
Abstract
The house mouse, Mus musculus, is a widely used animal model in biomedical research, with classical laboratory strains (CLS) being the most frequently employed. However, the limited genetic variability in CLS hinders their applicability in evolutionary studies. Wild-derived strains (WDS), on the other hand, provide a suitable resource for such investigations. This study quantifies genetic and phenotypic data of 101 WDS representing 5 species, 3 subspecies, and 8 natural Y consomic strains and compares them with CLS. Genetic variability was estimated using whole mtDNA sequences, the Prdm9 gene, and copy number variation at two sex chromosome-linked genes. WDS exhibit a large natural variation with up to 2173 polymorphic sites in mitogenomes, whereas CLS display 92 sites. Moreover, while CLS have two Prdm9 alleles, WDS harbour 46 different alleles. Although CLS resemble M. m. domesticus and M. m. musculus WDS, they differ from them in 10 and 14 out of 16 phenotypic traits, respectively. The results suggest that WDS can be a useful tool in evolutionary and biomedical studies with great potential for medical applications.
Similar content being viewed by others
Introduction
Progress in many areas of the life sciences would be inconceivable without appropriate biological models. For example, the Drosophila fruit fly was used to establish the fundamentals of genetics1,2, while yeast was used to discover DNA recombination mechanisms and gene-protein functions3. The laboratory mouse is the most prominent vertebrate model4. Mice are easy to keep, breed, and handle, they are also highly tolerant to inbreeding, and their normative biology is well understood5. A whole-genome sequence was published almost simultaneously with that of humans6, and large databases of well-annotated sequences, SNPs, and other markers with known positions in the genome are now available, making the laboratory mouse an ideal organism for tracing genotype-phenotype associations and inferring gene function e.g7. The ability to replenish breeding stocks from frozen embryos avoids the negative effects of mutation load and thus increases the reproducibility of experimental preclinical studies. Due to a relatively short generation time and the ability to reproduce throughout the year, it is possible to generate multiple laboratory stocks within a reasonable time. In fact, dozens of inbred ‘classical laboratory strains’ (CLS) have been established since the early 20th century. There are more than 200 CLS listed in8, indicating the availability of significant genetic diversity that can be harnessed when selecting the most suitable mouse model for a specific study.
Despite the indispensability of CLS for biomedical and life science studies, their applicability appears to be reaching its limits. First, their genome is an artificial mixture of three house mouse subspecies: Mus musculus domesticus, M. m. musculus, and M. m. castaneus9,10. Second, despite the high number of existing CLS, they display limited genetic variation. For example, they all essentially represent a single female as demonstrated by Ferris et al.11 and later confirmed by mitochondrial whole-genome sequences12,13,14 or hundreds of thousand SNP in over 150 CLS9. This finding poses a significant limitation when one wishes to apply CLS-based genotype-phenotype associations to human populations that exhibit high genetic variability and, subsequently, an increased level of (epi)genetic interactions.
Naturally, the highest variation in house mice occurs within wild populations. While geographically wide-scale whole genome sequencing of wild mice has not progressed as extensively as human studies (as exemplified by the 1000 Genomes Project or the Pangenome Reference project15,16), recent publications have summarized diversity across more than 310 mouse genomes17,18,19,20,21,22,23,24,25,26. This diversity is further expanded by introgressive hybridization observed in their contact zones, such as between M. m. castaneus and M. m. musculus22 or M. m. domesticus and M. m. musculus subspecies27,28,29,30. On the other hands, in contrast to inbred laboratory stocks, the high natural variation in wild mice conflicts with strict requirements for experimental reproducibility. Moreover, the inevitable presence of rare alleles in wild populations hampers genome-wide association studies (GWAS)31,32.
Inbred wild-derived strains (WDS) offer an excellent compromise between the two conflicting requirements of suitable variation and reproducibility. Unlike CLS, the geographic origin and pedigree of WDS are precisely known. Although WDS cannot fully capture natural variation, increasing the number of stocks can mitigate this limitation. In addition, WDS are thought to represent valuable resources suitable for GWAS, as rare alleles that might hinder association studies in wild mice are fixed for different variants in individual strains. Thus, by exploiting their phenotypic variation and genomic complexity, WDS provide an ultimate model system for understanding the genetic control of quantitative traits relevant to evolutionary and biomedical research. However, our knowledge of phenogenomic data is limited and confined to dozens of WDS held in different laboratories26,33,34,35,36.
In this study we report a generation of a unique world-wide largest public resource of WDS. To assess the extent of variation contributed by wild progenitors, we evaluated the genetic and phenotype variability in more than 100 WDS representing five species of the genus Mus and compared it with that of CLS. For genetic variation, we focused on components known to evolve rapidly due to higher mutation rates or genomic conflict. Specifically, we analyzed whole-genome mitochondrial (mtDNA) sequences, which have high mutation rates and are frequently used to delineate or break species boundaries37,38. From the nuclear genome, we examined partial sequences of the Prdm9 gene known to cause reproductive isolation between the M. m. domesticus C57BL/6J (CLS) and M. m. musculus PWD (WDS) strains39,40. Additionally, we assessed copy number variation in two sex chromosome-linked genes, Slx and Sly, that are involved in genomic conflict27,41. To investigate phenotypic variation, we analyzed a set of 16 phenotypic traits, encompassing external morphological characteristics, reproductive organ morphology in both sexes, and reproductive performance. We show that genetic variation is significantly higher in WDS compared to CLS, and its magnitude increases with divergence time. Similarly, the phenotypic diversity preserved in WDS substantially exceeds that observed in classical laboratory mice. Both datasets, i.e. genetic and phenotypic, are congruent, suggesting that CLS harbour only a tiny fraction of the house mouse variability and hence do not adequately represent mice in natural populations. Incorporating variation from WDS can, therefore, enhance the biological reality of the house mouse model. In conclusion, we advocate for including the variation preserved in WDS, which holds great potential to enhance the effectiveness of the house mouse model not only in evolutionary studies but also in biomedical research.
Results
Mouse repository
The repository has been established by merging four WDS resources previously based in Montpellier, France42,43, Plön, Germany19, Prague44 and Studenec, Czech Republic45,46 representing a major consolidation of wild-derived mouse strains into a single location. In total, it consists of 106 strains (94 WDS and 7 CLS), all currently kept (81 alive) or were kept in the past (25 became extinct due to reproductive failure or were removed from the breeding scheme). The WDS were classified into groups representing five species, one synanthropic species (M. musculus) with three subspecies (M. m. musculus, M. m. domesticus, M. m. castaneus), and four non-synanthropic species (M. caroli, M. macedonicus, M. spicilegus, M. spretus). In the following text, we will use the subspecies and species names of mouse taxa (e.g. musculus for M. m. musculus or caroli for M. caroli). For comparative purposes in this paper, we consider all CLS with mixed but predominantly domesticus genomes as a separate group. This spectrum of WDS captures over 5 million years of mouse evolution, as estimated from the split of caroli from the remaining species under study47. Detailed information on individual strains, including their geographic origin, chromosome number, mtDNA and Y (sub)specific ancestry, the presence of the meiotic driver, t-haplotype, generation of inbreeding, and reproductive ability over the last four years, is provided in Fig. 1 and Supplementary Table S1. Further details can be found at https://housemice.cz/en/strains. Importantly, while many strains have been included in previous studies9,25,42,46,48,49,50, this study provides new data on mitochondrial DNA, Prdm9, Slx/Sly, and various phenotypes. Moreover, several strains are newly introduced here for the first time, further highlighting the novelty and value of this repository.

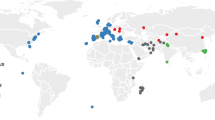
Geographic origin of WDS kept in Studenec; red: musculus, blue: domesticus, green: spretus, brown: macedonicus, light olive: spicilegus; circles: alive WDS, diamonds: extinct WDS. Small green dots indicate the position of the house mouse hybrid zone adapted from28,51. The numbers labelling localities correspond with those in Supplementary Table S1. WDS out of the depicted area are KTK (caroli from Thailand), CIM (castaneus from India), TAIG (castaneus from Taiwan), DKN and CKN (castaneus from Kenya), MDG (castaneus from Madagascar), BID, KAK, and TEH (musculus from Iran), KH (musculus from Kazakhstan), MPR (musculus from Pakistan), AH and AH7 (domesticus from Iran), and DOT (domesticus from Tahiti, French Polynesia). The map was created using the Free and Open Source QGIS (https://www.qgis.org/).
mtDNA variability
Extra-nuclear variation was assessed using 114 newly sequenced mitogenomes and 49 published whole mtDNA sequences (see Material and Methods and Supplementary Table S2). After alignment, the sequences were 16,516 bp long, all representing unique haplotypes, with a total of 3,907 polymorphic sites. Aligned sequences are available in Dataset 1 on DRYAD at https://doi.org/10.5061/dryad.r7sqv9skn.
Prior to estimating genetic variation, we assessed taxonomic identity and phylogenetic relationships of all the WDS. The maximum-likelihood tree revealed eight distinct clades, each representing a (sub)species. The Madagascan strain (MDG) carried a unique haplotype within the monophyletic M. musculus haplogroups (Fig. 2A, D), similar to the mtDNA lineage previously described as M. m. gentilulus52,53. All CLS formed a monophyletic group embedded within the domesticus WDS group, reflecting the fact that all CLS carry domesticus mtDNA (Fig. 2B). The castaneus clade consists of two deeply divergent clusters, one involving samples from Iran, and the second from Thailand, India, and Kenya (Fig. 2C). This divergence pattern can be attributed either to the presence of a 76-bp insert in the former cluster, or paraphyly of the subspecies as suggested earlier54,55,56, or both.

