
Abstract
Craniofacial microsomia (CFM) is the second most common congenital craniofacial deformity, presenting diverse clinical manifestations and treatments that may influence oral bacteria dysbiosis (OBD). However, research linking CFM to OBD is limited. Saliva samples were collected from 20 patients with CFM and 24 controls. We compared oral microflora and gene function using 16 S ribosomal RNA sequencing and metagenomics. We also evaluated the correlation between CFM clinical phenotypes and microbiota community structure. Patients with CFM demonstrated greater richness and evenness in their oral microflora. The dominant genera included several pathogenic species, such as Actinomyces, Fusobacterium, and Prevotella. Notably, the severity of CFM correlated positively with the abundance of Neisseria and Porphyromonas. Upregulated pathways were primarily linked to biotin and amino acid metabolism, such as Tryptophan metabolism and Lysine degradation, and further underscored the need for focused oral health interventions in this population. This study is the first to indicate that CFM patients exhibit unique oral bacterial dysbiosis, marked by a higher presence of opportunistic pathogens and increased pathways related to oral and systemic health. These findings highlight the importance of monitoring oral health in patients with CFM.
Similar content being viewed by others

Introduction
During embryonic development, the craniomaxillofacial region forms from various prominences, including the frontonasal, maxillary, mandibular, median nasal, and lateral nasal prominences. Disruptions in the development of the first and second pharyngeal arches during the initial six weeks can result in a range of deformities, leading to craniofacial microsomia (CFM). CFM is the second most common congenital craniofacial malformation, following cleft lip and palate, and is marked by underdevelopment of the mandible, maxilla, ear, facial nerve, and associated muscles. Additionally, it often presents with dental anomalies linked to dysplasia of the first pharyngeal arch1,2,3.
Children with craniofacial deformities are often at risk for poor oral health, primarily due to oral structural deficiencies (OSD)4. These structural abnormalities can lead to inadequate oral hygiene and disrupt the balance of the oral microenvironment, which may further compromise alveolar bone health, cause masseter dysfunction, delay the development of permanent teeth, and complicate orthodontic and orthognathic treatment plans5,6,7,8. In patients with CFM, OSD may manifest as abnormalities in the maxilla and mandible, dental agenesis, delayed tooth development, variations in tooth size, irregular tooth morphology, and enamel defects1. Additionally, obstructive sleep apnea syndrome (OSAS), a common condition associated with CFM, along with treatments like mandibular distraction osteogenesis, orthodontics, and orthognathic surgery, have all been linked to OBD. Despite these connections, the overall oral health status of CFM patients has yet to be thoroughly understood.
The oral microenvironment’s homeostasis includes various components, such as oral anatomy, microbiota, the mucOSASl immune system, and the epithelial barrier, all of which interact in a complex and dynamic manner. The oral microbiota, a crucial element of this homeostasis, is the second largest microbiota in the human body and comprises bacteria, fungi, viruses, and some low-abundance microorganisms. Among these, bacteria are the predominant component due to their diversity and abundance. Numerous endogenous and exogenous factors, including anatomical structure, oral hygiene practices, and OSAS, influence the oral bacterial population. These interactions and the balance of oral bacteria significantly impact both oral health and systemic diseases. Consequently, changes in the abundance, structure, and function of oral bacteria serve as vital indicators for assessing oral health status9,10,11,12.
Various clinical manifestations and treatments significantly impact oral health in patients with CFM. Consequently, it is essential to investigate the structural and functional changes in the oral microbiota of CFM patients to support the integration of oral hygiene therapies into future treatment plans. However, no comparative analysis currently exists between the oral flora of CFM patients and healthy controls. This study aims to examine the characteristics and changes in the oral bacteria of CFM patients using 16 S ribosomal RNA (rRNA) sequencing and to assess potential functional molecular changes through metagenomics. A combined multi-omics approach was employed to preliminarily characterize the oral microbiota in CFM and evaluate whether changes in the microbiota correlate with functional alterations.
Materials and methods
Subject selection
CFM patients were recruited from Maxillo-facial Surgery Center, Plastic Surgery Hospital, Chinese Academy of Medical Sciences, Peking Union Medical College, from November 2022 to April 2023. Healthy controls diagnosed with hypertrophic scarring were recruited from the Department of Scar & Wound Treatment of Plastic Surgery Hospital from October 2022 to April 2023.
Inclusion criteria were as follows: (1) patients diagnosed with CFM and classified according to the Pruzansky-Kaban Classification and healthy controls diagnosed with hypertrophic scarring; (2) aged 6–12 years old (the mixed dentition stage); (3) parents or guardians signed the informed consent form.
Exclusion criteria were as follows: (1) diagnosed with other systemic diseases reported except for CFM or hypertrophic scarring; (2) those who had taken probiotics, fluoride application treatment (didn’t include the use of fluoride toothpaste), or antibiotics for at least 3 months; (3) those who had received periodontal and/or OSAS treatment in the last 1 month; (4) particular dietary preference such as vegetarianism; (5) pet feeding; (6) those was discomfort on the day of sampling; (7) females who were in the physiological period.
All Subjects understood the nature of the experiment and provided informed consent before participating in the study. This study was approved by the ethical review board of the Plastic Surgery Hospital, Chinese Academy of Medical Sciences, Peking Union Medical College (approval no. 2021 − 199) on Month 12, 2021. All methods were performed in accordance with the relevant guidelines and regulations.
Sample collection
Non-stimulated saliva samples were collected from both CFM patients and healthy controls. Participants were instructed to refrain from brushing their teeth for 24 h and from eating or drinking for 1 h prior to sampling. After rinsing their mouths with 20 ml of physiological saline for 1 min, eight sterile cotton swabs were placed in their mouths for 2 min to absorb saliva, which was then transferred into a 5 ml sterilized Cryogenic Storage Tube. These tubes were frozen in liquid nitrogen and stored at −80 °C.
Demographic data for each participant, including age, gender, and body mass index (BMI), were recorded. The oral health status of participants was assessed by two authors during sample collection. One of the authors has a master’s degree in stomatology, while the other author is a specialist in oral and maxillofacial surgery. Additionally, all patients underwent sleep monitoring with respiratory poly-somnography(RP), with the obstructive apnea-hypopnea index (OAHI) and blood oxygen saturation (SpO2) levels documented.
16 S rRNA diversity sequencing
DNA extraction and amplification
Genomic DNA was extracted from saliva samples using the MagPure Soil DNA KF Kit and DNA concentrations and integrity were measured using the NanoDrop 2000 (Thermo Fisher Scientific, USA). Polymerase chain reaction (PCR) amplification of bacterial 16S rRNA genes was performed using barcoded primers and Takara Ex Taq (Takara) using extracted DNA stored in a −20°C refrigerator. V3-V4 regions of 16S rRNA genes were amplified using primers 343F (5’-TACGGRAGGCAGCAG-3’) and 798R (5’-AGGGTATCTAATCCT-3’)13.
Library construction and sequencing
PCR products were purified with AMPure XP beads (Agencourt) and re-amplified for quality assessment using agarose gel electrophoresis. AMPure XP beads were used to purify the final amplicon, and the Qubit dsDNA Assay Kit (Thermo Fisher Scientific, USA) was used to measure the DNA concentration of samples for sequencing using an Illumina NovaSeq 6000 platform with 250 base pairs (bp) paired-end reads (Illumina Inc.; OE Biotech Company, Shanghai, China).
Bioinformatic analysis
Sequencing and data processing were performed by OE Biotech Co., Ltd. (Shanghai, China). Raw sequencing data were in the FASTQ format. Preprocessing was then carried out using Cutadapt software to detect and remove adapters from the paired-end reads. DADA23 with QIIME24 (2020.11) default parameters were used to filter low-quality reads, denoise, merge, detect, and cut off the chimera reads after trimming paired-end reads. The QIIME2 package was used to select the representative read of each amplicon sequence variant and output the abundance table. With default parameters, a q2-feature classifier was used to classify all representative reads against the Silva database (Version 138). And the sequences were categorized based on their taxonomic group using the Human Oral Microbiome Database ( https://www.homd.org/).
A MagPure Soil DNA KF Kit was used to isolate total DNA from the sample. An agarose gel electrophoresis and NanoDrop2000 spectrophotometer (Thermo Fisher Scientific, Waltham, MA, USA) were used to measure DNA concentration and integrity. DNA was fragmented using an S220 Focused-ultrasonicator (Covaris, USA) and cleansed using Agencourt AMPure XP beads (Beckman Coulter Co., USA). Subsequently, libraries were constructed using the TruSeq Nano DNA LT Sample Preparation Kit (Illumina, San Diego, CA, USA) following the manufacturer’s instructions. The Metagenome was sequenced and analyzed by OE Biotech Co., Ltd. (Shanghai, China).
