
Abstract
Cardiomyopathies are the most common heritable heart diseases in cats and humans. This study aimed to identify novel genetic variants in cats with hypertrophic cardiomyopathy (HCM) and restrictive cardiomyopathy (RCM) using a targeted panel of genes associated with human cardiomyopathy. Cats were phenotyped for HCM/RCM by echocardiography ± necropsy. DNA was extracted from residual blood, and targeted next-generation sequencing was performed on two separate feline cohorts: an across-breed cohort (23 healthy cats and 21 HCM-affected pedigree or Domestic Shorthair cats), and a within-breed cohort of Birman pedigree cats (14 healthy, 8 HCM-affected, and 6 RCM-affected). Genome Analysis Toolkit was used for variant discovery. Genomic association analyses, including the covariates breed, age, and sex, were conducted to identify genetic variants of interest. We identified genetic variants associated with both HCM and RCM susceptibility in the sarcomeric genes ACTC1, ACTN2, MYH7, TNNT2 and the non-sarcomeric gene CSRP3 in the Birman pedigree cats. These findings suggest that, as proposed in humans, there is at least partial overlap in the genetic background between the HCM and RCM phenotypes in cats. These findings offer potential insights for comparative cardiac research and translational medicine.
Similar content being viewed by others
Introduction
Cardiomyopathies are diseases of the myocardium that can lead to a range of functional and structural abnormalities of the heart. Cardiomyopathies in both humans and cats are associated with elevated risks of congestive heart failure, cardioembolic events, and sudden cardiac death1,2,3. Subcategorization of cardiomyopathies is based on the underlying disease cause, cardiac morphology and function, clinical presentation, and associated signs.
Hypertrophic cardiomyopathy (HCM) is defined in both humans and cats as the presence of a hypertrophied left ventricle (LV) in the absence of abnormal loading conditions capable of producing a similar degree of hypertrophy, such as other cardiac or systemic diseases3,4. Human HCM is the most common genetic cardiovascular disease, affecting 0.2% of the global population (1 in 500 individuals)5. The inheritance pattern in 60% of cases is autosomal dominant6. Nevertheless, more recent findings propose that human HCM follows a more complex mode of inheritance7, with reports of genotype-positive/phenotype-negative individuals and a higher estimated population prevalence of 0.5% (1 in 200 individuals)8. Among domesticated animals, cats are particularly predisposed to HCM, with an estimated prevalence of 15% reported in the general cat population9.
In humans, HCM has a well-characterised genetic basis with more than 1500 variants described to date10. In up to 60% of human cases, HCM is caused by variants in genes encoding cardiac sarcomeric proteins that form the cardiac contractile unit; thus, HCM is generally considered a ‘disease of the sarcomere’11. Variants in the genes encoding beta-myosin heavy chain (MYH7) and myosin-binding protein C (MYBPC3) account for the majority of cases in humans (approximately 70% of patients with sarcomeric variants)11,12. Less commonly affected genes include cardiac troponin I (TNNI3) and cardiac troponin T (TNNT2), α-tropomyosin (TPM1), myosin light chain (regulatory MYL2 and essential MYL3) and cardiac actin (ACTC)11,12,13.
To date, four variants associated with feline HCM susceptibility have been reported within specific cat breeds. Three of these variants are exonic and located in sarcomeric genes: MYBPC3 in Maine Coon14 and Ragdoll cats15, and MYH7 in a non-pedigree domestic shorthair cat (DSH)16. The fourth variant, reported in Sphynx cats, is in the non-sarcomeric Alstrom syndrome 1 (ALMS1) gene17, which is associated with cardiac development and cell regulation18. One further intronic (splicing) variant within the sarcomeric TNNT2 gene was initially associated with HCM in the Maine Coon breed19. However, a more recent study found that this variant was present in high frequencies across multiple cat breeds, suggesting that it might not have a significant association with HCM20. A recent classification study applying modified ACMG guidelines classified the two MYBPC3 gene variants A31P and R820W as pathogenic; the MYH7 E188K variant as likely pathogenic, and the remaining variants of unknown significance21. Based on these findings, routine genetic testing is currently recommended only for specific variants in Maine Coon and Ragdoll breeds.