Maximum-likelihood trees for the whole mtDNA dataset (A) and separately for domesticus (B), castaneus (C), and musculus (D) strains. In the domesticus tree, the CLS cluster is shaded. The codes for individual samples indicate group, WDS name, mouse ID, and the number of generations of inbreeding when known.
The number of polymorphic sites and nucleotide diversity values can be found in Table 1. We detected 36× more sites and approximately a 100-fold nucleotide diversity in WDS compared to CLS. Interestingly, nucleotide diversity was more than twofold in musculus-derived WDS compared to domesticus-derived WDS.
The numbers of polymorphic sites between individual strains are provided in Supplementary Table S3, while the minimum and maximum numbers of polymorphic sites between the groups are summarized in Table 2. The most significant differentiation was observed between caroli and musculus mitogenomes, with 2,251 polymorphic sites, accounting for 13.8% of the mitogenome. The non-synanthropic mice (caroli, macedonicus, spicilegus, spretus) differed by more than 2,250 polymorphic sites from the synanthropic mice. Differences between house mouse WDS did not exceed 548 polymorphic sites. Despite the musculus WDS group having less than half the number of strains compared to the domesticus WDS group, variation within the former group was higher as shown in the diagonal of Table 2), confirming the results in Table 1. This difference is primarily attributed to the presence of a 75-bp long insertion in the control region in some musculus strains, absent in all domesticus WDS. Although some domesticus strains possess an 11-bp insert in another part of the control region, which is not present in musculus, the 75-bp segment significantly contributes to the higher variation. Indeed, when considering musculus WDS without the insert, domesticus WDS have up to 40 more polymorphic sites. The same argument applies to the castaneus strains.
A relatively high number of polymorphic sites found within CLS (N = 92) was caused by the strain NZB developed at the University of Dunedin in New Zealand57. When the NZB strain was excluded, the level of variation dropped substantially, reaching a maximum of 6 polymorphic sites between C3H and MA strains (Supplementary Table S3). Accordingly, the variation within the domesticus WDS and musculus WDS strains increased by a factor of 1.62 (24.83 when the NZB strain is excluded) and 2.63 (40.33 without NZB), respectively, compared to what was observed in the CLS strain.
The mtDNA dataset included 31 duplicated samples, which were utilized to assess consistency in two aspects: (i) between the newly sequenced and published mtDNAs in CLS and (ii) between mice of the same strain with substantial differences in the numbers of generations since the founding of the strain. While no differences were observed between our and published CLS mitogenomes, inconsistencies between sequences in samples with varying numbers of generations were detected in 12 out of 31 strains (Supplementary Table S4). Remarkably, half of these cases involved introgression between subspecies or even species such as from spretus to domesticus in STF strain (Supplementary Table S4.A).
Prdm9 gene
Prdm9 protein is a histone methyltransferase that initiates meiotic recombination by binding to allele specific DNA sequences via its highly polymorphic zinc finger domain58. To assess its variation, we sequenced the zinc finger (ZnF) domain of the Prdm9 gene in 98 samples (Supplementary Table S5). This dataset was supplemented with 12 CLS sequences from the study by Kono et al.59. Five Prdm9 sequences from CLS maintained in Studenec, along with the published sequences from the same CLS59, were used as a sequencing quality control. No differences in duplicated samples were detected.
By sequencing the Prdm9 ZnF domain, we identified 48 alleles among the 105 strains analysed (see Supplementary Table S5 for allelic designations, which provide information on the most amino-acid polymorphic sites across all ZnFs, following the nomenclature introduced by Mukaj et al.60). An overall comparison between M. musculus WDS and CLS revealed a significant distinction: while only two alleles were found in 12 CLS, as many as 38 alleles were present in 83 M. musculus WDS. This difference cannot be explained by the higher number of WDS (sevenfold compared to CLS, contrasting to 19× higher number of alleles). Allelic diversity detected in the domesticus and musculus WDS (A = 0.51 and A = 0.39, respectively) was 3.1–2.4× higher than in the CLS (A = 0.17). In contrast to mtDNA, the correspondence between the Prdm9 gene tree and species tree is weaker. Although most domesticus-derived strains, including all analyzed CLS, cluster within a single large group, several of them are interspersed among other clusters. A similar scattered pattern is also observed in WDS representing other (sub)species (Fig. 3), with most variable castaneus WDS dispersed across the entire phylogenetic tree.

Maximum-likelihood tree of the Prdm9 gene; blue: domesticus WDS (including CLS), red: musculus, magenta: castaneus, green: spretus, brown: macedonicus, dark green: spicilegus. The codes for individual samples indicate group, WDS name, mouse ID, and the number of generations of inbreeding.
Slx/Sly copy number variation (CNV)
We assessed CNV in two highly ampliconic genes, Slx (Sycp3 like X-linked) located on the X chromosome and Sly (Sycp3 like Y-linked) on the Y chromosome, in 53 strains (Supplementary Table S6). As shown in Table 3, musculus WDS revealed approximately a two times higher CN compared to domesticus WDS for both genes. Furthermore, Sly CN was, on average, approximately 2.5-fold higher than that of Slx. Interestingly, even though both CLS analyzed (C57BL/6J and C3Hb) predominantly possess domesticus-like genomes, their Sly CN is considerably higher than those in domesticus WDS. The higher Sly CN in CLS relative to domesticus WDS can be explained by the fact that these strains carry musculus-like Y chromosome61,62. However, the average Sly CN in CLS is still lower than that in musculus WDS (Table 3). Consequently, the two CLS appear intermediate between the musculus WDS and domesticus WDS clusters when Sly CN is plotted against Slx CN (Fig. 4). It is worth noting that two musculus WDS outliers deviate from the overall pattern: MAM and STUS with 21 and 54 Slx copies, respectively. These numbers are characteristic of domesticus WDS; however, X-linked SNPs in STUS9 and MAM exome42 confirm their musculus origin.

Scatterplot of Slx and Sly CN in musculus WDS, domesticus WDS, and CLS. Vertical and horizontal error bars show Poisson distribution-based errors from triplicate measures of Slx and Sly copy numbers, respectively.
Phenotype variability
We assessed phenotypic variation in nine traits recorded in 4,494 mice, representing 89 WDS of musculus, domesticus, castaneus, spretus, spicilegus, macedonicus, caroli, and five CLS (see Dataset 2–4 in DRYAD: https://doi.org/10.5061/dryad.r7sqv9skn). Our dataset included only individuals between 65 and 600 days old, since subadult and aged individuals are more likely to be infertile. We measured the following traits: body weight, spleen weight (recorded in 3,239 mice), body length, tail length, weight of both ovaries (measured in 2,131 females), sperm count, the weight of testes, left epididymis, and seminal vesicles (measured in 2,363 males). Sperm numbers were counted in a Bürker hematocytometer, with ten chambers per sample, following the methodology described in Vyskočilová et al.63.
Descriptive statistics of the morphometric data computed separately for males and females revealed high variation both across strains (Supplementary Table S7) and the groups (Supplementary Table S8). ANOVA detected significant differences between the groups for all measured traits, including sex*group and sex*strain interactions for external measurements (Supplementary Table S9). It is worth noting that males of Palearctic non-synanthropic species (macedonicus, spicilegus and spretus) displayed substantially higher testis weight and sperm count values, while ovary size showed lower differentiation among females across all groups (Supplementary Figure S15).
The analysis of morphological data, restricted to CLS, domesticus WDS, and musculus WDS (4,100 mice) revealed substantial differentiation between the groups both for males and females. Out of 39 comparative tests for the nine traits, only five were insignificant (4× for CLS and domesticus [males: testicular and epididymal weight; females: tail length and ovary weight], 1× for CLS and musculus [males: sperm count]) (Supplementary Table S10). Individual-based analyses of the morphospace, defined by the projection of two variables, confirmed a higher divergence of the three groups in external traits than in the reproductive characteristics (Fig. 5A-C, and 5a-c). Since CLS are predominantly derived from domesticus9, their phenotypic variation was expected to overlap with the morphospace defined by the latter group. Indeed, the contribution of CLS to the overall morphological variation was negligible, ranging from 0.0% (testis weight vs. sperm count) to 0.1% (body weight vs. relative tail length) (Fig. 5a and c). In contrast, WDS males and females exclusively contributed 57.7% and 52.7%, respectively, to the total variation defined by body weight and relative tail length; the remaining proportion of the morphospace was shared among CLS and WDS. This pattern differs from the reproductive traits, where most of the variation is shared between all three groups in males (59.3% in testis weight vs. sperm count, Fig. 5C and c) and between domesticus and musculus and all three mouse groups in females (each contributing 46.9% and 44.4%, respectively, in the morphospace defined by body weight and ovary weight [data not shown]). Supplementary Table S10 provides pairwise tests of the variance ratios for morphological traits among CLS, domesticus, and musculus groups. In six out of 22 comparisons, the variance ratios did not differ among groups, and in only two of the 14 significant comparisons was the ratio of variances higher in CLS (in both cases for seminal vesicles).

Distribution of phenotypic variation in mouse strains. Morphospaces defined by biplots of body weight vs. relative tail length (100*body length/tail length) in males (A) and females (B); testis weight vs. sperm count (C); and mean litter sizes vs. number of generations delivered per year (D). Small lettered Venn diagrams provide the numbers of overlapping and non-overlapping individuals in domesticus WDS (dom), musculus WDS (mus), and CLS for corresponding graphs labelled with capital letters (A–C).