Meanwhile, 150 bp paired-end reads were generated from the libraries on an Illumina Novaseq 6000 platform. Sequences in the FastQ file were trimmed and filtered using Fastp (v 0.20.1)14, low-quality primary groups were filtered out, and reads containing N bases were removed. The host pollution was controlled by aligning the post-filtered pair-end reads against the human genome using bowtie2 (v 2.2.9) and discarding the aligned reads. MEGAHIT (v 1.1.2) was used for genome assembly after obtaining valid reads15,16. Gaps inside the scaffold were used to break the scaffold into new contigs (Scaftigs), and new Scaftigs with a length > 500 bp were retained. Using Prodigal (v 2.6.3), open reading frames (ORFs) were predicted on assembled scaffolds and translated into amino acid sequences17. CDHIT (v 4.5.7) was used to construct non-redundant gene sets from all predicted genes. The clustering parameters had a 95% identity and 90% coverage. The longest gene represented each gene set. Then, clean reads of each sample were aligned against the non-redundant gene set (95% identity) using bowtie2 (v 2.2.9), and the abundant information of the gene in the corresponding gene set was analyzed. The species’ taxonomy was determined using the Non Redundant Protein (NR) database (https://www.ncbi.nlm.nih.gov/refseq/about/nonredundantproteins/), and gene abundance was used to calculate the abundance of the species. Abundance statistics were performed at Domain, Kingdom, Phylum, Class, Order, Family, Genus, and Species levels to construct abundance profiles on the corresponding taxonomy level. The amino acid sequence representing the gene set was compared and annotated using DIAMOND (v 0.9.7) with NR, Kyoto Encyclopaedia of Genes and Genomes (KEGG) (https://www.genome.jp/kegg/), eggNOG (http://eggnog-mapper.embl.de/), SWISS-PROT (https://www.ebi.ac.uk/uniprot/), and GO (https://www.geneontology.org/) databases at an e-value of 1e-518,19,20. Gene sets were compared with the CAZy database (https://www.cazy.org/) using hmmscan (v3.1) to obtain the carbohydrate-active enzyme corresponding to the gene. The carbohydrate activity was calculated by summing the gene abundances corresponding to the carbohydrate-active enzyme abundance21.
Statistical analysis
Normally distributed data were compared using the independent samples t-test; otherwise, the Wilcoxon rank-sum test was adopted. In 16 S rRNA sequencing, QIIME2 software was used to analyze alpha and beta diversity. The Shannon index was used to estimate microbial diversity in samples22,23. PERMANOVA was used for principal coordinate analysis (PCoA) analysis that performed to estimate beta diversity using an unweighted Unifrac distance matrix. A T-test was used to analyze the differences in diversity and taxonomic composition between the two groups. Spearman correlation analysis was applied to measure the correlation between the clinical characteristics of patients and microbial structure. In metagenome sequencing, the taxonomic or functional abundance spectrum was analyzed using R software (version 3.2.0), and an equidistant matrix of PCoA was calculated and analyzed. The Student’s t-test was used to analyze significant differences between groups. Taxonomic and functional abundance spectra were compared using linear discriminant analysis effect size (LEfSe). All data were analyzed using R software. P < 0.05 was considered statistically significant.
Results
Baseline demographic and clinical characteristics
A total of 44 participants were recruited, including 20 in the CFM group and 24 in the Ctrl group. The baseline demographic and clinical characteristics for both groups are presented in (Table 1). There were no significant differences in age, gender, or BMI between the CFM and Ctrl groups. In assessment of oral disease, the two clinicians exhibited an exceptional degree of consistency in the evaluation of occlusal relationships (kappa = 1, P<0.05), while other diagnoses had a good consistency with a kappa value of 0.688, P<0.05. In CFM group, gingival inflammation, gingival bleeding, and abnormal tooth morphology demonstrated a greater frequency of occurrence, with no statistical significance. The incidence of dental caries and abnormal occlusal relationship in the CFM group was significantly elevated, with P of 0.041 and 0.037, respectively.
16 S rRNA gene sequencing of the 44 samples produced 3,518,300 raw reads, with an average of 79,961 sequences per sample. After preprocessing, 2,969,017 high-quality sequences were retained, averaging 67,477 sequences per sample. A total of 2,789 amplicon sequence variants were identified across the samples, encompassing 15 phyla, 26 classes, 73 orders, 126 families, 247 genera, and 479 species.
Additionally, metagenomic sequencing was conducted on samples from 6 CFM patients and 6 healthy controls. The effective data size for each sample ranged from 11.96 to 15.93 G, with N50 values for contigs between 454 and 938 bp. After removing redundancy, 1,499,952 open reading frames (ORFs) were identified in the non-redundant gene catalog. Annotation rates for these non-redundant genes compared to the KEGG, NR, and CAZy databases were 94.15%, 51.21 and 1.69%, respectively.
Normally distributed data were expressed as mean ± SD (standard deviations) and non-normally distributed data were expressed as median interquartile range. The occlusal relationship was recorded according to the Angl’s classification of malocclusion, Class I occlusion is considered normal, while Class II and Class III occlusions are classified as abnormal.
BMI body mass index, OHAI obstructive apnea/hypopnea index, SpO2 oxygen saturation.
P<0.05 was considered statistically significant.
Diversity, composition, and comparison of the salivary microbiome
The rarefaction curve indicated that a plateau had been reached (Fig. S1), suggesting that all samples were sequenced to an adequate depth, reflecting the microbial content of the saliva samples.
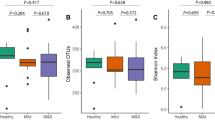
The Shannon index revealed a significant difference between the CFM group and the Ctrl group (P = 0.024) (Fig. 1A). To further analyze variations in the salivary microbial community structure, principal coordinates analysis (PCoA) was conducted using the unweighted UniFrac distance. The first axis (PC1) accounted for 9.56% of the variability, while the second axis (PC2) explained 9.23%. The analysis showed significant differences in salivary microbial communities between the CFM and Ctrl groups (P = 0.001), highlighting notable disparities in their phylogenetic structures (Fig. 1B).
The relative abundance of the top 10 phyla is summarized in (Fig. 1C). Pseudomonadota (36.46%), Bacillota (31.34%), and Bacteroidota (20.07%) were the three most abundant phyla in the Ctrl group. Pseudomonadota (31.82%), Bacteroidota (23.34%), Bacillota (20.82%), Actinomycetota (11.35%), and Fusobacteriota (10.34%) were the five most abundant phyla in the CFM group. Detailed statistics are presented in (Supplementary Table 1). Actinomycetota (Ctrl = 0.04, CFM = 0.11, P < 0.001), Campilobacterota (Ctrl = 0.006, CFM = 0.02, P = 0.013); Bacillota (Ctrl = 0.31, CFM = 0.21, P = 0.012), and Desulfobacterota (Ctrl = 0.0007, CFM = 0.0002, P = 0.002) differed significantly between the two groups (Fig. 2A).

Comparative analysis of salivary bacterial composition in individuals diagnosed with CFM and Ctrl. group. (A) Boxplot diagram of alpha-diversity. (B) Comparison of beta diversity (PCoA). (C,D) The top 15 higher relative abundance of salivary bacteria at the phylum (C) and genus (D) level.
The top 15 most abundant taxa are shown in (Fig. 1D). Streptococcus (24.20%), Haemophilus (18.23%), and Neisseria (11.63%) were significantly enriched in the Ctrl group. Neisseria (18.07%) and Streptococcus (14.34%) were highly abundant in the CFM group. Detailed statistics are shown in (Supplementary Table 2). The top 10 most abundant genera are shown in (Fig. 2B). Corynebacterium (Ctrl = 0.01, CFM = 0.05, P < 0.001), Capnocytophaga (Ctrl = 0.03, CFM = 0.07, P = 0.002), Rothia (Ctrl = 0.01, CFM = 0.03, P = 0.021), Campylobacter (Ctrl = 0.01, CFM = 0.02, P = 0.013), Actinomyces (Ctrl = 0.02, CFM = 0.04, P = 0.022), and Prevotella (Ctrl = 0.05, CFM = 0.09, P = 0.048) were the most predominant genera in the CFM group. Haemophilus (Ctrl = 0.18, CFM = 0.07, P < 0.001), Gemella (Ctrl = 0.03, CFM = 0.01, P < 0.001), Streptococcus (Ctrl = 0.24, CFM = 0.14, P = 0.008), and Porphyromonas (Ctrl = 0.06, CFM = 0.03, P = 0.04) were the most abundant genera in the Ctrl group. The relative abundance of bacterial genera was assessed via metagenomic sequencing, and the resultant data are summarized in the (Supplementary Table 3).
Based on the relative abundance of community structure, Neisseria and Porphyromonas were positively correlated with the Pruzansky-Kaban classification, with a Spearman’s rank correlation coefficient (rs) of 0.66 (P = 0.0016) and 0.52 (P = 0.019), respectively. Prevotella and Kingella were negatively correlated with the Pruzansky-Kaban classification, with the rs is -0.57 (P = 0.009) and − 0.45 (P = 0.046), respectively. Campylobacter, Leptotrichia, and Kingella were positively correlated with SpO2, with the rs is 0.54 (P = 0.014), 0.53 (P = 0.03), and 0.48 (P = 0.016), respectively. Staphylococcus significantly negatively correlated with SpO2, with the rs is 0.46 (P = 0.04). Meanwhile, no correlation was found between microbial community structure and BMI and OAHI (Fig. 2C).