In humans, HCM is most commonly associated with exonic variants in sarcomeric genes22. These variants cause variable degrees of left ventricular hypertrophy (LVH) and different cardiomyopathy phenotypes within the same family23,24. Individuals sharing the same causative variant may variously exhibit HCM, RCM or dilated cardiomyopathy (DCM) phenotypes23,24. This pleiotropy leading to varying disease severity/expression in cardiomyopathies suggests that factors beyond the sarcomeric variant itself may influence disease expression, such as modifier genes, environmental factors, and epigenetic modifiers13,25. The non-coding genome is an emerging area of study for HCM, with regulatory variants now identified in human HCM26,27. For most variants, the exact underlying molecular mechanisms linking genotype to phenotype remain unclear.
There are numerous similarities between human and feline HCM, highlighting the potential value of using cats as a model to study the human disease. In both species, the disease is spontaneous, exhibiting similar natural histories and wide phenotypic spectra. They also share genetic homology: Orthologous variants associated with HCM in humans have been identified in cats. Specifically, the R820W variant in the MYBPC3 gene in Ragdolls and the E188K variant in the MYH7 gene in a DSH, correspond to known human HCM variants16,28. Studies of feline HCM could uncover further shared genetic variants, disease mechanisms and therapeutic targets. A better understanding of the genetic factors causing disease and influencing disease severity in cats could aid with diagnosis and management of the disease.
Our main aim was to identify novel genetic variants for HCM susceptibility within and across cat breeds. Our secondary aim was to investigate different forms of cardiomyopathies (HCM and RCM) within a cohort of related and unrelated Birman cats to identify family-specific variants and explore possible co-occurrence of cardiomyopathies within feline families.
HCM has been reported to affect both pedigree and non-pedigree cats, with some breeds displaying a greater predisposition to the disease than others29. We initially investigated the disease across multiple pedigree and non-pedigree cats to capture associations across a spectrum of cat breeds. In the second study we focussed on cats of the Birman breed, since Birmans are recognised as a breed with a familial tendency to develop HCM (unpublished observations). We primarily focussed on investigating HCM, since this is the most common cardiomyopathy observed in cats, however we also included Birman cats with RCM. We aimed to determine whether shared HCM/RCM phenotype causative variants exist in Birman cats as they do in humans23,24.
Results
Descriptive statistics of the studied cat populations
Descriptive statistics of the clinical and echocardiographic characteristics of the Across-breeds cat cohort (n = 44) and Birman cat cohort (n = 28) are summarised in Tables 1 and 2, respectively. Due to sample limitations, we included two DSH control cats with a left ventricular wall thickness (LVWT) = 5.5 mm. Both cats were from a geriatric population (≥ 9 years old) with no previous signs of heart disease, all other cats included in the control population had an LVWT < 5.5 mm.
Genetic variant discovery and statistical analyses
The 18 candidate genes studied are listed in Table 3.
Study 1: Across-breeds
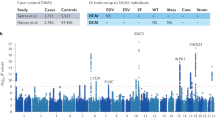
Targeted next-generation sequencing analysis revealed the presence of a total of 4025 variants, single nucleotide variants (SNVs) (3400) and indels (666) in the 18 candidate genes in the Across-breeds cohort. Details of the identified genetic variants are presented in Fig. 1.

Statistical analysis of variants identified in the Across-breeds cat cohort using targeted next-generation sequencing of 18 cardiomyopathy-related genes, annotated with the Ensembl Variant Effect Predictor tool. The gene selection was based on human cardiomyopathy literature.
Chi-squared comparisons
The annotation of the identified variants detected eight high-impact variants, including the known Ragdoll missense variant R820W in MYBPC315, which was included as a positive control. However, none of these variants had a significantly different prevalence between cases and controls when analysed both within and across the cat breeds.