Reproduction performance
Reproductive ability was estimated in 92 WDS and 8 CLS. In total, the dataset consisted of 112,997 offspring born to 10,471 mothers in 21,427 litters recorded in Studenec studbooks between 2000 and 2024 (see Dataset 5 in DRYAD: https://doi.org/10.5061/dryad.r7sqv9skn). Reproductive performance was characterized by litter size, newborn mortality (calculated as the proportion of stillborn or cannibalized mice across all litters), and the number of generations produced per year. We also estimated the time since a WDS was established from wild progenitors until completing 20 generations of strict brother-sister mating, i.e., the generation at which a strain of mice can be considered inbred64. These data were summarized across the groups and individual stocks.
Table 4 lists reproductive parameters summarized for each group of mice (note that, based on exome data, gentilulus is grouped with the castaneus MDG strain42). Excluding caroli and spicilegus WDS with fewer than three strains per taxon, litter size, mortality, and the number of generations delivered within a single year showed significant differences among the groups (ANOVA, P < 0.0182 for all variables). CLS were the only stocks that could produce, on average, more than six pups per litter and four generations per year (Table 4). Furthermore, along with domesticus WDS, CLS were characterized by newborn mortality below 10%. In this regard, CLS exhibit reproductive characteristics that are highly suitable for maintaining and efficiently breeding, reaffirming their significance as an ideal model in biomedical research. The non-synanthropic species exhibited the lowest reproductive performance among all the groups. This fact can have significant implications when planning and preparing experiments using newly derived strains: achieving the desired level of inbreeding would take roughly five years in CLS or their derivates (consomics, congenics), and approximately seven years in musculus-derived WDS. However, the process could extend to about 12 and 15 years for macedonicus and spicilegus strains, respectively.
High variation in the reproductive parameters is also present among individual strains (Supplementary Table S11). For example, the difference between the minimum and maximum litter sizes is 4.00-fold, ranging from 3.05 (musculus: MDH) to 12.20 (CLS: CD-1). CD-1 (outbred mice derived from a group of Swiss albinos) was the only strain with an average litter size exceeding 10 (as indicated by the rightmost outlier in Fig. 5D). Similarly, higher inter-strain variability was observed in the rate of reproduction, with a 4.72-fold difference between the minimum and maximum numbers of generation produced per year, ranging from 1.11 (spicilegus: ZBP) to 5.23 (domesticus: SWI). Although we cannot test it, these traits may have both genetic and environmental components, and, as a result, their values can vary between different laboratory settings.
All three reproductive parameters differed between musculus WDS, domesticus WDS (excluding CD-1 mice), and CLS (ANOVA: litter size: P = 0.0011; newborn mortality P = 0.0418, generations/year: P = 0.0448). Pairwise comparisons between these three groups showed significant differences in four of the nine comparisons (Supplementary Table S12): musculus WDS had a lower litter size than domesticus WDS and CLS. Additionally, CLS were the only strains capable of producing over four generations per year; however, they were significantly different only from musculus WDS (Table 4, Supplementary Table S12). Despite our best efforts to maintain all strains in optimal conditions, some strains ceased reproduction in various generations. The extinction rate observed across 104 strains was 22.1% (Supplementary Table S1).
Discussion
Any model organism would benefit from incorporating natural variation, to assess how accurately laboratory stocks represent the species they are meant to emulate and provide insight into molecular processes and their variability. Such an integration would significantly enhance the relevance of model systems for the study of complex trait variations, including diseases65.
In this study, we examined genetic and phenotypic variation across 100 wild-derived mouse strains (WDS) to understand the complexity of this vertebrate model. We compared this data to classical laboratory strains (CLS), which are often used as representatives of the house mouse in comparative evolutionary studies and biomedicine e.g66,67,68. Our analysis highlights important distinctions between CLS and their wild-derived counterparts, which are crucial for determining the future utility of both groups in biological and medical sciences.
However, it is important to acknowledge that while we provide a comprehensive comparison, the interpretation of our results may be influenced by the diversity of wild-derived strains sampled. The management of mouse stocks, including breeding practices and colony maintenance, may also impact genetic and phenotypic variation, introducing potential caveats when comparing CLS and WDS. Differences in environmental history, genetic drift, and selective pressures in laboratory settings versus natural populations could further shape the observed variation. Future studies should consider these factors when designing comparative analyses and interpreting evolutionary or biomedical relevance.
By addressing these caveats, we aim to encourage more rigorous approaches in the use of both CLS and WDS as models in biological research, ensuring that their distinct genetic and phenotypic traits are leveraged appropriately.
WDS versus CLS
Given their different sources and histories, higher genetic variability in WDS compared to CLS is expected. However, the magnitude of this difference, as summarized in Supplementary Table S13, is stunning. For example, WDS mitogenomes harbour approximately 90 times higher nucleotide diversity than CLS. None of the mitogenomes is shared between CLS and WDS. The allelic diversity in the Prdm9 gene, which plays an important role in musculus/domesticus hybrid male sterility40,69, is approximately three times higher in domesticus and musculus WDS than in CLS. Of the two Prdm9 alleles present in CLS, only one (dom3) was detected among WDS, while the other allele (dom2) is private for CLS (but present in wild domesticus in North America and North Europe—Emil Parvanov and Jiří Forejt, personal communication 202170). Prdm9 primarily defines the positions of recombination hotspots during meiosis70. Consequently, the presence of only two alleles in CLS generally limits the mapping of quantitative trait loci in laboratory crosses. The presence of 36 Prdm9 alleles in musculus and domesticus WDS, along with the substantial inter-strain/inter-subspecific variation documented here, may surmount this limitation and provide rich material for testing genotype-phenotype associations.
Copy number variation is an important driver of genome and phenotype evolution71. It is worth noting that the proportion of the autosomes and the X chromosome affected by CNV was found to be lower in CLS (estimated size of 1.7 Mb, representing 0.065% of the mouse genome) compared to WDS (3.8 Mb, representing 0.14% of the genome)72. In this study, we analyzed CNV in Slx and Sly genes, whose disrupted balance is associated with infertility and sex ratio bias in house mice73,74,75,76. Recently, it has been shown that an arms race between these genes significantly affects the dynamics of the hybrid zone between musculus and domesticus27. Interestingly, the number of gene copies of CLS Sly appeared intermediate between the musculus-derived and domesticus-derived strains (Fig. 4; Table 3). Since both CLS analyzed in this study possess predominantly domesticus-like autosomal genes and musculus-like Y chromosomes9,61,62, we should expect Y-linked Sly CN to fall within the musculus range. Because of their hybrid origin, CLS resemble domesticus populations close to the hybrid zone centre, possessing introgressed musculus Y chromosomes27,28,41 rather than genetically pure domesticus populations. Moreover, due to the presence of chromosomal fusions and whole-arm reciprocal translocations, the western house mouse (domesticus) has become a well-known model for the study of karyotype evolution77. While only the standard karyotype with 2N = 40 acrocentric chromosomes occurs in CLS, 12 of 40 domesticus WDS karyotypes display reduced diploid numbers ranging between 22 and 38 chromosomes (Supplementary Table S1).
Morphological variation mirrors genetic variability. Notably, CLS are almost entirely embedded within the morphospace defined by WDS. On the other hand, when comparing CLS with domesticus and musculus WDS across individual traits, we observe significant differences in most external morphological and reproductive traits (summarized in Supplementary Table S13). As anticipated, due to their genomic composition, the morphological differentiation between CLS and musculus WDS is higher than that between CLS and domesticus WDS (Supplementary Table S13). Possibly because of long-term artificial selection in captivity, CLS outperform WDS in terms of reproductive parameters (Table 4), which highlights their suitability as models for biomedical research. The phenotypic differentiation between WDS and CLS observed here is congruent with the study of Takada et al.36 comparing 10 WDS derived from domesticus, castaneus, and molossinus subspecies and one CLS (C57BL/6). In that study, significant differences were found in 62—68% of 21 measured morphological and physiological traits36.
In summary, the genetic data corroborate previous analyses that have found higher diversity in wild mice compared to CLS e.g9,36,42,62,78. Simultaneously, Sly CN and the numerous significant differences in external morphology and reproductive characteristics, suggest CLS are not simply domesticated representatives of M. m. domesticus, despite the overwhelming portion of domesticus genome they carry9. It seems the genetic and morphological differences documented here reflect not only the complex history of CLS but also their long-term adaptation to laboratory conditions, influenced by genetically conditioned behavioural changes fixed during domestication25. In recognizing their unnatural genetic constitution, CLS have been suggested to be called ‘Mus laboratorius’33 or ‘Mus gemischus’79. Here we added a phenotypic dimension to this perspective.
WDS versus wild mice
While we revealed significantly higher variation in WDS compared to CLS, an important question to consider is to what extent WDS truly represent wild mice. For instance, in comparing CNV as a quantitative trait, we found that differentiation in Slx and Sly copy number between domesticus and musculus WDS mirror what has recently been reported from the European house mouse hybrid zone27. When examining mtDNA, the most comprehensive data has been gathered by analyzing cytochrome b (mt-Cytb) and D-loop sequences. In domesticus, seven (mt-Cytb) and 11 (D-loop) haplogroups have been described80,81,82, five mt-Cytb haplogroups were found in musculus83. WDS displayed six mt-Cytb haplogroups in domesticus and the same number in musculus strains. These numbers suggest that while WDS preserve variation from the major representatives of deeply diverged haplogroups, they may encounter difficulties in capturing natural variation on local scales within these haplogroups. For example, an analysis of whole mitogenome sequences in 98 wild mice, mostly from the eastern Palearctic, revealed the presence of 90 haplotypes56. Similarly, while we identified 48 variants in the Prdm9 gene in 99 WDS, 225 different alleles were identified in 632 wild and wild-derived animals59,84,85,86,87, and our unpublished data.