To identify potential biomarkers, the LEfSe method was used to analyze differences in the composition of the microbial communities between the two groups. The cladogram of salivary microbial structure and dominant bacteria was generated based on a linear discriminant analysis (LDA) threshold of 3, which indicated the most significant differences between CFM and Ctrl groups. Neisseriaceae was the most predominant at the family level and Prevotella, Capnocytophaga, Corynebacterium, Leptotrichia, Actinomyces, Fusobacterium, Campylobacter, Rothia, and Kingella were the most predominant at the genus level in the CFM group. Gemellaceae was the most predominant at the family level and Pelomonas, Gemella, Porphyromonas, and Streptococcus were dominant at the genus level in the Ctrl group (Fig. 2D,E).
According to the Pruzansky-Kaban classification, CFM patients were divided into Type I-IIA and Type IIB-III groups, patients with or without temporomandibular joint (TMJ) dysfunction. The difference in the salivary microbiome of both groups was compared to explore the relationship between the disease severity and microbial community structure. The top three genera with the highest average abundance were Prevotella (0.16), Neisseria (0.09), and Leptotrichia (0.07) in Type I-IIA and Neisseria (0.23), Haemophilus (0.09), and Prevotella (0.06) in Type IIB-III. The top 3 statistically significantly differential genera between the two groups included Faecalibacterium (0.0001, 0.0006, P = 0.001), Neisseria (0.09, 0.23, P = 0.004), and Porphyromonas (0.01, 0.05, P = 0.009) (Fig. 2F, G).

The diversity, abundance, and distribution of salivary bacteria were compared between the CFM and Ctrl. groups. (A) Boxplot of the differential phyla between the CFM and Ctrl. groups. (B) Boxplot of the top 10 differential genera between the CFM and Ctrl. groups. (C) Heat map matrix of Spearman correlation between high-abundance genera and clinical characterization of CFM patients. (D) The contribution of different species to differences between group CFM and Ctrl. (E) The differential species annotation analysis between group CFM and Ctrl. (F) The contribution of different species to differences between group TypeI-IIA and TypeIIB-III in CFM.The initial data indicate that the average abundance of Lachnoclostridium in the Type I-IIA group is 0, while in the Type IIB-III group is 0.00010579. (G) The contribution of different species to differences between group TypeI-IIA and TypeIIB-III in CFM.
Functional comparison of the salivary microbiome between CFM and Ctrl groups
The distribution of gene numbers between the two groups is displayed in Fig. S2A, which revealed a significant difference between CFM and Ctrl (P = 0.016). Hierarchical clustering analysis was performed to evaluate the similarities and differences in gene abundances of all samples and the results showed that CFM and Ctrl groups had a high intra-group similarity (Fig. S2B).
KEGG annotations of amino acid sequences obtained from the two groups are shown in Fig S2C, with metabolism-associated genes accounting for the most significant proportion. Carbohydrate metabolism was annotated to 77,286 genes, making it the most functionally rich category, followed by amino acid metabolism, with 63,843 annotated genes, and Metabolism of cofactors and vitamins, Energy metabolism, Nucleotide metabolism, and Glycan biosynthesis and metabolism, with relatively abundant genes. Other metabolic pathways with relatively abundant genes included Genetic Information Processing, Environmental Information Processing, and Cellular Processes.
Based on the annotation of differentially-expressed KEGG ORTHOLOGY (KO) between CFM and Ctrl groups at level 3, the top 30 KO relative abundances were selected for clustering information based on the P-value to draw their functional heatmaps (Fig. 3A), revealing significant differences in functional abundance between the two groups.
Thirty-four functional pathways differed significantly between the two groups, and the top 10 functional pathways with the highest abundance were selected to generate the boxplots (Fig. 3B). Functional pathways with significant differences in the CFM group were Porphyrin and chlorophyll metabolism (0.013), Sulfur metabolism (0.003), Biotin metabolism (0.042), Histidine metabolism (0.040), Plant-pathogen interaction (0.019), Lysine degradation (0.005), Tryptophan metabolism (0.031), Salmonella infection (0.015), and Fluid shear stress and atherosclerosis (0.034). Biofilm formation-Escherichia coli (0.003) had a higher functional abundance and statistical significance in the Ctrl group. Functional biomarkers with significant differences between groups based on an LDA score of 2 are shown in (Fig. 3C).

Functional annotation and difference analysis between the CFM and Ctrl. groups. (A) Heatmap of differentiated functions between group CFM and Ctrl. (B) Boxplot of significant difference function between group CFM and Ctrl. (C) Functional biomarkers with significant differences between group CFM and Ctrl.
Discussion
The human oral microbiota plays a crucial role in protecting against potential pathogens by occupying specific ecological niches without causing harm24. When the balance of this microbial community is disrupted, it can lead to a dysbiotic state characterized by an altered microbial structure and a decrease in pH within the oral cavity. This acidic environment fosters the growth of pathogenic bacteria, resulting in inflammation and tissue damage25. The pathogenic effects observed are not solely due to individual bacterial species; rather, they stem from changes in the overall community structure, which are linked to various systemic and oral diseases. Systemically, dysbiosis has been associated with conditions such as osteoporosis, cancer, cardiovascular diseases, gastrointestinal disorders, and diabetes9,26,27,28. In the oral context, shifts in microbial composition can lead to increased levels of pathogenic bacteria, exacerbating immune-mediated inflammatory responses. Moreover, oral microbiota dysbiosis contributes to heightened levels of specific cytokines and inflammatory mediators, promoting the degradation of alveolar bone and periodontal tissues. This degradation can result in alveolar bone resorption, adversely affecting both masticatory function and aesthetic appearance. Dysbiosis in the oral cavity is implicated in several conditions, including dental caries, pulpitis, periodontitis, and head and neck tumors28,29,30,31,32,33.
Individuals born with craniofacial disorders, which affect the growth and function of teeth and jaws, are often at increased risk for poor oral health34. This elevated risk stems from difficulties in effectively cleaning structurally abnormal oral cavities, along with the constant exposure of dental enamel and gum tissue to the oral environment due to these deformities. As a result, these factors can promote plaque formation and accumulation, heightening susceptibility to oral diseases.
However, in the realm of craniofacial deformities, the role of OBD has primarily been examined in the context of cleft lip and palate (CLP) treatments. Research indicates that the structural issues associated with CLP can lead to OBD, while subsequent treatments can influence the oral microbiome35,36,37. However, many features of CFM remain unexplored regarding their interaction with OBD, highlighting an area that warrants further investigation.
CFM is the second most common craniofacial anomaly after cleft lip and palate, with an incidence rate of 1 in 3500 to 1 in 5600 live births38,39. The etiology of CFM is linked to disruptions in the development of the first and second pharyngeal arches during the first six weeks of gestation, resulting in a range of craniofacial anomalies that are recognized as contributing factors to OBD40,41. Underdeveloped mandible is typical manifestition of CFM with the incident rate is 89-100%44. This underdevelopment, along with hypoplasia of the maxilla and mandible, increases the likelihood of malformations in oral structures and malocclusion2,42. Patients with CFM are more likely to experience poorly developed teeth (6.7–33.3%) and delayed tooth eruption (20.5–54.3%) compared to normal individuals, whose respective rates are 4.5–13.3 and 3.4–4.3%43,44,45. Additionally, CFM patients have a significantly higher incidence of OSAS, with rates around 71%44,45,46,49,50. Notably, OBD in pediatric patients with OSAS has been documented47,48,49 attributed to recurrent upper airway collapses, reduced nasal airflow, snoring, and mouth breathing during sleep50. These factors compromise oral hygiene, impair salivary function, and affect blood oxygen levels, as well as alter airway humidity, temperature, and pH51,52,53,54.Moreover, the oral cavity of OSAS patients is more exposed to dust, particles, and airborne microorganisms, disrupting microbial balance55,56. These disruptions lead to increased secretion of pro-inflammatory cytokines, which can mediate respiratory inflammation and edema, further inducing or exacerbating the severity of OSAS.