Comparisons using the Chi-squared (χ²) test of the allelic and genotypic frequencies of the identified variants with a predicted moderate or modifier effect on the encoded protein identified a total of 160 variants with a significant (P ≤ 0.05, nominal P-value) difference between HCM cases and controls. 157 of these variants were intronic: 91/157 had a higher prevalence in cases, with 8/157 being present only in cases (see Supplementary Table 1 for full list of variants with a statistically significant difference). Four of these variants remained significant after correcting for multiple testing: one intronic variant (X:101006474 T > C, P = 0.0002) in lysosomal associated membrane protein (LAMP2) gene, one 3’UTR (F1:42194903 G > GGT, P = 0.025) located in TNNT2 gene and two missense variants (B3:76167563 C > T L88F, P = 0.003; D1:76804158 T > C I45V, P = 0.004) located within the novel gene ENSFCAG00000040035 (overlapping MYH7) and cysteine and glycine rich protein (CSRP3) gene respectively. The intronic SNV in LAMP2 was only present in control cats. The 3’UTR variant located in TNNT2 had a significantly higher genotypic frequency in cases. According to the Softberry FPROM human promoter predictor analysis close to this 3’UTR SNV there are two promoters. However, this SNV did not directly overlap with these promoter predictions. Moreover, this 3’UTR SNV exhibited a high effect allelic frequency (EAF) of 0.60 in the general cat population, as evidenced by data from the 99Lives Cat Whole Genome Sequencing (WGS) Initiative database (https://cvm.missouri.edu/research/feline-genetics-and-comparative-medicine-laboratory/99-lives/)30.
The two missense variants were present in both case and control cats with a higher prevalence in controls and were located within the novel gene ENSFCAG00000040035 (overlapping MYH7) (B3:76167563 L88F) and CSRP3 gene (D1:7680415 I45V) respectively. The CSRP3 gene missense variant has a reported ‘sorting intolerant from tolerant’ (SIFT) score of 0.34, which is considered ‘tolerated’. Analysis of the 99Lives cat WGS database revealed that both variants were prevalent in the general cat population.
Genomic association analysis
The genomic association analysis for the Across-breeds cohort which included age, sex and breed as covariates, identified one intronic variant in TNNT2 (F1:42199381 CA > C) significantly associated with HCM (P = 0.0006). This variant was exclusively present in affected individuals (n = 5). The Softberry NSITE analysis predicted that this variant affected the motifs that were able to bind to the nearby transcription factor binding sites (TFBS), with three additional motifs identified in the intronic sequence compared to the control cat population sequence (see supplementary Table 8). The Manhattan plot presenting the results of the genomic association analysis for HCM susceptibility in the Across-breeds cat cohort is shown in Fig. 2. Moreover, this intronic variant had a low frequency (EAF = 0.026) in the general cat population, as represented in the 99Lives cat WGS database analysis.

Manhattan plot presenting the results of the genomic association analysis for hypertrophic cardiomyopathy susceptibility in the Across-breeds cat cohort. Genomic location (x-axis) is plotted against -log10(P-value) (y-axis). The red line indicates the significance threshold (P ≤ 0.05) after multiple testing correction.
Study 2: Birman cats
The targeted next-generation sequencing analysis of the Birman cat cohort revealed the presence of 2298 genetic variants, SNVs (1921) and indels (395) across the candidate genes. Details of the identified genetic variants are presented in Fig. 3.

Statistical analysis of variants identified in the Birman pedigree cat cohort using targeted next-generation sequencing of 16 cardiomyopathy-related genes, annotated with the Ensembl Variant Effect Predictor tool. The gene selection was based on human cardiomyopathy literature.
Chi-squared comparisons
The annotation of the genetic variants revealed five high-impact variants within the Birman cohort. However, their frequency was not significantly different between case and control cats. When variants with a predicted moderate or modifier effect were compared using the χ² test, 177 variants were identified with significantly different frequencies (P ≤ 0.05, nominal P-value) between cases (HCM and RCM) and controls. Among these variants was a missense SNV located in the ENSFCAG00000040035 gene, resulting in a glutamic acid to glutamine substitution at position 22 (B3:76167365 G > C E22Q, P = 0.048). This variant was more prevalent in cases (heterozygous in five HCM and three RCM). Notably, this missense variant is synonymous with a variant in the MYH7 gene and overlaps with the microRNA miR-208B (ENSFCAG00000018043), previously implicated in heart disease31. The variant exhibited a higher frequency in the case population (EAF = 0.29) compared to both the control Birman population (EAF = 0.14) and the general cat population (EAF = 0.20), as represented by the 99Lives cat WGS database. Moreover, one 3’UTR SNV (D1:76785090, C > A, P = 0.03) within CSRP3 gene was identified only in control cats. This variant exhibited comparable frequencies (EAF = 0.14) in both the control Birman population and the 99Lives WGS general cat population. Nevertheless, the Softberry analysis did not reveal any further overlapping of this 3’UTR SNV with promoter regions. Fourteen intronic variants located in actin alpha cardiac muscle 1 (ACTC1), MYH7 and TNNT2 genes were more prevalent in cases (P ≤ 0.05). Specifically, variants in the ACTC1 gene exhibited an EAF of 0.18 in cases, while being absent in controls. Analysis of the 99Lives cat WGS database revealed these variants had an EAF of < 0.05 in the general cat population.