The documented diversity preserved in WDS can be a valuable resource for investigating genotype-phenotype variation in evolutionary contexts. For example, the observed variation of Prdm9 alleles in WDS can provide insight into our understanding of the recombination machinery associated with the dynamics of formation and erosion of Prdm9-dependent binding sites88. Furthermore, negative interactions of specific alleles in intersubspecific mouse hybrids can result in meiotic arrest and male sterility40,60,69,89,90. As mentioned earlier, the WDS described in Supplementary Table S1 preserve approximately 20% of currently known natural Prdm9 allelic variation. With this limited number of alleles, we can still observe the extent and intensity of negative interactions among 36 alleles in 1296 F1 hybrids within and between domesticus and musculus WDS. Importantly, since some alleles are shared among multiple strains (e.g., the sterility-causing allele dom3 in 8 domesticus WDS), it becomes possible to assess the effects of genetic background and the evolution of sterility in natural populations.
While there are numerous genetic comparisons between wild-derived mouse strains (WDS) and wild mice, comparative morphometric studies between these two groups are scarce. For example, a study employed four sperm size parameters to assess whether WDS can serve as a proxy for exploring evolutionary processes related to post-copulatory selection91. For this purpose, 28 wild-caught and four WDS derived from domesticus and musculus subspecies were utilized. The subspecies exhibited significant differences in sperm head length and midpiece length, and these differences were consistent for wild mice and wild-derived strains when pooled over genomes. However, when the inbred strains were individually analyzed, their strain-specific values sometimes significantly deviated from subspecies-specific values obtained from wild mice. This study by Albrechtová et al.91, therefore, suggests that future experiments should involve a larger number of strains to account for natural variation and avoid confounding results due to reduced variability and founder effects within individual stocks. In this respect, we believe that the documented genetic variation in the resources of WDS presented in this paper (and maintained in other laboratories) can effectively capture the majority of genetic and morphological diversity observed in their wild counterparts.
WDS cross-contamination
An important consideration when using wild-derived mouse strains (WDS) for modelling evolutionary processes is ensuring that these strains represent natural, not artificially introduced, genetic variation. In a study by Yang et al.9, traces of intersubspecific contamination were reported in the WDS under investigation. They proposed that introgression in these WDS could have resulted from a combination of cross-contamination from CLS, gene flow in the wild (pronounced especially in WDS captured near hybrid zones), or breeding with other wild-derived mice in laboratory settings. We analyzed potential cross-contamination in our WDS panel by comparing mitogenomes of the same strains sampled at different times during their existence. Inconsistencies in these duplicated samples were observed in 38.7% of 31 cases (Supplementary Table S4), indicating the presence of introgression from other genomes. It is worth noting that some cross-contaminations can be attributed to the breeding scheme used to maintain mouse stocks. For instance, certain WDS were initially kept through breeding among distantly related pairs (outbreeding) at the University of Montpellier, a practice that inherently maintains much genetic polymorphism. Subsequently, these outbred stocks turned to brother-sister mating and are now considered inbred. However, without complete information on the original parental individuals, it remains unclear which alleles were fixed in subsequent generations. This change in the breeding scheme can explain at least the intrasubspecific shifts between mtDNA haplogroups. On the other hand, intersubspecific mtDNA shifts, like the presence of a domesticus mitogenome with an 11-bp insertion in the D-loop of two musculus WDS from Bulgaria, may suggest unintended cross-contamination. The fact that this + 11bp variant is widespread in northern Germany and Scandinavia and matches a haplotype of a Danish domesticus WDS further supports its origin in the Bulgarian strains. Similarly, two castaneus strains from Kenya (CKN and CKS), were found to share the same musculus haplotype with the musculus-derived MBK strain from Bulgaria. A possible explanation is that during the inbreeding process, which inevitably leads to the fixation of deleterious variants, there is a strong advantage for the offspring of an unintentional interstrain cross restoring fertility. Such events can potentially occur in mouse facilities where stocks are kept in the same room.
To conclude this section, although we have detected between-generation inconsistencies in mitogenomes in 15.2% of the analyzed 79 WDS (Supplementary Table S4), the majority of these strains remain a valuable source of natural variation. They have the potential to complement and expand research that has traditionally relied heavily on CLS. Given the time-consuming nature of strict inbreeding, which can span from 3.8 to 18 years to obtain an inbred strain, we believe it is essential to initiate the documentation and characterization of mouse resources developed and maintained in various laboratories worldwide. Making these resources available to the scientific community can have a significant impact. In a prospective view, since WDS can provide reproducible genotypes similar to CLS, they can be integrated into Phenome projects once they have been transferred to specific-pathogen-free facilities.
Relevance of WDS to human and biomedical research
From an evolutionary perspective, the genotype-phenotype variation preserved in mouse WDS is undoubtedly very important. However, it can also serve as a surrogate for studying human populations in both evolutionary and biomedical contexts. The synanthropic bond between house mice and humans began to evolve approximately 10,000 years ago with the onset of human agriculture92,93. This association has allowed mice to inhabit new human-associated ecological niches and spread across the globe33,34,94. The colonization of new environments has driven adaptations to varying local conditions, as observed along a latitudinal gradient on the eastern coast of North America20. These adaptations to local conditions are expected to result in global intrasubspecific and intersubspecific divergence. Indeed, an analysis of more than 150 whole-genome sequences of wild mouse populations sampled worldwide revealed that a significant fraction of genetic variation is private to individual populations95, which can limit GWAS studies of their associations. Interestingly, the analysis also detected strong signals of positive selection in many genes associated with human diseases95. The genetic variation can be significantly extended when using long-read whole genome sequencing, as exemplified in a study of 11 domesticus WDS by Dumont et al.26.
Among the traits examined in this study, mtDNA holds significant importance due to its known role in causing several inherited diseases in humans96. CLS are frequently utilized in these studies97. Here, we document that the variability in mtDNA within our mouse WDS exceeds that observed in humans. To illustrate this point, we compared our finding to a dataset comprising 560 maternally unrelated human individuals of European, African, and Asian ancestry published by Elson et al.98. A pairwise haplotype comparison within this human dataset revealed a maximum difference of 260 polymorphic sites, which is similar to the variation observed within intrasubspecific musculus and domesticus WDS, but lower than what was found in intersubspecific domesticus/musculus WDS comparisons (548 polymorphic sites; see Table 2). Such comparisons hold the potential to become a cornerstone of biomedical research, where the haplotype diversity detected in WDS can be harnessed, for example, in the context of tissue transplantation. The introduction of new nuclear DNA–mtDNA interacting systems may accelerate the onset of metabolic disorders in the recipient organism as reviewed in99. Understanding the mechanisms governing competitive mtDNA segregation and identifying which tissues can tolerate heteroplasmy is essential for making more accurate predictions in these interactions. Using a mouse model, it was demonstrated that mtDNA segregation in heteroplasmic mice depends on the genetic divergence between a donor and a recipient and is also tissue-dependent, implying potential complications in human therapies100. Therefore, the concept of haplotype matching has been proposed as an approach to mitigate these issues in the context of mitochondrial replacement therapies100,101.
A similar pattern emerges when comparing the variation in the Prdm9 gene, which also governs the distribution of recombination hotspots in humans102. Alleva et al.103 studied Prdm9 allelic diversity in 720 individuals from seven worldwide human populations and detected 69 alleles (i.e., approximately 0.10 per individual), compared to 38 alleles observed here in 83 M. musculus WDS (i.e., approximately 0.46 per strain). While our focus here has been on individual representatives of mouse genomes, recent whole-genome analyses of laboratory stocks (including three WDS) have identified that the most variable regions of the mouse genome are enriched with genes relevant to disease and infection response104. Returning to humans, a new pangenome human reference map that aligns 47 genome assemblies from genetically diverse individuals promises to enhance our understanding of genomics and our ability to predict, diagnose and treat diseases16.
Relevance of WDS for preclinical testing
Drug discovery and development is a long, costly, and high-risk process spanning over 10–15 years, with an average cost of over $1–2 billion for each new drug to gain approval for clinical use105. Despite rigorous optimization of drug candidates during the preclinical stage, nine of ten candidates allowed to proceed to clinical studies fail during one of three phases of clinical trials in the drug approval process106. New approaches, such as machine learning or open-source cross-sector collaboration, are being employed to reduce the failure rate of medications in preclinical or clinical settings. However, significant effort is still directed toward enhancing the rigour and reproducibility of testing107,108.
Due to high demands on reproducibility, CLS are the dominant animal models used in drug testing and developing therapies for human disease. On the other hand, the fact that mouse WDS exhibit dramatically higher genetic and phenotypic variation than CLS and that their genetic variation is comparable to that found in human populations can greatly enhance their relevance in medical research, for example, in preclinical testing of the effectivity of biomolecules in in vivo pharmacology. When WDS are integrated into preclinical biomolecule testing and complement the battery of tests performed on CLS, the effectiveness of molecules can be assessed across a broad spectrum of genotypes. Molecules that exhibit specific responses within a limited range of genetic variability can be identified and excluded from further evaluation. Consequently, the risk of failure in clinical trials can be significantly reduced, potentially saving up to 90% of the associated costs.