Mainstream treatments for CFM include mandibular distraction osteotomy (MDO), orthodontics, and orthognathic surgery57. In the treatment process of CFM, there are many factors may interact with OBD. During the treatment of CFM, various factors may influence OBD. MDO is typically performed during the mixed dentition period, when children experience significant skeletal growth. This procedure helps lengthen the mandible, create an open bite, lower the maxillary occlusal plane, and enhance both occlusal alignment and facial symmetry58,59. The active growth of alveolar bone on the affected side during MDO is crucial for the downward movement of the maxillary occlusal plane, thereby affecting the stability of the occlusal plane60,61,62,63,64. Orthodontics and orthognathic surgery often serve as supplementary treatments for patients facing poor occlusion and aesthetic issues during the permanent dentition stage65. At this stage, factors such as the alveolar bone are vital for dental implants, orthodontic treatments, periodontal therapy, and restoring oral function61,66,67,68,69,70. In summary, numerous factors may interact with OBD in CFM patients, yet the relationship between CFM and OBD remains unclear. Establishing this association is essential for optimizing treatment protocols. This study aims to compare the structure and function of the oral microbiota in CFM patients and normal individuals to confirm the presence of OBD in those with CFM.
This study included children with oral diseases in the control group to more effectively contrast the impact of CFM on OBD, recognizing that healthy children without CFM can also develop oral diseases. The control group consisted of patients with scars from our hospital, making routine craniofacial X-rays or CBCT examinations unnecessary. Given the potential health risks and ethical considerations related to radiological examinations in the control group, such procedures were not performed. Oral health assessments focused on dental caries, gingival inflammation and bleeding, abnormal occlusal relationships, and tooth morphology anomalies, all determined through clinical observations by the authors during sample collection. The evaluation of occlusal relationships followed Angle’s classification system. The prevalence of oral diseases was 85% in the CFM group and 50% in the Ctrl group, with statistically significant differences observed in the rates of dental caries and malocclusion between the two groups. Preliminary data suggested a higher prevalence of oral diseases in CFM patients. However, given that diagnoses predominantly rely on clinical observations, further research on oral microbiome is needed to substantiate the association between CFM and oral health issues.
For sample selection and timing, previous research indicates that saliva is the most suitable specimen for analyzing oral biofilm microbiota, as it causes minimal harm to the host71,72. Additionally, the oral microbiota of young children undergoes significant changes in composition and diversity, stabilizing around age two73. Therefore, this study focused on children in the mixed dentition period and utilized saliva for analysis.
The Shannon dilution curve confirmed the adequacy of the sequenced data, showcasing excellent sequencing quality and robust research findings. Compared to the control group, the CFM group displayed enhanced species richness and evenness. Beta diversity analysis revealed significant differences in microbial community structure between the two groups, with between-group variations surpassing within-group variations. Numerous phyla and genera showed statistical differences, indicating the presence of resident bacteria in the oral cavity, including some classified as opportunistic pathogens74,75. These findings preliminarily suggest that patients with CFM exhibit oral bacterial dysbiosis.
Alterations in oral bacterial composition have been observed in patients with OSAS and obesity76,77. To assess whether oral bacterial dysbiosis in CFM patients is primarily influenced by OSAS or obesity, we conducted a Spearman correlation analysis. This analysis examined the relationships among CFM Pruzansky-Kaban classification, OAHI, SpO2, BMI, and oral microbial structure78. The Pruzansky-Kaban system categorizes patients into Types I, IIa, IIb, and III. Type I features mild hypoplasia of the mandibular ramus, while Type IIa maintains a normal fossa-condyle relationship and TMJ function. Type IIb exhibits a disrupted fossa-condyle relationship, impairing TMJ function, and Type III involves complete degeneration of the ramus with no discernible TMJ79. Given the TMJ’s critical role in maintaining oral function, its presence or absence was a criterion for patient classification. Patients were divided into Type I-IIa and Type IIb-III. According to the 2020 Guidelines for the Diagnosis and Treatment of Obstructive Sleep Apnea in Chinese Children, OSAS severity is graded based on OAHI: mild (1–5 events/h), moderate (5–10 events/h), and severe (over 10 events/h)80. Our results indicated that the relative abundance of Neisseria and Porphyromonas increased with the severity of CFM deformity, while Staphylococcus abundance rose as SpO2 decreased, suggesting it is a facultative anaerobic organism. Certain species, such as S. aureus and S. epidermidis, are opportunistic pathogens found in the oral cavity, associated with oral mucositis and gingivitis. Notably, there was no significant correlation between OAHI, BMI, and bacterial structure. Previous studies have reported a decrease in oral microbiota diversity among individuals with moderate to severe OSAS81. In contrast, our study found a significant increase in both richness and evenness of the oral microbiota in patients, suggesting a potential link between oral microbiome diversity and CFM.
In addition to Neisseria and Porphyromonas, several other genera displayed significant differences in abundance within the CFM group, including Corynebacterium, Capnocytophaga, Rothia, Actinomyces, and Prevotella. These genera are integral to the oral microbiome and help maintain microbial balance. However, certain species may become pathogenic when their populations increase or when the host’s immune system is weakened.
Neisseria, was the most prevalent genus in the CFM group, accounting for 18.07% of the microbial community compared to 11.63% in the control group. Its abundance correlates positively with the severity of CFM. An excessively high level of Neisseria can trigger abnormal immune responses and inflammation, creating a cycle that further encourages Neisseria colonization82,83. This relationship underscores the importance of monitoring oral health in CFM patients, as higher Neisseria levels indicate increased inflammation.
Both Porphyromonas and Prevotella can metabolize nitrogenous compounds into cytotoxic substances such as short-chain fatty acids, sulfide compounds, and ammonia. These metabolites can induce tissue inflammation by modulating immune responses and even promote cell apoptosis, contributing to the development and progression of periodontal diseases84,85. Increased levels of Prevotella are associated with periodontal disease, abscesses, and oral tumors86,87,88. Interestingly, Porphyromonas was more abundant in the control group, likely because this group included children not screened for oral diseases. In the CFM group, Porphyromonas levels were higher in patients classified as Type IIb-III compared to those in Type I-IIa. It is important to note that not all Porphyromonas species are pathogenic. Porphyromonas gingivalis, in particular, is recognized as a key pathogen in periodontal disease, as it ferments sugars and produces acids that promote microbial adhesion and enamel demineralization89,90,91,92,93. Metagenomic sequencing revealed mean abundances of P. gingivalis of 0.0003 in the control group and 0.0005 in the CFM group, with no statistically significant difference observed.
Increased levels of Capnocytophaga have been linked to prepubertal periodontitis94,95, gingivitis96, and oral cancer97,98. Corynebacterium is primarily aerobic and usually non-pathogenic; however, it can opportunistically invade tissues or cause infections such as granulomatous lymphadenitis, pneumonia, and endocarditis in immunocompromised individuals99,100,101. Actinomyces, which showed higher abundance in CFM patients, are facultative anaerobes that thrive in low-oxygen environments and can act as opportunistic pathogens in the oral cavity. Their metabolic processes play a significant role in maintaining acid-base balance, as they convert sugars and amino acids into acids and ammonia. Actinomyces are also known to produce hydrogen sulfide and methyl mercaptan, both of which are associated with oral malodor, dental caries, and periodontal disease85,102. Campylobacter thrives in microaerobic conditions and is known to cause gastrointestinal infections103. Certain species of Rothia, a Gram-positive bacterium, can act as opportunistic pathogens in the oral cavity and pharynx, occasionally leading to severe infections such as septicemia and endocarditis104,105,106.
Streptococcus was found in greater abundance in the control group. Streptococcus mutans, a Gram-positive, anaerobic microorganism, is known for producing acid and extracellular glucan polymers. This bacterium thrives in acidic environments, contributing to tooth decay by dissolving hydroxyapatite in enamel and dentin. Interestingly, S. mutans has also been detected in the saliva of individuals with good oral health, indicating its role in the resident oral microbiome. Our study revealed that the average abundance of S. mutans was 0.00012 in the CFM group and 0.000068 in the control group, with no significant statistical difference observed. It is well known that S. mutans plays a significant role in the etiology and progression of dental caries, that may explain the higher incidence of dental caries in CFM. Future research should aim for larger sample sizes in metagenomic sequencing to clarify its distributions.
Following the identification of OBD in CFM patients, we selected six CFM patients and six controls for metagenomic sequencing. Analysis revealed no significant differences between the two groups in the areas of Cellular Processes, Environmental Information Processing, Genetic Information Processing, Human Diseases, Metabolism, and Organismal Systems. However, at level 3, the CFM group showed significantly increased pathway activity in Porphyrin and chlorophyll metabolism, Sulfur metabolism, Biotin metabolism, Histidine metabolism, Plant-pathogen interaction, Lysine degradation, Tryptophan metabolism, Salmonella infection, Fluid shear stress, and atherosclerosis compared to the control group.
Tryptophan is an essential amino acid that is absorbed through the intestinal lining and is primarily used for protein synthesis. Approximately 10–20% of tryptophan is metabolized by bacteria107,108,109. One key metabolic pathway for tryptophan is the indole pathway, where it is converted into indole and its derivatives by periodontal plaque microorganisms such as Fusobacterium, Prevotella, and Porphyromonas. The metabolites produced through this pathway can contribute to conditions like periodontitis and halitosis110. Sulfur metabolism is also significant in regulating energy metabolism in humans. Sulfur-metabolizing bacteria, including Prevotella and Fusobacterium, play a crucial role in this process111,112,113,114,115,116,117. Physiological levels of sulfur metabolites have anti-inflammatory properties and can inhibit harmful pathogens. However, excessive levels may cause DNA damage, disrupt the intestinal mucus layer, promote inflammation, and increase the risk of colorectal cancerr85.
