu. Rev. Nutr. 39, 21–44 (2019).
Lim, M. Y. C. & Ho, H. K. Pharmacological modulation of cholesterol 7α-hydroxylase as a therapeutic strategy for hypercholesterolemia. Biochem. Pharmacol. 220 (2024).
Li, T. G. et al. Overexpression of cholesterol 7α-hydroxylase promotes hepatic bile acid synthesis and secretion and maintains cholesterol homeostasis. Hepatology 53 (3), 996–1006 (2011).
Wruck, W. & Adjaye, J. Meta-analysis reveals up-regulation of cholesterol processes in non-alcoholic and down-regulation in alcoholic fatty liver disease. World J. Hepatol. 9 (8), 443–454 (2017).
Wu, J. et al. Characterization of porcine ENO3: Genomic and cDNA structure, polymorphism and expression. Genet. Sel. Evol. 40 (5), 563–579 (2008).
Lu, D. et al. ENO3 promoted the progression of NASH by negatively regulating ferroptosis via elevation of GPX4 expression and lipid accumulation. Ann. Transl. Med. 9(8). (2021).
Agius, L. Glucokinase and molecular aspects of liver glycogen metabolism. Biochem. J. 414, 1–18 (2008).
Bechmann, L. P. et al. Glucokinase links Kruppel-like factor 6 to the regulation of hepatic insulin sensitivity in nonalcoholic fatty liver disease. Hepatology 55 (4), 1083–1093 (2012).
Wang, Y. et al. MicroRNA-34a mediates high-fat-induced hepatic insulin resistance by targeting ENO3. Nutrients 15(21). (2023).
Dali-Youcef, N. et al. Interleukin-32 contributes to human nonalcoholic fatty liver disease and insulin resistance. Hepatol. Commun. 3 (9), 1205–1220 (2019).
Oh, S. et al. Glycerol 3-phosphate dehydrogenases (1 and 2) in cancer and other diseases. Exp. Mol. Med. 56 (5), 1066–1079 (2024).
Swierczynski, J. et al. Enhanced glycerol 3-phosphate dehydrogenase activity in adipose tissue of obese humans. Mol. Cell. Biochem. 254 (1–2), 55–59 (2003).
Arrese, M. et al. Innate immunity and inflammation in NAFLD/NASH. Dig. Dis. Sci. 61 (5), 1294–1303 (2016).
Adams, V. R. et al. Myeloid cell MHC I expression drives CD8(+) T cell activation in nonalcoholic steatohepatitis. Front. Immunol. 14, 1302006 (2023).
Torres-Hernandez, A. et al. γδ T cells promote steatohepatitis by orchestrating innate and adaptive Iimmune programming. Hepatology 71 (2), 477–494 (2020).
Li, F. et al. The microbiota maintain homeostasis of liver-resident γδT-17 cells in a lipid antigen/CD1d-dependent manner. Nat. Commun. 7, 13839 (2017).
Wang, F. et al. Activated natural killer cell promotes nonalcoholic Steatohepatitis through mediating JAK/STAT pathway. Cell. Mol. Gastroenterol. Hepatol. 13 (1), 257–274 (2022).
Heymann, F. & Tacke, F. Immunology in the liver–from homeostasis to disease. Nat. Rev. Gastroenterol. Hepatol. 13 (2), 88–110 (2016).
Chackelevicius, C. M. et al. Th17 involvement in nonalcoholic fatty liver disease progression to non-alcoholic steatohepatitis. World J. Gastroenterol. 22 (41), 9096–9103 (2016).
Rau, M. et al. Progression from nonalcoholic fatty liver to nonalcoholic steatohepatitis is marked by a higher frequency of Th17 cells in the liver and an increased Th17/resting regulatory T cell ratio in peripheral blood and in the liver. J. Immunol. 196 (1), 97–105 (2016).
Park, S. L. et al. Tissue-resident memory CD8(+) T cells promote melanoma-immune equilibrium in skin. Nature 565 (7739), 366–371 (2019).
Zundler, S. et al. Hobit- and Blimp-1-driven CD4(+) tissue-resident memory T cells control chronic intestinal inflammation. Nat. Immunol. 20 (3), 288–300 (2019).
Koda, Y. et al. CD8(+) tissue-resident memory T cells promote liver fibrosis resolution by inducing apoptosis of hepatic stellate cells. Nat. Commun. 12 (1), 4474 (2021).
Funding
This work was supported by the Natural Science Foundation of Henan Provincial (NO. 242300421499).
Author information
Authors and Affiliations
Contributions
Haoran Xie and Junjun wang analyzed data and wrote the manuscript. Qiuyan Zhao proposed the research conceptualization and revised the manuscript. The final manuscript was read and confirmed by all authors.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Xie, H., Wang, J. & Zhao, Q. Identification of potential metabolic biomarkers and immune cell infiltration for metabolic associated steatohepatitis by bioinformatics analysis and machine learning. Sci Rep 15, 16596 (2025). https://doi.org/10.1038/s41598-025-86397-x
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-86397-x
Keywords
This article is cited by
-
Opportunities and challenges of artificial intelligence in hepatology
npj Gut and Liver (2026)
-
Identification of lipid metabolism-related signature in nonalcoholic fatty liver: evidences from transcriptomics and single cell RNA-sequencing analysis
European Journal of Medical Research (2025)