Materials and methods
Mice
All mice are housed in a conventional breeding facility (i.e., without nanofilter barrier or specific-pathogen-free conditions) of the Institute of Vertebrate Biology, Czech Academy of Sciences, in Studenec. They are maintained under standard conditions, with a light/dark regime of 14/10 hours, with temperatures of 23 ± 1 °C during summer (April-September) and 22 ± 1 °C during winter (October-March), respectively. The relative humidity is maintained within 40–70%. Mice have access to food pellets (Myška 1, VKS Podhledští Dvořáci, Hamry, Czechia) and tap water ad libitum. Mice are weaned at 20 days of age and housed in brother–sister pairs in EURO IIL cages with a floor area of 530 cm2 made of transparent polycarbonate (Tecniplast, Varese, Italy). The cages are equipped with bedding material (sifted sawdust, Happy Horses, Martinsberg, Austria), shredded paper for nest building and nest houses made of red transparent polycarbonate (Tecniplast). The facility is authorized for the use of experimental animals (licences 61974/2017-MZE-17214 and MZE-50144/2022–13143), as well as for the breeding and supply of experimental animals to third parties (62065/2017-MZE-17214 and MZE-50151/2022–13143). These licences are in compliance with the corresponding regulations and standards of the European Union, as specified in Council Directive 86/609/EEC.
mtDNA
Genomic DNA was isolated from frozen (− 80 °C) or alcohol-preserved muscle or spleen tissues using DNeasy Blood & Tissue Kits (Qiagen). High-quality DNA aliquots were sequenced at the Edinburgh Genomics facility using HiSeq X technology. Two-paired 150 bp reads were merged and mapped against the C57BL/6 mitogenome (GeneBank NC_005089)12 using the Geneious Prime software (Biomatters: www.geneious.com). The fine-tuning iteration was set to 5. In the cases where the presence of the 75-bp insertion in the control region109 was indicated by a peak of increased read coverage in the respective region, the sample was re-mapped against de novo assembled mitogenome of an M. m. musculus individual (Bot360) from Botosani, Romania, which is known to carry the insert110. The average coverage was 225 ± 151 reads per sample (individual data are available in Supplementary Table S2). Consensus sequences were generated with the sequence matching threshold set to 65%, and ‘N’ was called in a sequence when the coverage was less than 3. These data were supplemented with 49 published WDS and CLS mitogenomes12,13,14,111,112,113.
All sequences were aligned using Clustal Omega114 implemented in the Geneious Prime (Biomatters Ltd. www.geneious.com). Maximum likelihood phylogenetic trees were inferred using the GTR + G + I model115 selected with jModelTest116,117. Six discrete categories118 were used to approximate the continuous gamma distribution. In addition, an extensive subtree pruning and regrafting procedure119 was applied to improve searching for the best tree. The weakest stringency of optimization with respect to branch lengths and improvements in log-likelihood values (branch swap filter) was used to maximize the explored search space. The MEGA11 software120 was employed for the analysis.
Prdm9 sequencing
We followed the protocols published by Buard et al.84 and Kono et al.59. The ZnF array was amplified using PrimeSTAR HS DNA Polymerase (Takara Bio) and primers Prdm9-F (TGAGATCTGAGGAAAGTAAGAG) and Pdrm9-R (TCCTGTAATTGTTGAGATGTGG); 20 µl of the total volume included 30 ng of genomic DNA and 0.5 µM of each primer. The PCR conditions were as follows: after 30 s at 98 °C, 28 cycles were carried out, including 10 s at 98 °C, 15 s at 63 °C, and 2 min at 68 °C. The PCR product’s size was determined using electrophoresis in 1% agarose gel (Seakem). In cases where mice were heterozygous and carried alleles of different lengths, the corresponding bands were excised from the gel and purified using ethanol precipitation. A second round of PCR was performed using sequencing primers: Prdm9seqF (CTCAGAACAGGCCAGACAACA) and Prdm9seqR (TTGTTGAGATGTGGTTTTATTGCT). Sanger sequencing was conducted by Eurofins Genomics (Olomouc, Czechia) from both ends of the purified PCR products using the sequencing primers. It is important to note that many PCR products could not be sequenced up to their ends in both directions due to the repetitive nature of ZnF arrays. In such cases, sequencing was repeated until high-quality electropherograms were obtained.
Assembly of the forward and reverse sequences and the translation of DNA sequences into amino acids were performed using Geneious Prime. To define individual Prdm9 alleles, triplets of amino acids located in the most variable positions, specifically positions − 1, + 3, and + 6 of each ZnF, were utilized, as described by Oliver et al.121. The sequences were aligned with ClustalW. A maximum-likelihood tree was inferred in MEGA11120 using default values.
Slx/Slycopy number variation
The digital droplet PCR (ddPCR) method122 was employed to estimate copy number in two ampliconic genes, Slx (Sycp3 like X-linked) and Sly (Sycp3 like Y-linked), in 53 strains. A custom-designed PrimeTime qPCR assays (IDT®, Coralville, Iowa, USA) were used. Two assays were run in duplex reactions, where each well contained one primer pair and probe designed for the target gene (either Slx or Sly) and one for the Tert gene. Tert was used as a reference since it is known to be consistently present in two copies in diploid organisms123. The assay sequences and PCR conditions were identical to those used in Baird et al.27.
The ddPCR reactions were carried out in the Genomics Core Facility at the Central European Institute of Technology (CEITEC) in Brno using the QX100/200 Droplet Digital PCR System (Bio-Rad, Hercules, CA, USA). For each sample, measurements were performed in triplicate. Quantasoft™ Software (Bio-Rad) was used to estimate the error and merge the three obtained values into a single outcome representing the number of gene copies. Being a quantitative trait, CNV may also exhibit laboratory-based systematic effects. Therefore, we conducted ddPCR of Sly in two laboratories: in Plön, Germany, and Brno, Czechia. Figure 6 indicates that the data from different laboratories are almost identical, documenting the reliability of the CN estimates and the technique as such.

Inter-laboratory comparison of Sly CN obtained at the Max-Planck Institute for Evolutionary Biology, Plön, and at CEITEC Masaryk University, Brno. The measures are highly correlated (Pearson’s product-moment correlation = 0.977). A linear model fit with a standard error envelope is depicted in grey. Vertical and horizontal error bars represent Poisson distribution-based errors from triplicate measures of Sly copy numbers in either laboratory.
Statistical analyses
The analysis of variance (ANOVA) was used to detect variability in the morphological traits. The factors compared in the models were group, strain, sex, and their interactions. The analyses were based on two models. The first model tested global differentiation within the whole dataset comprising eight groups, 90 strains, and, where appropriate, sex. The second model was confined to the three most representative groups: musculus WDS, domesticus WDS, and CLS (2,162 males and 1,938 females). This dataset was lower for splenic weights and consisted of 1,529 males and 1,430 females. All statistical analyses were conducted in the R statistical language R Core124 and run in the R Studio environment RStudio125. Pairwise comparisons between group means were tested using the ‘emmeans’ package (https://CRAN.R-project.org/package=emmeans).
Two reproductive traits deviated from normal distribution. Litter size was normalized by excluding one outlier with an exceptionally large litter size (CD-1 with 12.2 young in a litter). For the mortality rate, a normal distribution was achieved using a square root transformation. The third variable describing the rate of reproduction (the average number of generations delivered per year) displayed normal distribution.
Moreover, we aimed to determine the proportion of variation in a morphospace defined by length or mass variables specific to each group and the proportion shared between groups. For each group, we first computed the subset of points on the convex hull of the set of points specified in the biplot projection of two length/mass variables using the ‘hull’ algorithm126. We then calculated the numbers of points falling into individual polygons and their intersections using the ‘point.in.polygon’ function from the ‘sp’ package in R. These analyses included three groups of mice: musculus WDS, domesticus WDS, and CLS. The ‘ggvenn’ R library was used to visualize data in the form of Venn diagrams. Regarding morphometric traits, we used data for body weight and tail/body length ratio, which is known to be higher in domesticus than in musculus127,128. We also separately analysed morphological variation defined by testis weight and sperm count in males and ovary weight and body mass in females. Reproductive ability was investigated in a similar way for all eight groups.
Data availability
Sequences of 114 complete mtDNA genomes were submitted to GenBank with accession codes OR840712– OR840825. Aligned mitogenome sequences are available on DRYAD at https://doi.org/10.5061/dryad.r7sqv9skn. Znf array sequences of the Prdm9 gene of 98 mice can be accessed at GenBank under OQ054372–OQ054469. Raw phenotypic data are available on DRYAD at https://doi.org/10.5061/dryad.r7sqv9skn. WDS are registered at Mouse Genome Informatics http://www.informatics.jax.org/strain/summary. Further data on this mouse repository can be obtained at https://housemice.cz/en/strains/.
References
Yamaguchi, M. & Yoshida, H. in In Drosophila Models for Human Diseases (eds Yamaguchi, M.) 1–10 (Springer, 2018).
Morgan, T. H., Sturtevant, A. H., Muller, H. J. & Bridges, C. B. The Mechanisms of Mendelian Heredity (Henry Holt & Co., 1915).
Botstein, D. & Fink, G. R. Yeast: An experimental organism for 21st century biology. Genetics 189, 695–704. https://doi.org/10.1534/genetics.111.130765 (2011).
Fox, J. G. et al. The Mouse in Biomedical Research. Vol. 1. History, Wild Mice, and Genetics (Elsevier, 2007).
Fox, J. G. et al. The Mouse in Biomedical Research. Vol. 3. Normative Biology, Husbandry, and Models (Elsevier, 2007).
Waterston, R. H. et al. Initial sequencing and comparative analysis of the mouse genome. Nature 420, 520–562. https://doi.org/10.1038/nature01262 (2002).
Eppig, J. T. Mouse Genome Informatics (MGI) resource: genetic, genomic, and biological knowledgebase for the laboratory mouse. ILAR J. 58, 17–41. https://doi.org/10.1093/ilar/ilx013 (2017).