Biotin, or vitamin B7, is essential for metabolism as it acts as a cofactor for various enzymes. Deficiencies in biotin production by bacteria can disrupt microbial community dynamics, host metabolic processes, and inflammatory responses118,119,120. Most organisms within the Bacteroidota and Pseudomonadota are responsible for biotin biosynthesis121,122. Biotin has anti-inflammatory effects by inhibiting NF-κB activation, and its deficiency can lead to increased inflammation through the secretion of pro-inflammatory cytokines123,124.
Histidine, another essential amino acid, plays a vital role in the human digestive tract, serving as a nutrient source for various gut bacteria. Research indicates that histidine and its metabolites have diverse physiological effects, with increased levels associated with the inhibition of inflammation and oxidative stress, particularly through compounds like histamine125,126. Similarly, lysine, an essential amino acid, is degraded through two distinct pathways. Increased levels of Fusobacterium nucleatum, noted in CFM patients, are linked to lysine fermentation127. Elevated lysine degradation has been shown to hinder the effective removal of toxins, further complicating the microbiome’s balance128,129.
This study compared the saliva microbiome and function of patients with CFM to that of controls using 16 S rRNA and metagenomic sequencing. The findings indicated significant differences in the diversity, composition, and functional profiles of oral microbial communities between CFM patients and controls during the mixed dentition stage. Notably, CFM patients exhibited a higher abundance of bacterial genera, primarily consisting of opportunistic pathogens. Previous research has associated the overgrowth of these bacteria with various oral health issues, including halitosis, dental caries, periodontitis, pulpitis, oral infections, periodontal abscesses, and oral tumors. Additionally, certain genera have been linked to systemic conditions such as gastrointestinal infections, tumors, pneumonia, pharyngitis, skin infections, sepsis, and endocarditis. Particularly, Neisseria and Porphyromonas were positively correlated with CFM severity and may contribute to cellular inflammation when present in high quantities, potentially exacerbating oral inflammation. The CFM group also showed an enrichment of pathogenic bacteria like Prevotella and S. mutans. Several functional pathways, including Lysine degradation and Tryptophan metabolism, were upregulated in CFM patients compared to controls, which may heighten the risk of various diseases as suggested by existing literature. This research highlights the significance of oral health in CFM by identifying key bacterial genera and functional alterations.
However, the study has limitations. The small sample size reflects the rarity of CFM, with an incidence rate of 1 in 3,500 to 1 in 5,600. Additionally, factors such as dietary habits and geographical location were not accounted for, potentially affecting the generalizability of the findings across different regions or ethnic groups. Future research will explore the effects of MDO and oral hygiene on the oral microbiota of CFM patients. We aim to investigate the meanings and mechanisms behind the identified microbial biomarkers, employing 16 S rRNA sequencing, metagenomics, and metabolomics in a larger cohort.
Conclusion
In conclusion, our findings indicate significant oral bacterial imbalances in CFM patients, characterized by an increased presence of opportunistic pathogens. Specifically, Neisseria and Porphyromonas showed a positive correlation with CFM severity. The functional pathways predominantly upregulated in CFM included Tryptophan metabolism, Sulfur metabolism, Biotin metabolism, Histidine metabolism, and Lysine degradation. This study provides clear evidence supporting the existence of oral bacterial disorders in CFM patients and underscores the importance of maintaining oral hygiene in this population. In the next step of research, oral hygiene care will be included as part of the treatment sequence. The preoperative and postoperative oral microbiota structures of CFM patients will be compared to examine changes in the microbiota and their relationship with surgical outcomes.
Data availability
The datasets supporting the conclusions of this article are available in the Bioproject database repository.[unique persistent identifier and hyperlink to datasets of macrogenomic sequencing in https://www.ncbi.nlm.nih.gov/bioproject/PRJNA1118211], with ID 1118211.[unique persistent identifier and hyperlink to datasets of 16s rRNA sequencing in https://www.ncbi.nlm.nih.gov/bioproject/PRJNA1118189], with ID 1118189.
References
Elsten, E. E. C. M. et al. Dental anomalies in craniofacial microsomia: a systematic review. Orthod. Craniofac. Res. 23 (1), 16–26. https://doi.org/10.1111/ocr.12351 (2020).
Maruko, E., Hayes, C., Evans, C. A., Padwa, B. & Mulliken, J. B. Hypodontia in hemifacial microsomia. Cleft Palate Craniofac. J. 38 (1), 15–19 (2001).
Ongkosuwito, E. M., De Gijt, P., Wattel, E., Carels, C. E. L. & Kuijpers-Jagtman, A. M. Dental development in hemifacial microsomia. J. Dent. Res. 89 (12), 1368–1372. https://doi.org/10.1177/0022034510378425 (2010).
Cheng, L. L., Moor, S. L. & Ho, C. T. C. Predisposing factors to dental caries in children with cleft lip and palate: a review and strategies for early prevention. Cleft Palate-Craniofacial J. 44 (1), 67–72. https://doi.org/10.1597/05-112 (2007).
Meaney, S., Anweigi, L., Ziada, H. & Allen, F. The impact of hypodontia: a qualitative study on the experiences of patients. Eur. J. Orthod. 34 (5), 547–552 (2012).
Rakhshan, V. Congenitally missing teeth (hypodontia): a review of the literature concerning the etiology, prevalence, risk factors, patterns and treatment. Dent. Res. J. 12 (1), https://pubmed.ncbi.nlm.nih.gov/25709668 (2015).
Park, M. K. et al. Prevalence of delayed tooth development and its relation to tooth agenesis in Korean children. Arch. Oral Biol. 73, 243–247. https://doi.org/10.1016/j.archoralbio.2016.10.024 (2017).
Badrov, J., Lauc, T., Nakaš, E. & Galić, I. Dental age and tooth development in orthodontic patients with agenesis of permanent teeth. Biomed. Res. Int. 2017, 8683970. https://doi.org/10.1155/2017/8683970 (2017).
Mark Welch, J. L., Ramírez-Puebla, S. T. & Borisy, G. G. Oral microbiome geography: micron-scale habitat and niche. Cell. Host Microbe. 28 (2), 160–168. https://doi.org/10.1016/j.chom.2020.07.009 (2020).
Zhang, Y. et al. Human oral microbiota and its modulation for oral health. Biomed. Pharmacother. 99, 883–893. https://doi.org/10.1016/j.biopha.2018.01.146 (2018).
Lamont, R. J., Koo, H. & Hajishengallis, G. The oral microbiota: dynamic communities and host interactions. Nat. Rev. Microbiol. 16 (12), 745–759. https://doi.org/10.1038/s41579-018-0089-x (2018).
Chhaliyil, P., Fischer, K. F., Schoel, B. & Chhalliyil, P. A Novel, simple, frequent oral cleaning method reduces damaging bacteria in the dental microbiota. J. Int. Soc. Prev. Commun. Dent. 10 (4), 511–519. https://doi.org/10.4103/jispcd.JISPCD_31_20 (2020).
Nossa, C. W. Design of 16S rRNA gene primers for 454 pyrosequencing of the human foregut microbiome. WJG 16 (33), 4135. https://doi.org/10.3748/wjg.v16.i33.4135 (2010).
Chen, S., Zhou, Y., Chen, Y. & Gu, J. Fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34 (17), i884–i890. https://doi.org/10.1093/bioinformatics/bty560 (2018).
Li, D. et al. MEGAHIT v1.0: a fast and scalable metagenome assembler driven by advanced methodologies and community practices. Methods 102 https://doi.org/10.1016/j.ymeth.2016.02.020 (2016).
Li, D., Liu, C. M., Luo, R., Sadakane, K. & Lam, T. W. MEGAHIT: an ultra-fast single-node solution for large and complex metagenomics assembly via succinct de bruijn graph. Bioinformatics 31 (10), 1674–1676. https://doi.org/10.1093/bioinformatics/btv033 (2015).
Hyatt, D. et al. Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinform. 11, 119. https://doi.org/10.1186/1471-2105-11-119 (2010).
Buchfink, B., Xie, C. & Huson, D. H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods 12 (1), 59–60. https://doi.org/10.1038/nmeth.3176 (2015).
Powell, S. et al. eggNOG v4.0: nested orthology inference across 3686 organisms. Nucleic Acids Res. 42 D231–D239. https://doi.org/10.1093/nar/gkt1253 (2014).
Kanehisa, M. et al. From genomics to chemical genomics: new developments in KEGG. Nucleic Acids Res. 34, D354–D357 (2006).
Cantarel, B. L. et al. The carbohydrate-active EnZymes database (CAZy): an expert resource for glycogenomics. Nucleic Acids Res. 37 (Database issue), D233–D238. https://doi.org/10.1093/nar/gkn663 (2009).