When cases were restricted to cats with HCM, the χ² comparisons revealed 153 variants that were significantly different (P ≤ 0.05) between cases and controls. Of these 153 variants, 117 had a higher prevalence in cases, with 78 intronic variants present only in cats with HCM (mainly spanning the genes ACTC1 and actinin alpha 2 (ACTN2)).
Full details of the identified variants in the HCM and RCM analyses and the HCM analyses are presented in Supplementary Tables 3 and 4, respectively. The majority of variants in the ACTN2 gene were identified exclusively in cases with an EAF of 0.13, while being absent in controls. Analysis of the 99Lives cat WGS database revealed these variants had a low EAF of < 0.05 in the general cat population.
Genomic association analyses
-
I.
Genomic Analysis: HCM Cases (n = 8) and Controls (n = 14).
Genomic association analysis, restricted to HCM cases, and including age and sex as covariates, identified 24 intronic variants with a significant association with HCM. The top intronic variant for each gene associated to cases were located within MYH7 (B3:76168426 G > A), ACTN2 (D2:12374576 C > T) and ACTC1 (B3:70080970 T > TC) genes. One intronic variant in ACTN2 (D2:12407434 GGGGT > G) showed a significant association with controls. Analysis of the 99Lives cat WGS database revealed that variants more prevalent in Birman HCM cases had lower EAFs in the general cat population. For MYH7 and ACTN2, the case EAFs were 0.38 and 0.31 respectively compared to a general population EAF of 0.13 for both variants. The ACTC1 variant exhibited a case EAF of 0.18 and a general cat population EAF of < 0.05. The Manhattan plot presenting these results is shown in Fig. 4.

Manhattan plot presenting the results of the genomic association analysis for hypertrophic cardiomyopathy in Birman cats. Genomic location (x-axis) is plotted against -log10(P-value) (y-axis). The red line indicates the significance threshold (P ≤ 0.05) after multiple testing correction.
-
II.
Genomic Analysis: HCM and RCM Cases (n = 14) and Controls (n = 14).
The genomic association analysis of the merged HCM and RCM phenotypes identified ten intronic variants with a statistically significant difference (P ≤ 0.05) between cases and controls, nine within CSRP3 and one within MYH7 gene. Analysis of the 99Lives cat WGS database revealed nine of the variants more prevalent in the Birman control population had a high EAF (> 0.55) in the general cat population. The one SNV (CSRP3 D1:76784404 G > A) more prevalent in Birman cases (EAF = 0.82) had a lower frequency (EAF = 0.26) in the general cat population. The Manhattan plot presenting these results is shown in Fig. 5.

Manhattan plot presenting the results of the genomic association analysis for hypertrophic cardiomyopathy and restrictive cardiomyopathy in Birman cats. Genomic location (x-axis) is plotted against -log10(P-value) (y-axis). The red line indicates the significance threshold (P ≤ 0.05) after multiple testing correction.
Discussion
We aimed to identify novel genetic variants associated with HCM and RCM in Birman cats, as well as across pedigree breeds and DSH cats. We performed two studies using a targeted cardiomyopathy gene panel on meticulously phenotyped cats. Although there are a few shared genetic variants associated with HCM resistance or susceptibility across cat breeds, the genetic architecture of the disease seems to be breed-specific.