Festing, M. F. W. in In Genetic Variants and Strains of the Laboratory Mouse (eds Lyon, M. F. et al.) 1537–1576 (Oxford University Press, 1996).
Yang, H. et al. Subspecific origin and haplotype diversity in the laboratory mouse. Nat. Genet. 43, 648–655. https://doi.org/10.1038/ng.847 (2011).
Takada, T. et al. The ancestor of extant Japanese fancy mice contributed to the mosaic genomes of classical inbred strains. Genome Res. 23, 1329–1338. https://doi.org/10.1101/gr.156497.113 (2013).
Ferris, S. D., Sage, R. D. & Wilson, A. C. Evidence from mtDNA sequences that common laboratory strains of inbred mice are descended from a single female. Nature 295, 163–165. https://doi.org/10.1038/295163a0 (1982).
Bayona-Bafaluy, M. P. et al. Revisiting the mouse mitochondrial DNA sequence. Nucleic Acids Res. 31, 5349–5355. https://doi.org/10.1093/nar/gkg739 (2003).
Goios, A., Pereira, L., Bogue, M., Macaulay, V. & Amorim, A. mtDNA phylogeny and evolution of laboratory mouse strains. Genome Res. 17, 293–298. https://doi.org/10.1101/gr.5941007 (2007).
Yu, X. et al. Dissecting the effects of mtDNA variations on complex traits using mouse conplastic strains. Genome Res. 19, 159–165. https://doi.org/10.1101/gr.078865.108 (2009).
Auton, A. et al. A global reference for human genetic variation. Nature 526, 68–74. https://doi.org/10.1038/nature15393 (2015).
Liao, W. W. et al. A draft human pangenome reference. Nature 617, 312–324. https://doi.org/10.1038/s41586-023-05896-x (2023).
Halligan, D. L., Oliver, F., Eyre-Walker, A., Harr, B. & Keightley, P. D. evidence for pervasive adaptive protein evolution in wild mice. PLoS Genet. 6, e1000825. https://doi.org/10.1371/journal.pgen.1000825 (2010).
Davies, R. W. Factors Influencing Genetic Variation in Wild Mice. PhD thesis, University of Oxford (2015).
Harr, B. et al. Genomic resources for wild populations of the house mouse, Mus musculus and its close relative Mus spretus. Sci. Data 3, 160075. https://doi.org/10.1038/sdata.2016.75 (2016).
Phifer-Rixey, M. et al. The genomic basis of environmental adaptation in house mice. PLoS Genet. 14, e1007672. https://doi.org/10.1371/journal.pgen.1007672 (2018).
Payseur, B. A. & Jing, P. Genomic targets of positive selection in giant mice from Gough Island. Mol. Biol. Evol. 38, 911–926. https://doi.org/10.1093/molbev/msaa255 (2021).
Fujiwara, K. et al. Insights into Mus musculus population structure across Eurasia revealed by whole-genome analysis. Genome Biol. Evol. 14, evac068. https://doi.org/10.1093/gbe/evac068 (2022).
Morgan, A. P. et al. Population structure and inbreeding in wild house mice (Mus musculus) at different geographic scales. Heredity 129, 183–194. https://doi.org/10.1038/s41437-022-00551-z (2022).
Lawal, R. A. et al. Taxonomic assessment of two wild house mouse subspecies using whole-genome sequencing. Sci. Rep. 12, 20866. https://doi.org/10.1038/s41598-022-25420-x (2022).
Liu, M. et al. Whole-genome sequencing reveals the genetic mechanisms of domestication in classical inbred mice. Genome Biol. 23, 203. https://doi.org/10.1186/s13059-022-02772-1 (2022).
Dumont, B. L. et al. Into the wild: a novel wild-derived inbred strain resource expands the genomic and phenotypic diversity of laboratory mouse models. PLoS Genet. 20, e1011228. https://doi.org/10.1371/journal.pgen.1011228 (2024).
Baird, S. J. E., Hiadlovská, Z., Daniszová, K., Piálek, J. & Macholán, M. A gene copy number arms race in action: X,Y-chromosome transmission distortion across a species barrier. Evolution 77, 1330–1340. https://doi.org/10.1093/evolut/qpad051 (2023).
Macholán, M. et al. Widespread introgression of the Mus musculus musculus Y chromosome in Central Europe. BioRxiv. https://doi.org/10.1101/2019.12.23.887471 (2019).
Macholán, M. et al. A reappraisal of mitochondrial DNA introgression in the Mus musculus musculus/M. m. domesticus hybrid zone suggests ancient north-european associations between mice and humans. Zool. J. Linn. Soc. 1, 1 (2024).
Staubach, F. et al. Genome patterns of selection and introgression of haplotypes in natural populations of the house mouse (Mus musculus). PLoS Genet. 8, e1002891. https://doi.org/10.1371/journal.pgen.1002891 (2012).
Tam, V. et al. Benefits and limitations of genome-wide association studies. Nat. Rev. Genet. 20, 467–484. https://doi.org/10.1038/s41576-019-0127-1 (2019).
Flint, J. & Eskin, E. Genome-wide association studies in mice. Nat. Rev. Genet. 13, 807–817 (2012).
Guénet, J. L. & Bonhomme, F. Wild mice: an ever-increasing contribution to a popular mammalian model. Trends Genet. 19, 24–31. https://doi.org/10.1016/S0168-9525(02)00007-0 (2003).
Phifer-Rixey, M. & Nachman, M. W. Insights into mammalian biology from the wild house mouse Mus musculus. Elife 4, e05959. https://doi.org/10.7554/eLife.05959 (2015).
Harper, J. M. Wild-derived mouse stocks: an underappreciated tool for aging research. Age 30, 135–145. https://doi.org/10.1007/s11357-008-9057-0 (2008).
Takada, T. et al. MoG+: a database of genomic variations across three mouse subspecies for biomedical research. Mamm. Genome 33, 31–43. https://doi.org/10.1007/s00335-021-09933-w (2022).
Seixas, F. A., Boursot, P. & Melo-Ferreira, J. The genomic impact of historical hybridization with massive mitochondrial DNA introgression. Genome Biol. 19, 91. https://doi.org/10.1186/s13059-018-1471-8 (2018).
Macholán, M. et al. A reappraisal of mitochondrial DNA introgression in the Mus musculus musculus/M. m. domesticus hybrid zone suggests ancient north-european associations between mice and humans. Zool. J. Linn. Soc. 202, 110. https://doi.org/10.1093/zoolinnean/zlae110 (2024).
Forejt, J. & Iványi, P. Genetic studies on male sterility of hybrids between laboratory and wild mice (Mus musculus L). Genet. Res. 24, 189–206. https://doi.org/10.1017/S0016672300015214 (1974).
Mihola, O., Trachtulec, Z., Vlcek, C., Schimenti, J. C. & Forejt, J. A mouse speciation gene encodes a meiotic histone H3 methyltransferase. Science 323, 373–375. https://doi.org/10.1126/science.1163601 (2009).
Macholán, M. et al. Genetic conflict outweighs heterogametic incompatibility in the mouse hybrid zone? BMC Evol. Biol. 8, 271. https://doi.org/10.1186/1471-2148-8-271 (2008).
Chang, P. L. et al. Whole exome sequencing of wild-derived inbred strains of mice improves power to link phenotype and genotype. Mamm. Genome 28, 416–425. https://doi.org/10.1007/s00335-017-9704-9 (2017).
Bonhomme, F. & Guénet, J. L. in In Genetic Variants and Strains of the Laboratory Mouse (eds Lyon, M. F. et al.) Vol. 2, 1577–1596 (Oxford University Press, 1996).
Gregorová, S. & Forejt, J. PWD/Ph and PWK/Ph inbred mouse strains of Mus m. musculus subspecies—A valuable resource of phenotypic variations and genomic polymorphisms. Folia Biol. Praha 46, 31–41 (2000).
Piálek, J. et al. Development of unique house mouse resources suitable for evolutionary studies of speciation. J. Hered. 99, 34–44. https://doi.org/10.1093/jhered/esm083 (2008).
Martincová, I., Dureje, L., Kreisinger, J., Macholán, M. & Piálek, J. Phenotypic effects of the Y chromosome are variable and structured in hybrids among house mouse recombinant lines. Ecol. Evol. 9, 6124–6137. https://doi.org/10.1002/ece3.5196 (2019).
Rudra, M., Chatterjee, B. & Bahadur, M. Phylogenetic relationship and time of divergence of Mus terricolor with reference to other Mus species. J. Genet. 95, 399–409. https://doi.org/10.1007/s12041-016-0654-x (2016).
Stopková, R. et al. Variation in mouse chemical signals is genetically controlled and environmentally modulated. Sci. Rep. 13, 8573. https://doi.org/10.1038/s41598-023-35450-8 (2023).
Flachs, P. et al. Prdm9 incompatibility controls oligospermia and delayed fertility but no selfish transmission in mouse intersubspecific hybrids. PLoS ONE 9, e95806. https://doi.org/10.1371/journal.pone.0095806 (2014).
Vošlajerová Bímová, B. et al. Sperm quality, aggressiveness and generation turnover may facilitate unidirectional Y chromosome introgression across the European house mouse hybrid zone. Heredity 125, 200–211. https://doi.org/10.1038/s41437-020-0330-z (2020).
Ďureje, L., Macholán, M., Baird, S. J. E. & Piálek, J. The mouse hybrid zone in Central Europe: from morphology to molecules. Folia Zool. 61, 308–318. https://doi.org/10.25225/fozo.v61.i3.a13.2012 (2012).