Hill, T. C. J., Walsh, K. A., Harris, J. A. & Moffett, B. F. Using ecological diversity measures with bacterial communities. FEMS Microbiol. Ecol. 43 (1). https://doi.org/10.1111/j.1574-6941.2003.tb01040.x (2003).
Chao, A. & Bunge, J. Estimating the number of species in a stochastic abundance model. Biometrics 58 (3), 531–539 (2002).
Bowen, W. H., Burne, R. A., Wu, H. & Koo, H. Oral biofilms: pathogens, matrix, and polymicrobial interactions in microenvironments. Trends Microbiol. 26 (3), 229–242. https://doi.org/10.1016/j.tim.2017.09.008 (2018).
Chimenos-Küstner, E., Giovannoni, M. L. & Schemel-Suárez, M. Dysbiosis as a determinant factor of systemic and oral pathology: importance of microbiome. Med. Clin. (Barc). 149 (7), 305–309. https://doi.org/10.1016/j.medcli.2017.05.036 (2017).
Tuganbaev, T., Yoshida, K. & Honda, K. The effects of oral microbiota on health. Science 376 (6596), 934–936. https://doi.org/10.1126/science.abn1890 (2022).
Tonelli, A., Lumngwena, E. N. & Ntusi, N. A. B. The oral microbiome in the pathophysiology of cardiovascular disease. Nat. Rev. Cardiol. 20 (6), 386–403. https://doi.org/10.1038/s41569-022-00825-3 (2023).
Radaic, A. The oralome and its dysbiosis: new insights into oral microbiome-host interactions. Comput. Struct. Biotechnol. J. (2021).
Santos, A. L. et al. Comparing the bacterial diversity of acute and chronic dental root canal infections. PLoS One. 6 (11), e28088. https://doi.org/10.1371/journal.pone.0028088 (2011).
Schulze-Schweifing, K., Banerjee, A. & Wade, W. G. Comparison of bacterial culture and 16S rRNA community profiling by clonal analysis and pyrosequencing for the characterization of the dentine caries-associated microbiome. Front. Cell. Infect. Microbiol. 4, 164. https://doi.org/10.3389/fcimb.2014.00164 (2014).
Hathaway-Schrader, J. D. & Novince, C. M. Maintaining homeostatic control of periodontal bone tissue. Periodontol 86 (1), 157–187 https://doi.org/10.1111/prd.12368 (2021).
Yang, F. et al. Saliva microbiomes distinguish caries-active from healthy human populations. ISME J. 6 (1). https://doi.org/10.1038/ismej.2011.71 (2012).
Antiabong, J. F., Boardman, W., Moore, R. B., Brown, M. H. & Ball, A. S. The oral microbial community of gingivitis and lumpy jaw in captive macropods. Res. Vet. Sci. 95 (3). https://doi.org/10.1016/j.rvsc.2013.08.010 (2013).
Cheng, L. L., Moor, S. L. & Ho, C. T. C. Predisposing factors to dental caries in children with cleft lip and palate: a review and strategies for early prevention. Cleft Palate Craniofac. J. 44 (1), 67–72 (2007).
Suzuki, A. et al. A longitudinal study of the presence of dental anomalies in the primary and permanent dentitions of cleft lip and/or palate patients. Cleft Palate Craniofac. J. 54 (3), 309–320. https://doi.org/10.1597/15-186 (2017).
Wu, Q., Li, Z., Zhang, Y., Peng, X. & Zhou, X. Dental caries and periodontitis risk factors in cleft lip and palate patients. Front. Pediatr. 10, 1092809. https://doi.org/10.3389/fped.2022.1092809 (2023).
Liu, L. et al. Investigating oral microbiome profiles in children with cleft lip and palate for prognosis of alveolar bone grafting. PLoS One 11 (5), e0155683. https://doi.org/10.1371/journal.pone.0155683 (2016).
Chen, Q., Zhao, Y., Shen, G. & Dai, J. Etiology and pathogenesis of hemifacial microsomia. J. Dent. Res. 97 (12), 1297–1305. https://doi.org/10.1177/0022034518795609 (2018).
Hartsfield, J. K. Review of the etiologic heterogeneity of the oculo-auriculo-vertebral spectrum (hemifacial microsomia). Orthod. Craniofac. Res. 10 (3), 121–128 (2007).
Birgfeld, C. B. & Heike, C. Craniofacial microsomia. Semin Plast. Surg. 26 (2). https://doi.org/10.1055/s-0032-1320067 (2012).
Li, X., Liu, Y., Yang, X., Li, C. & Song, Z. The oral microbiota: community composition, influencing factors, pathogenesis, and interventions. Front. Microbiol. 13, 895537. https://doi.org/10.3389/fmicb.2022.895537 (2022).
Netchine, I., Rossignol, S., Azzi, S. & Le Bouc, Y. Epigenetic anomalies in childhood growth disorders. Nestle Nutr. Inst. Workshop Ser. 71, 65–73. https://doi.org/10.1159/000342568 (2013).
Silvestri, A., Natali, G. & Fadda, M. T. Dental agenesis in hemifacial microsomia. Pediatr. Dent. 18 (1), 48–51 (1996).
Caron, C. J. J. M. et al. Craniofacial and extracraniofacial anomalies in craniofacial microsomia: a multicenter study of 755 patients’. J. Craniomaxillofac. Surg. 45 (8), 1302–1310. https://doi.org/10.1016/j.jcms.2017.06.001 (2017).
Ma, L. et al. yong, jun,. Analysis of obstructive sleep apnoea in craniofacial microsomia based on polysomnography. Cleft Palate Craniofac. J. 10556656231221654. https://doi.org/10.1177/10556656231221654 (2023).
Caron, C. J. J. M. et al. Obstructive sleep apnoea in craniofacial microsomia: analysis of 755 patients. Int. J. Oral Maxillofac. Surg. 46 (10), 1330–1337. https://doi.org/10.1016/j.ijom.2017.05.020 (2017).
Xu, H. et al. Pediatric obstructive sleep apnea is associated with changes in the oral microbiome and urinary metabolomics profile: a pilot study. J. Clin. Sleep. Med. 14 (9), 1559–1567. https://doi.org/10.5664/jcsm.7336 (2018).
Jia, P. et al. Analysis of the salivary microbiome in obstructive sleep apnea syndrome patients. Can. J. Infect. Dis. Med. Microbiol. 1–7. https://doi.org/10.1155/2020/6682020 (2020).
Nizam, N., Basoglu, O. K., Tasbakan, M. S., Lappin, D. F. & Buduneli, N. Is there an association between obstructive sleep apnea syndrome and periodontal inflammation? Clin. Oral Invest. 20 (4), 659–668. https://doi.org/10.1007/s00784-015-1544-y (2016).
Oksenberg, A., Froom, P. & Melamed, S. Dry mouth upon awakening in obstructive sleep apnea. J. Sleep. Res. 15 (3), 317–320 (2006).
Boink, M. A. et al. Saliva-derived host defense peptides histatin1 and LL-37 increase secretion of antimicrobial skin and oral mucosa chemokine CCL20 in an IL-1α-independent manner. J. Immunol. Res. 2017, 3078194. https://doi.org/10.1155/2017/3078194 (2017).
Frenkel, E. S. & Ribbeck, K. Salivary mucins in host defense and disease prevention. J. Oral Microbiol. 7, 29759. https://doi.org/10.3402/jom.v7.29759 (2015).
Ginsburg, I., Kohen, R. & Koren, E. Saliva: a solubilizer of lipophilic antioxidant polyphenols. Oral Dis. 19 (3), 321–322. https://doi.org/10.1111/odi.12038 (2013).
Frenkel, E. S. & Ribbeck, K. Salivary mucins promote the coexistence of competing oral bacterial species. ISME J. 11 (5), 1286–1290. https://doi.org/10.1038/ismej.2016.200 (2017).
Man, W. H., de Steenhuijsen Piters, W. A. A. & Bogaert, D. The microbiota of the respiratory tract: gatekeeper to respiratory health. Nat. Rev. Microbiol. 15 (5), 259–270. https://doi.org/10.1038/nrmicro.2017.14 (2017).
Zhang, X. et al. Metabolomics and microbiome profiling as biomarkers in obstructive sleep apnoea: a comprehensive review. Eur. Respir Rev. 30 (160). https://doi.org/10.1183/16000617.0220-2020 (2021).
Efunkoya, A. A., Bamgbose, B. O., Adebola, R. A., Adeoye, J. B. & Akpasa, I. O. Maxillomandibular distraction osteogenesis. J. Craniofac. Surg. 25 (5), 1787–1792. https://doi.org/10.1097/SCS.0000000000000907 (2014).
Weichman, K. E. et al. Early distraction for mild to moderate unilateral craniofacial microsomia: long-term follow-up, outcomes, and recommendations. Plast. Reconstr. Surg. 139 (4), 941e–953e. https://doi.org/10.1097/PRS.0000000000003223 (2017).