Moreover, our studies identified high-impact variants within sarcomeric genes that were present at similar frequencies in both HCM cases and healthy controls. Similarly, several missense variants were present in both cases and controls, with no significant differences between the groups. These findings indicate that studies with appropriate sample sizes of cases and controls are required for the identification of truly causative HCM variants in cats. Nevertheless, we did identify a missense variant located on a novel gene ENSFCAG00000040035, which overlaps with MYH7 and a microRNA (miR-208). This missense variant (E22Q) had a significantly higher prevalence in HCM cases (P = 0.048) in our Birman cat cohort. The function of ENSFCAG00000040035 gene is unknown. In contrast, MYH7 is a sarcomeric gene that encodes beta-myosin heavy chain; a key protein component of the sarcomere involved in cardiac contractile function32. Of increased interest is the presence of this variant within miR-208 (overlapping with ENSFCAG00000040035 and MYH7), which is expressed in cardiac tissue and regulates the production of beta-myosin heavy chain during cardiomyocyte development33. MicroRNAs (miRNA) are non-coding RNA sequences which are crucial in biochemical pathway regulation including during cardiac development34. According to previous studies, genetic variants in miRNA genes can have profound effects on miRNA functionality at all levels, including miRNA transcription, maturation, and target specificity, and as such they can also contribute to disease35,36,37. Therefore miR-208 might prove to be an important biomarker in feline cardiac disease, especially since it has already been linked to myocardial infarction, hypertensive heart disease38,39 and dilated cardiomyopathy in humans31. Relationships between miRNAs and cardiac conditions such as left ventricular hypertrophy and fibrosis have already been shown across studies40,41, including evidence for other miRNAs that overlap with the MYH7 gene serving as biomarkers for human HCM42,43. Nevertheless, the role of microRNAs in feline HCM remains largely unknown. There is only one previous study that compared serum miRNA profiles between a healthy control cat cohort (n = 12) and cats with advanced HCM and documented clinical signs (ACVIM classification stage C) (n = 11)44. In that study, seven human HCM-associated miRNAs (causing cardiac tissue damage and disturbed blood flow) were upregulated in the HCM feline cohort44. These findings suggest that miRNAs might also play a role in feline HCM and could potentially be useful biomarkers as in humans. Notably, the miR-208 was not among the upregulated miRNAs within their feline cohort, suggesting our miR-208 variant may be specific to Birman pedigree cats. The missense variant (E22Q) in ENSFCAG0000004003 exhibited a higher prevalence in the 99Lives general cat population compared to our Birman study control group. This finding suggests the variant may be specifically associated with HCM and RCM in Birmans, comparable to breed-specific variants previously identified in Maine Coons and Ragdolls. The lower frequency in the Birman controls relative to the general population could indicate a breed-specific effect. Alternatively, the higher frequency within the 99Lives database may reflect the inclusion of cats predisposed to cardiomyopathies, given the lack of comprehensive echocardiographic phenotyping for the majority of samples in this database. Further studies in Birman cats are needed to validate these results and confirm the potential role of miR-208 and/or ENSFCAG00000040035 in feline HCM susceptibility.
Recent studies suggest intronic variants might play a more significant role in HCM susceptibility than previously thought45,46. Whole genome sequencing studies of human HCM have identified pathogenic intronic variants that disrupt splicing and TFBS45. In our study, we identified several intronic variants significantly associated with HCM susceptibility in cats, both across cat breeds and in the Birman cohort. Of particular interest was the intronic variant in TNNT2 (position F1:42199381) which was present only in HCM cases in the Across-breeds cat study and had a very low frequency in the general cat population. Softberry analysis predicted that this intronic variant affects the number of motifs in or around the TFBS which has the potential to influence the binding of transcription factors, and consequently gene regulation. Several variants in TNNT2 gene are associated with human HCM47. TNNT2 encodes cardiac troponin-T (cTnT) a cardiac specific protein forming part of the troponin complex associated with thin filaments. This protein plays a crucial role in cardiomyocyte contraction and relaxation through its interaction with actin and tropomyosin48. Further studies are needed to confirm whether this intronic variant plays a regulatory role and contributes to HCM susceptibility since Softberry tools currently provide predictions based on the human (and not the feline) genome assembly. Moreover, we found several intronic variants associated with HCM susceptibility both in the Across-breeds and Birman cat studies, however it is difficult to assess if they have a pathological impact. Further genomic and functional genomic studies with larger sample sizes are needed to shed light on the role of intronic variants in feline HCM susceptibility.