Prager, E. M., Orrego, C. & Sage, R. D. Genetic variation and phylogeography of central Asian and other house mice, including a major new mitochondrial lineage in Yemen. Genetics 150, 835–861. https://doi.org/10.1093/genetics/150.2.835 (1998).
Duplantier, J. M., Orth, A., Catalan, J. & Bonhomme, F. Evidence for a mitochondrial lineage originating from the Arabian peninsula in the Madagascar house mouse (Mus musculus). Heredity 89, 154–158. https://doi.org/10.1038/sj.hdy.6800122 (2002).
Bibi, S. et al. Mitochondrial genetic diversity and phylogeography of Mus musculus castaneus in Northern Punjab, Pakistan. Zool. Sci. 34, 490–497. https://doi.org/10.2108/zs170086 (2017).
Rajabi-Maham, H. et al. The south-eastern house mouse Mus musculus castaneus (Rodentia: Muridae) is a polytypic subspecies. Biol. J. Linn. Soc. 107, 295–306. https://doi.org/10.1111/j.1095-8312.2012.01957.x (2012).
Li, Y. et al. House mouse Mus musculus dispersal in East Eurasia inferred from 98 newly determined complete mitochondrial genome sequences. Heredity 126, 132–147. https://doi.org/10.1038/s41437-020-00364-y (2021).
Bielschowsky, M. & Goodall, C. M. Origin of inbred NZ mouse strains. Cancer Res. 30, 834–836 (1970).
Grey, C., Baudat, F. & de Massy, B. PRDM9, a driver of the genetic map. PLoS Genet. 14, e1007479 (2018).
Kono, H. et al. Prdm9 polymorphism unveils mouse evolutionary tracks. DNA Res. 21, 315–326. https://doi.org/10.1093/dnares/dst059 (2014).
Mukaj, A. et al. Prdm9 intersubspecific interactions in hybrid male sterility of house mouse. Mol. Biol. Evol. 37, 3423–3438. https://doi.org/10.1093/molbev/msaa167 (2020).
Bishop, C. E., Boursot, P., Baron, B., Bonhomme, F. & Hatat, D. Most classical Mus musculus domesticus laboratory mouse strains carry a Mus musculus musculus Y chromosome. Nature 315, 70–72. https://doi.org/10.1038/315070a0 (1985).
Morgan, A. P. & de Villena, F. P.-M. Sequence and structural diversity of mouse Y chromosomes. Mol. Biol. Evol. 34, 3186–3204. https://doi.org/10.1093/molbev/msx250 (2017).
Vyskočilová, M., Pražanová, G. & Piálek, J. Polymorphism in hybrid male sterility in wild-derived Mus musculus musculus strains on proximal chromosome 17. Mamm. Genome 20, 83–91. https://doi.org/10.1007/s00335-008-9164-3 (2009).
Silver, L. M. Mouse Genetics (Oxford University Press, 1995).
Gasch, A. P., Payseur, B. A. & Pool, J. E. The power of natural variation for model organism biology. Trends Genet. 32, 147–154. https://doi.org/10.1016/j.tig.2015.12.003 (2016).
Fitzpatrick, J. L., Kahrl, A. F. & Snook, R. R. SpermTree, a species-level database of sperm morphology spanning the animal tree of life. Sci. Data 9, 30. https://doi.org/10.1038/s41597-022-01131-w (2022).
Mestas, J. & Hughes, C. C. W. Of mice and not men: differences between mouse and human immunology. J. Immunol. 172, 2731–2738. https://doi.org/10.4049/jimmunol.172.5.2731 (2004).
Gage, M. J. G. Mammalian sperm morphometry. Proc. R. Soc. Lond. Ser. B Biol. Sci. 265, 97–103. https://doi.org/10.1098/rspb.1998.0269 (1998).
Forejt, J., Jansa, P. & Parvanov, E. Hybrid sterility genes in mice (Mus musculus): a peculiar case of PRDM9 incompatibility. Trends Genet. 37, 1095–1108. https://doi.org/10.1016/j.tig.2021.06.008 (2021).
Paigen, K. & Petkov, P. M. PRDM9 and its role in genetic recombination. Trends Genet. 34, 291–300. https://doi.org/10.1016/j.tig.2017.12.017 (2018).
Saitou, M. & Gokcumen, O. An evolutionary perspective on the impact of genomic copy number variation on human health. J. Mol. Evol. 88, 104–119. https://doi.org/10.1007/s00239-019-09911-6 (2020).
Locke, M. E. O. et al. Genomic copy number variation in Mus musculus. BMC Genom. 16, 497. https://doi.org/10.1186/s12864-015-1713-z (2015).
Cocquet, J. et al. A genetic basis for a postmeiotic X versus Y chromosome intragenomic conflict in the mouse. PLoS Genet. 8, e1002900. https://doi.org/10.1371/journal.pgen.1002900 (2012).
Rathje, C. C. et al. Differential sperm motility mediates the sex ratio drive shaping mouse sex chromosome evolution. Curr. Biol. 29, 3692–3698. https://doi.org/10.1016/j.cub.2019.09.031 (2019).
Kruger, A. N. et al. A neofunctionalized X-linked ampliconic gene family is essential for male fertility and equal sex ratio in mice. Curr. Biol. 29, 3699–3706. https://doi.org/10.1016/j.cub.2019.08.057 (2019).
Moretti, C. et al. Battle of the sex chromosomes: competition between X and Y chromosome-encoded proteins for partner interaction and chromatin occupancy drives multicopy gene expression and evolution in muroid rodents. Mol. Biol. Evol. 37, 3453–3468. https://doi.org/10.1093/molbev/msaa175 (2020).
Piálek, J., Hauffe, H. C. & Searle, J. B. Chromosomal variation in the house mouse. Biol. J. Linn. Soc. 84, 535–563. https://doi.org/10.1111/j.1095-8312.2005.00454.x (2005).
Sigmon, J. S. et al. Content and performance of the MiniMUGA genotyping array: a new tool to improve rigor and reproducibility in mouse research. Genetics 216, 905–930. https://doi.org/10.1534/genetics.120.303596 (2020).
Didion, J. P., de Villena, F. P.-M. Deconstructing Mus gemischus: Advances in understanding ancestry, structure, and variation in the genome of the laboratory mouse. Mamm. Genome 24, 1–20. https://doi.org/10.1007/s00335-012-9441-z (2013).
Bonhomme, F. et al. in Evolution of the House Mouse. Cambridge Studies in Morphology and Molecules: New Paradigms in Evolutionary Biology (eds Macholán, M. et al.) 278–296 (Cambridge University Press, 2012).
Bonhomme, F. et al. Genetic differentiation of the house mouse around the Mediterranean basin: matrilineal footprints of early and late colonization. Proc. R. Soc. B Biol. Sci. 278, 1034–1043. https://doi.org/10.1098/rspb.2010.1228 (2011).
García-Rodríguez, O. et al. Cyprus as an ancient hub for house mice and humans. J. Biogeogr. 45, 2619–2630. https://doi.org/10.1111/jbi.13458 (2018).
Fornuskova, A., Bryja, J., Vinkler, M., Macholán, M. & Piálek, J. Contrasting patterns of polymorphism and selection in bacterial-sensing toll-like receptor 4 in two house mouse subspecies. Ecol. Evol. 4, 2931–2944. https://doi.org/10.1002/ece3.1137 (2014).
Buard, J. et al. Diversity of Prdm9 zinc finger array in wild mice unravels new facets of the evolutionary turnover of this coding minisatellite. PLoS ONE 9, e85021. https://doi.org/10.1371/journal.pone.0085021 (2014).
Vara, C. et al. PRDM9 diversity at fine geographical scale reveals contrasting evolutionary patterns and functional constraints in natural populations of house mice. Mol. Biol. Evol. 36, 1686–1700. https://doi.org/10.1093/molbev/msz091 (2019).
AbuAlia, K. F. N. et al. Natural variation in the zinc-finger-encoding exon of Prdm9 affects hybrid sterility phenotypes in mice. Genetics 226, iyae004. https://doi.org/10.1093/genetics/iyae004 (2024).
Marín-García, C. et al. Multiple genomic landscapes of recombination and genomic divergence in wild populations of house mice—the role of chromosomal fusions and Prdm9. Mol. Biol. Evol. 41, msae063. https://doi.org/10.1093/molbev/msae063 (2024).
Smagulova, F., Brick, K., Pu, Y. M., Camerini-Otero, R. D. & Petukhova, G. V. The evolutionary turnover of recombination hot spots contributes to speciation in mice. Genes Dev. 30, 266–280. https://doi.org/10.1101/gad.270009.115 (2016).
Davies, B. et al. Re-engineering the zinc fingers of PRDM9 reverses hybrid sterility in mice. Nature 530, 171–176. https://doi.org/10.1038/nature16931 (2016).
Davies, B. et al. Altering the binding properties of PRDM9 partially restores fertility across the species boundary. Mol. Biol. Evol. 38, 5555–5562. https://doi.org/10.1093/molbev/msab269 (2021).
Albrechtová, J., Albrecht, T., Ďureje, L., Pallazola, V. A. & Piálek, J. Sperm morphology in two house mouse subspecies: do wild-derived strains and wild mice tell the same story? PLoS ONE 9, e0120484. https://doi.org/10.1371/journal.pone.0120484 (2014).
Weissbrod, L. et al. Origins of house mice in ecological niches created by settled hunter-gatherers in the Levant 15,000 y ago. Proc. Natl. Acad. Sci. U.S.A. 114, 4099–4104. https://doi.org/10.1073/pnas.1619137114 (2017).
Cucchi, T. et al. in Evolution of the House Mouse. Cambridge Studies in Morphology and Molecules: New Paradigms in Evolutionary Biology (eds Macholán, M. et al.) 65–93 (Cambridge University Press, 2012).