Zhang, R. S. et al. Early mandibular distraction in craniofacial microsomia and need for orthognathic correction at skeletal maturity: a comparative long-term follow-up study. Plast. Reconstr. Surg. 142 (5), 1285–1293. https://doi.org/10.1097/PRS.0000000000004842 (2018).
Melsen, B., Bjerregaard, J. & Bundgaard, M. The effect of treatment with functional appliance on a pathologic growth pattern of the condyle. Am. J. Orthod. Dentofac. Orthop. 90 (6), 503–512 (1986).
Shibazaki-Yorozuya, R. et al. Three-dimensional longitudinal changes in craniofacial growth in untreated hemifacial microsomia patients with cone-beam computed tomography. Am. J. Orthod. Dentofac. Orthop. 145 (5), 579–594. https://doi.org/10.1016/j.ajodo.2013.09.015 (2014).
Chow, A. et al. Cephalometric evaluation of the craniofacial complex in patients treated with an intraoral distraction osteogenesis device: a long-term study. Am. J. Orthod. Dentofac. Orthop. 134 (6), 724–731. https://doi.org/10.1016/j.ajodo.2007.01.029 (2008).
Ko, E. W. C., Chen, P. K. T. & Lo, L. J. Comparison of the adult three-dimensional craniofacial features of patients with unilateral craniofacial microsomia with and without early mandible distraction. Int. J. Oral Maxillofac. Surg. 46 (7), 811–818. https://doi.org/10.1016/j.ijom.2017.03.002 (2017).
Meazzini, M. C., Mazzoleni, F., Gabriele, C. & Bozzetti, A. Mandibular distraction osteogenesis in hemifacial microsomia: long-term follow-up. J. Craniomaxillofac. Surg. 33 (6), 370–376 (2005).
Aiyar, A. et al. Management of unilateral craniofacial microsomia with orthopaedic functional appliances: a systematic literature review. Orthod. Craniofac. Res. https://doi.org/10.1111/ocr.12729 (2023).
Li, Y., Ling, J. & Jiang, Q. Inflammasomes in alveolar bone loss. Front. Immunol. 12, 691013. https://doi.org/10.3389/fimmu.2021.691013 (2021).
Hathaway-Schrader, J. D. & Novince, C. M. Maintaining homeostatic control of periodontal bone tissue. Periodontology 2000. 86 (1), 157–187. https://doi.org/10.1111/prd.12368 (2021).
Lee, K. M., Kim, Y. I., Park, S. B. & Son, W. S. Alveolar bone loss around lower incisors during surgical orthodontic treatment in mandibular prognathism. Angle Orthod. 82 (4), 637–644. https://doi.org/10.2319/081711-526.1 (2012).
Ona, M. & Wakabayashi, N. Influence of alveolar support on stress in periodontal structures. J. Dent. Res. 85 (12), 1087–1091 (2006).
Irie, K., Novince, C. M. & Darveau, R. P. Impact of the oral commensal flora on alveolar bone homeostasis. J. Dent. Res. 93 (8), 801–806. https://doi.org/10.1177/0022034514540173 (2014).
Bonne, N. J. & Wong, D. T. Salivary biomarker development using genomic, proteomic and metabolomic approaches. Genome Med. 4 (10), 82. https://doi.org/10.1186/gm383 (2012).
Yoshizawa, J. M. et al. Salivary biomarkers: toward future clinical and diagnostic utilities. Clin. Microbiol. Rev. 26 (4), 781–791. https://doi.org/10.1128/CMR.00021-13 (2013).
Gomez, A. & Nelson, K. E. The oral microbiome of children: development, disease, and implications beyond oral health. Microb. Ecol. 73 (2), 492–503. https://doi.org/10.1007/s00248-016-0854-1 (2017).
Pathak, J. L., Yan, Y., Zhang, Q., Wang, L. & Ge, L. The role of oral microbiome in respiratory health and diseases. Respir Med. 185, 106475. https://doi.org/10.1016/j.rmed.2021.106475 (2021).
Varoni, E. M., Rimondini, L., Oral & Microbiome Oral health and systemic health: a multidirectional link. Biomedicines 10 (1). https://doi.org/10.3390/biomedicines10010186 (2022).
Wu, Y., Chi, X., Zhang, Q., Chen, F. & Deng, X. Characterization of the salivary microbiome in people with obesity. PeerJ 6, e4458. https://doi.org/10.7717/peerj.4458 (2018).
Ma, T. et al. Characterization of the oral and gut microbiome in children with obesity aged 3 to 5 years. Front. Cell. Infect. Microbiol. 13, 1102650. https://doi.org/10.3389/fcimb.2023.1102650 (2023).
Saito, T., Shimazaki, Y. & Sakamoto, M. Obesity and periodontitis. N Engl. J. Med. 339 (7), 482–483 (1998).
Hunt, J. A. & Hobar, P. C. Common craniofacial anomalies: the facial dysostoses. Plast. Reconstr. Surg. 110 (7). https://pubmed.ncbi.nlm.nih.gov/12447054 (2002).
Zhao, J. et al. Association between mild or moderate obstructive sleep apnea-hypopnea syndrome and cognitive dysfunction in children. Sleep. Med. 50, 132–136. https://doi.org/10.1016/j.sleep.2018.04.009 (2018).
Shamriz, O. et al. Microbiota at the crossroads of autoimmunity. Autoimmun. Rev. 15 (9), 859–869. https://doi.org/10.1016/j.autrev.2016.07.012 (2016).
Iaffaldano, L. et al. Oropharyngeal microbiome evaluation highlights Neisseria abundance in active celiac patients. Sci. Rep. 8 (1), 11047. https://doi.org/10.1038/s41598-018-29443-1 (2018).
D’Argenio, V. et al. Metagenomics reveals dysbiosis and a potentially pathogenic N. flavescens strain in duodenum of adult celiac patients. Am. J. Gastroenterol. 111 (6), 879–890. https://doi.org/10.1038/ajg.2016.95 (2016).
Niederman, R., Zhang, J. & Kashket, S. Short-chain carboxylic-acid-stimulated, PMN-mediated gingival inflammation. Crit. Rev. Oral Biol. Med. 8 (3), 269–290 (1997).
Kurita-Ochiai, T. et al. Butyric acid induces apoptosis in inflamed fibroblasts. J. Dent. Res. 87 (1), 51–55 (2008).
Tanaka, S. et al. The relationship of Prevotella intermedia, Prevotella nigrescens and Prevotella melaninogenica in the supragingival plaque of children, caries and oral malodor. J. Clin. Pediatr. Dent. 32 (3), 195–200 (2008).
Thomas, R. M. & Jobin, C. The microbiome and cancer: is the Oncobiome mirage real? Trends Cancer. 1 (1), 24–35 (2015).
Schwabe, R. F. & Jobin, C. The microbiome and cancer. Nat. Rev. Cancer. 13 (11), 800–812. https://doi.org/10.1038/nrc3610 (2013).
Takahashi, N. & Nyvad, B. The role of bacteria in the caries process: ecological perspectives. J. Dent. Res. 90 (3), 294–303. https://doi.org/10.1177/0022034510379602 (2011).
Torlakovic, L. et al. Microbial community succession on developing lesions on human enamel. J. Oral Microbiol. 4 https://doi.org/10.3402/jom.v4i0.16125 (2012).
Belstrøm, D., Paster, B. J., Fiehn, N. E., Bardow, A. & Holmstrup, P. Salivary bacterial fingerprints of established oral disease revealed by the human oral microbe identification using next generation sequencing (HOMINGS) technique. J. Oral Microbiol. 8, 30170. https://doi.org/10.3402/jom.v8.30170 (2016).
Paju, S. et al. Detection of multiple pathogenic species in saliva is associated with periodontal infection in adults. J. Clin. Microbiol. 47 (1), 235–238. https://doi.org/10.1128/JCM.01824-08 (2009).
Lemos, J. A. et al. The biology of Streptococcus mutans. Microbiol. Spectr. 7 (1). https://doi.org/10.1128/microbiolspec.GPP3-0051-2018 (2019).
Page, R. C. et al. Clinical and laboratory studies of a family with a high prevalence of juvenile periodontitis. J. Periodontol. 56 (10), 602–610 (1985).
Watanabe, K. Prepubertal periodontitis: a review of diagnostic criteria, pathogenesis, and differential diagnosis. J. Periodontal Res. 25 (1), 31–48 (1990).
Page, R. C. & Gingivitis J. Clin. Periodontol ;13(5):345–359. (1986).
Zhu, W. et al. Capnocytophaga gingivalis is a potential tumor promotor in oral cancer. Oral Dis. 30 (2), 353–362. https://doi.org/10.1111/odi.14376 (2024).
Hayes, R. B. et al. Association of oral microbiome with risk for incident head and neck squamous cell cancer. JAMA Oncol. 4 (3), 358–365. https://doi.org/10.1001/jamaoncol.2017.4777 (2018).