Macholán, M., Baird, S. J. E., Munclinger, P. & Piálek, J. Evolution of the House Mouse (Cambridge University Press, 2012).
Lawal, R. A., Arora, U. P. & Dumont, B. L. Selection shapes the landscape of functional variation in wild house mice. BMC Biol. 19, 17. https://doi.org/10.1186/s12915-021-01165-3 (2021).
Taylor, R. W. & Turnbull, D. M. Mitochondrial DNA mutations in human disease. Nat. Rev. Genet. 6, 389–402. https://doi.org/10.1038/nrg1606 (2005).
Tyynismaa, H. & Suomalainen, A. Mouse models of mitochondrial DNA defects and their relevance for human disease. EMBO Rep. 10, 137–143. https://doi.org/10.1038/embor.2008.242 (2009).
Elson, J. L., Turnbull, D. M. & Howell, N. Comparative genomics and the evolution of human mitochondrial DNA: assessing the effects of selection. Am. J. Hum. Genet. 74, 229–238. https://doi.org/10.1086/381505 (2004).
Lechuga-Vieco, A. V., Justo-Méndez, R. & Enríquez, J. A. Not all mitochondrial DNAs are made equal and the nucleus knows it. IUBMB Life 73, 511–529. https://doi.org/10.1002/iub.2434 (2021).
Burgstaller, J. P. et al. mtDNA segregation in heteroplasmic tissues is common in vivo and modulated by haplotype differences and developmental stage. Cell Rep. 7, 2031–2041. https://doi.org/10.1016/j.celrep.2014.05.020 (2014).
Kang, E. et al. Mitochondrial replacement in human oocytes carrying pathogenic mitochondrial DNA mutations. Nature 540, 270–275. https://doi.org/10.1038/nature20592 (2016).
Myers, S. et al. Drive against hotspot motifs in primates implicates the PRDM9 gene in meiotic recombination. Science 327, 876–879. https://doi.org/10.1126/science.1182363 (2010).
Alleva, B., Brick, K., Pratto, F., Huang, M. N. & Camerini-Otero, R. D. Cataloging human PRDM9 allelic variation using long-read sequencing reveals PRDM9 population specificity and two distinct groupings of related alleles. Front. Cell. Dev. Biol. 9, 675286. https://doi.org/10.3389/fcell.2021.675286 (2021).
Lilue, J. T., Shivalikanjli, A., Adams, D. J. & Keane, T. M. Mouse protein coding diversity: what’s left to discover? PLoS Genet. 15, e1008446. https://doi.org/10.1371/journal.pgen.1008446 (2019).
Sun, D., Gao, W., Hu, H. & Zhou, S. Why 90% of clinical drug development fails and how to improve it? Acta Pharm. Sin. B 12, 3049–3062. https://doi.org/10.1016/j.apsb.2022.02.002 (2022).
Dowden, H. & Munro, J. Trends in clinical success rates and therapeutic focus. Nat. Rev. Drug Discov. 18, 495–496. https://doi.org/10.1038/d41573-019-00074-z (2019).
Hinkson, I. V., Madej, B. & Stahlberg, E. A. Accelerating therapeutics for opportunities in medicine: a paradigm shift in drug discovery. Front. Pharmacol. 11, 770. https://doi.org/10.3389/fphar.2020.00770 (2020).
Zeiss, C. J. & Johnson, L. K. Bridging the gap between reproducibility and translation: data resources and approaches. ILAR J. 58, CP3. https://doi.org/10.1093/ilar/ilx017 (2017).
Prager, E. M., Tichy, H. & Sage, R. D. Mitochondrial DNA sequence variation in the Eastern house mouse, Mus musculus: comparison with other house mouse mice and report of 75-bp tandem repeat. Genetics 143, 427–446. https://doi.org/10.1093/genetics/143.1.427 (1996).
Macholán, M., Vyskočilová, M., Bejcek, V. & Štastný, K. Mitochondrial DNA sequence variation and evolution of Old World house mice (Mus musculus). Folia Zool. 61, 284–307. https://doi.org/10.25225/fozo.v61.i3.a12.2012 (2012).
Gregorová, S. et al. Mouse consomic strains: exploiting genetic divergence between Mus m. musculus and Mus m. domesticus subspecies. Genome Res. 18, 509–515. https://doi.org/10.1101/gr.7160508 (2008).
Loveland, B., Wang, C. R., Yonekawa, H., Hermel, E. & Lindahl, K. F. Maternally transmitted histocompatibility antigen of mice: a hydrophobic peptide of a mitochondrially encoded protein. Cell 60, 971–980. https://doi.org/10.1016/0092-8674(90)90345-F (1990).
Zheng, J. et al. mtDNA sequence, phylogeny and evolution of laboratory mice. Mitochondrion 17, 126–131. https://doi.org/10.1016/j.mito.2014.07.006 (2014).
Sievers, F. et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 7, 539. https://doi.org/10.1038/msb.2011.75 (2011).
Tavaré, S. Some probabilistic and statistical problems in the analysis of DNA sequences. Lect. Math. Life Sci. 17, 57–86 (1986).
Darriba, D., Taboada, G. L., Doallo, R. & Posada, D. jModelTest 2: more models, new heuristics and parallel computing. Nat. Methods 9, 772–772. https://doi.org/10.1038/nmeth.2109 (2012).
Guindon, S. & Gascuel, O. A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst. Biol. 52, 696–704. https://doi.org/10.1080/10635150390235520 (2003).
Yang, Z. Maximum likelihood phylogenetic estimation from DNA sequences with variable rates over sites: approximate methods. J. Mol. Evol. 39, 306–314. https://doi.org/10.1007/BF00160154 (1994).
Felsenstein, J. Inferring Phylogenies (Sinauer Associates Inc., 2004).
Tamura, K., Stecher, G. & Kumar, S. MEGA11: molecular evolutionary genetics analysis version 11. Mol. Biol. Evol. 38, 3022–3027. https://doi.org/10.1093/molbev/msab120 (2021).
Oliver, P. L. et al. Accelerated evolution of the Prdm9 speciation gene across diverse metazoan taxa. PLoS Genet. 5, e1000753. https://doi.org/10.1371/journal.pgen.1000753 (2009).
Vogelstein, B. 7 Kinzler, K. W. Digital PCR. Proc. Natl. Acad. Sci. U.S.A. 96, 9236–9241 https://doi.org/10.1073/pnas.96.16.9236 (1999).
Pezer, Z., Harr, B., Teschke, M., Babiker, H. & Tautz, D. Divergence patterns of genic copy number variation in natural populations of the house mouse (Mus musculus domesticus) reveal three conserved genes with major population-specific expansions. Genome Res. 25, 1114–1124. https://doi.org/10.1101/gr.187187.114 (2015).
Team, R. R: A Language and Environment for Statistical Computing (2020).
Team, R. RStudio: Integrated Development for R (2019).
Eddy, W. F. Algorithm 523: CONVEX, a new convex hull algorithm for planar sets. ACM Trans. Math. Softw. 3, 411–412. https://doi.org/10.1145/355759.355768 (1977).
Auffray, J. C. et al. in Evolution of the House Mouse. Cambridge Studies in Morphology and Molecules: New Paradigms in Evolutionary Bio (eds Macholán, M. et al.) 1–34 (Cambridge University Press, 2012).
Marshall, J. T. & Sage, R. D. in Biology of the House Mouse Symposium of the Zoological Society of London (ed Berry, R. J.) 15–25 (Academic Press, 1981).
Acknowledgements
We want to express our gratitude to Stuart J. E. Baird for providing hybrid indices of strains. The construction of the breeding facility in Studenec was made possible through funding from the Czech Academy of Sciences. The long-term housing of mouse strains was supported by the Institute of Vertebrate Biology, with additional funding from Czech Science Foundation Grants No. 16–23773S, 17–04364S, 19–12774S, 21–28491S, 22–29928S as well as CAS within the program of the Strategy AV 21. We appreciate the access to the National Grid Infrastructure MetaCentrum, provided under the programme ‘Projects of Large Infrastructure for Research, Development, and Innovations’ (LM2010005). Core Facility Genomics of CEITEC Masaryk University (supported by the NCMG research infrastructure LM2023067 funded by MEYS CR) is gratefully acknowledged for providing the equipment used for CNV estimations.
Author information
Authors and Affiliations
Contributions
J.P. designed the study, collected mice, was responsible for morphological and reproductive performance analyses, and wrote the initial version of the manuscript. Ľ.Ď. collected mice, derived chromosomal and morphological data. Z.H. assessed CNV. J.K. designed statistical analyses, T.A., A.B. D.Č., J.GdB, and K.J. sequenced mtDNA. H.H., JanaP., I.M. and I.P. took care of mice. L.R. heads the breeding facility, took care of mice and genotyped mice for the presence of the t-haplotype. A.O. and F.B. were responsible for a mouse repository in Montpellier, and C.P. and D.T. provided mice from their collection in Plön. B.V.B. collected mice and genotyped Y chromosome. J.F. is responsible for a mouse repository in Prague and wrote the manuscript. M.M. analysed sequence data and wrote the manuscript. P.K. sequenced the Prdm9 gene, designed routine genotyping of strains at diagnostic markers and wrote the manuscript. All authors have read and approved the final version of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Piálek, J., Ďureje, Ľ., Hiadlovská, Z. et al. Phenogenomic resources immortalized in a panel of wild-derived strains of five species of house mice. Sci Rep 15, 12060 (2025). https://doi.org/10.1038/s41598-025-86505-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-025-86505-x