Kinoshita, S., Udaka, S. & Shimono, M. Studies on the amino acid fermentation. Part 1. Production of L-glutamic acid by various microorganisms. J. Gen. Appl. Microbiol. 50 (6), 331–343 (2004).
Poetsch, A., Haussmann, U. & Burkovski, A. Proteomics of corynebacteria: from biotechnology workhorses to pathogens. Proteomics 11 (15), 3244–3255. https://doi.org/10.1002/pmic.201000786 (2011).
Costa, J. J., Michel, J. L., Rappuoli, R. & Murphy, J. R. Restriction map of corynebacteriophages beta c and beta vir and physical localization of the diphtheria tox operon. J. Bacteriol. 148 (1), 124–130 (1981).
Washio, J., Sato, T., Koseki, T. & Takahashi, N. Hydrogen sulfide-producing bacteria in tongue biofilm and their relationship with oral malodour. J. Med. Microbiol. 54 (Pt 9), 889–895. https://doi.org/10.1099/jmm.0.46118-0 (2005).
Zilbauer, M., Dorrell, N., Wren, B. W. & Bajaj-Elliott, M. Campylobacter jejuni-mediated disease pathogenesis: an update. Trans. R Soc. Trop. Med. Hyg. 102 (2), 123–129 (2008).
Kaasch, A. J., Saxler, G. & Seifert, H. Septic arthritis due to Rothia mucilaginosa. Infection 39 (1), 81–82. https://doi.org/10.1007/s15010-010-0065-5 (2011).
Uchibori, S., Tsudukibashi, O., Goto, H., Kobayashi, T. & Aida, M. A novel selective medium for the isolation and distribution of Rothia dentocariosa in oral cavities. J. Microbiol. Methods. 91 (1), 205–207. https://doi.org/10.1016/j.mimet.2012.07.004 (2012).
Kobayashi, T., Uchibori, S., Tsuzukibashi, O., Goto, H. & Aida, M. A selective medium for Rothia mucilaginosa and its distribution in oral cavities. J. Microbiol. Methods. 91 (3), 364–365. https://doi.org/10.1016/j.mimet.2012.09.011 (2012).
Dong, F. & Perdew, G. H. The aryl hydrocarbon receptor as a mediator of host-microbiota interplay. Gut Microbes. 12 (1), 1859812. https://doi.org/10.1080/19490976.2020.1859812 (2020).
Barik, S. The uniqueness of tryptophan in biology: Properties, metabolism, interactions and localization in proteins. Int. J. Mol. Sci. 21 (22). https://doi.org/10.3390/ijms21228776 (2020).
Palego, L., Betti, L., Rossi, A. & Giannaccini, G. Tryptophan biochemistry: structural, nutritional, metabolic, and medical aspects in humans. J. Amino Acids. 2016, 8952520. https://doi.org/10.1155/2016/8952520 (2016).
Takahashi, N. Oral microbiome metabolism: from who are they? To what are they doing? J. Dent. Res. 94 (12), 1628–1637. https://doi.org/10.1177/0022034515606045 (2015).
Yoshida, Y. et al. Production of hydrogen sulfide by two enzymes associated with biosynthesis of homocysteine and lanthionine in Fusobacterium nucleatum subsp. Nucleatum ATCC 25586. Microbiol. (Reading). 156 (Pt 7), 2260–2269. https://doi.org/10.1099/mic.0.039180-0 (2010).
Marciano, D. et al. Biochemical characterization of serine acetyltransferase and cysteine desulfhydrase from Leishmania major. Mol. Biochem. Parasitol. 173 (2), 170–174. https://doi.org/10.1016/j.molbiopara.2010.06.004 (2010).
Dharmani, P., Strauss, J., Ambrose, C., Allen-Vercoe, E. & Chadee, K. Fusobacterium nucleatum infection of colonic cells stimulates MUC2 mucin and tumor necrosis factor alpha. Infect. Immun. 79 (7), 2597–2607. https://doi.org/10.1128/IAI.05118-11 (2011).
Takahashi, Y. et al. Streptococcus anginosus l-cysteine desulfhydrase gene expression is associated with abscess formation in BALB/c mice. Mol. Oral Microbiol. 26 (3), 221–227. https://doi.org/10.1111/j.2041-1014.2010.00599.x (2011).
Strauss, J. et al. Invasive potential of gut mucosa-derived Fusobacterium nucleatum positively correlates with IBD status of the host. Inflamm. Bowel Dis. 17 (9), 1971–1978. https://doi.org/10.1002/ibd.21606 (2011).
Castellarin, M. et al. Fusobacterium nucleatum infection is prevalent in human colorectal carcinoma. Genome Res. 22 (2), 299–306. https://doi.org/10.1101/gr.126516.111 (2012).
Kostic, A. D. et al. Genomic analysis identifies association of Fusobacterium with colorectal carcinoma. Genome Res. 22 (2), 292–298. https://doi.org/10.1101/gr.126573.111 (2012).
Yoshii, K., Hosomi, K., Sawane, K. & Kunisawa, J. Metabolism of dietary and microbial vitamin B Family in the regulation of host immunity. Front. Nutr. 6, 48. https://doi.org/10.3389/fnut.2019.00048 (2019).
Hayashi, A. et al. Intestinal dysbiosis and biotin deprivation induce alopecia through overgrowth of Lactobacillus murinus in mice. Cell. Rep. 20 (7), 1513–1524. https://doi.org/10.1016/j.celrep.2017.07.057 (2017).
Magnúsdóttir, S., Ravcheev, D., De Crécy-Lagard, V. & Thiele, I. Systematic genome assessment of B-vitamin biosynthesis suggests co-operation among gut microbes. Front. Genet. 6 https://doi.org/10.3389/fgene.2015.00148 (2015).
MetaHIT Consortium (additional members) et al. Enterotypes of the human gut microbiome. Nature 473 (7346), 174–180. https://doi.org/10.1038/nature09944 (2011).
Magnúsdóttir, S., Ravcheev, D., de Crécy-Lagard, V. & Thiele, I. Systematic genome assessment of B-vitamin biosynthesis suggests co-operation among gut microbes. Front. Genet. 6, 148. https://doi.org/10.3389/fgene.2015.00148 (2015).
Agrawal, S., Agrawal, A. & Said, H. M. Biotin deficiency enhances the inflammatory response of human dendritic cells. Am. J. Physiol. Cell. Physiol. 311 (3), C386–C391. https://doi.org/10.1152/ajpcell.00141.2016 (2016).
Elahi, A. et al. Biotin deficiency induces Th1- and Th17-mediated proinflammatory responses in human CD4 + T lymphocytes via activation of the mTOR signaling pathway. J. Immunol. 200 (8), 2563–2570. https://doi.org/10.4049/jimmunol.1701200 (2018).
Cacabelos, R., Torrellas, C., Fernández-Novoa, L. & López-Muñoz, F. Histamine and immune biomarkers in cns disorders. Mediat. Inflamm. 2016, 1–10. https://doi.org/10.1155/2016/1924603 (2016).
Smolinska, S., Winiarska, E., Globinska, A., Jutel, M. & Histamine A mediator of intestinal disorders—A review. Metabolites 12 (10), 895. https://doi.org/10.3390/metabo12100895 (2022).
Loesche, W. J. & Gibbons, R. J. Amino acid fermentation by Fusobacterium nucleatum. Arch. Oral Biol. 13 (2), 191–202 (1968).
Wang, X. et al. Aberrant gut microbiota alters host metabolome and impacts renal failure in humans and rodents. Gut 69 (12), 2131–2142. https://doi.org/10.1136/gutjnl-2019-319766 (2020).
Rubin, A. L. et al. The use of L-lysine monomydrochloride in combination with mercurial diuretics in the treatment of refractory fluid retention. Circulation 21, 332–336 (1960).
Acknowledgements
The authors thank the patients diagnosed with CFM and control group volunteers who participated in our study. We also thank Ouyi Biotechnology Company Limited for its technical services and valuable suggestions for bioinformatics analysis.
Author information
Authors and Affiliations
Contributions
TXJ, ZZY, and ZTY conceived and designed the study. ZTY, MLK, and LZF collected patient samples and clinical information. LBY and ZSBG performed data analysis and chart compilation. ZTY wrote the manuscript, with editing by LW and YL. TXJ and LW assisted with manuscript revisions. All authors have reviewed and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval and consent to participate
This study was approved by the ethical review board of the Plastic Surgery Hospital, Chinese Academy of Medical Sciences, Peking Union Medical College (Approval No. 2021 − 199) on Month 12, 2021. All participants voluntarily consented to participate in the study, providing informed consent. Their decision did not impact the treatment protocol.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Zang, T., Zhang, Z., Liu, W. et al. Structural and functional changes in the oral microbiome of patients with craniofacial microsomia. Sci Rep 15, 5400 (2025). https://doi.org/10.1038/s41598-025-86537-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-025-86537-3