We investigated RCM alongside HCM in Birman cats following previous unpublished observations of cats within the same family being diagnosed with different cardiomyopathy phenotypes, as has been observed in affected human families49. There are significant similarities in disease presentation between HCM and RCM, with both conditions leading to diastolic dysfunction and increased end-diastolic pressure49, predisposing cats to heart failure. RCM in cats shares similar histological features with HCM; thus, it has been proposed that RCM could be part of the HCM spectrum50. An intronic variant in the CSRP3 gene was significantly associated with the combined group of HCM and RCM cases. The CSRP3 gene encodes the muscle LIM protein (MLP) of functional importance for calcium handling and signalling in cardiomyocytes51. Variants within this gene have been identified as causative for both human HCM and dilated cardiomyopathy52,53. Within our Birman cat cohort, the intronic variant was more prevalent in the control population and may have a protective role. Our results indicate the potential presence of shared protective variants against the phenotypic expression of RCM/HCM phenotype within Birman cats. To identify whether our intronic variant is playing a protective role and understand the exact mechanism behind this, further molecular and functional studies would need to be conducted, alongside validation in another case-control study of Birman cats. Future studies on Birman cats should include related individuals exhibiting varying cardiomyopathies. Our Birman cohort included related cats, however these consisted of only four cats in the control population and two HCM cats.
Future research with larger sample sizes across multiple breeds, including Birmans, will further elucidate the role of specific genes and genetic variants in HCM susceptibility. Increasing the sample size will enable separate analyses to be conducted within each individual breed and among the DSH population, potentially revealing additional genetic variants of interest. Furthermore, incorporating genes of recent interest, such as junctophilin 2 (JPH2)54, within future analyses could provide valuable insights into the genetic basis of HCM.
Conclusion
This study identified several genetic variants with a predicted high or moderate impact located in sarcomere candidate genes for feline HCM. However, these were not unique to affected cats, therefore were not considered causative of HCM. These findings support the need for genetic studies of adequate sample size to properly assess the role of genetic variants with a predicted high impact. Moreover, we identified several intronic variants with a significant association with HCM susceptibility or protection, highlighting that non-coding variants may also play a role in feline HCM. Furthermore, in Birman cats, we identified an exonic variant in a gene of unknown function (ENSFCAG00000040035) overlapping with MYH7 and a myocardium related microRNA (miR-208). Further studies are needed to characterise the role of this variant in HCM susceptibility within this breed. Our findings suggest that feline HCM likely follows a complex polygenic or oligogenic inheritance pattern. The shared genetic variants between HCM and RCM in Birman cats indicate a breed-specific phenotypic overlap. These results highlight important genomic regions for further investigation. Future research should focus on functional characterisation of identified variants and exploration of microRNA roles in feline HCM.
Materials and methods
Ethical approval
Ethical approval was received by the Clinical Research Ethical Review Board of the Royal Veterinary College (URN 2019 1942-3, URN 2016 1515, URN 2015 − 1378), the study followed all relevant guidelines and regulations. The study is reported in adherence to ARRIVE (https://arriveguidelines.org) guidelines.
Study population
Cats were recruited into the study following routine or invited echocardiographic screening for heart disease or following post-mortem examination conducted at the Royal Veterinary College (RVC). Of the cats included in the study total (n = 72), all were initially diagnosed with HCM through ante-mortem echocardiographic measurements. Subsequently 13 of these cats (two from the Birman cohort and 11 from the Across-breeds cohort) had their HCM diagnosis confirmed at necropsy. Cats without evidence of HCM or other cardiac disease (confirmed via echocardiography) and above the age of 9 years old were used as controls. Cats of any age with a confirmed HCM phenotype were used as cases. Among the Birman cohort, two control cats were littermates, two control cats shared the same dam, and two HCM cats shared the same dam.
Our work included two studies: (i) An Across-breeds cat study, totalling 44 phenotyped cats (controls = 23, HCM = 21) representing 21 non-pedigree cats (DSH) and pedigree breeds (4 Bengal, 8 British shorthair, 1 British longhair, 6 NFC, 3 Ragdoll, and 1 Maine coon). Among the HCM cases we included a Ragdoll cat confirmed homozygous for the HCM-associated MYBPC3 variant (R820W); (ii) A Birman pedigree cat study, totalling 28 phenotyped Birman cats (controls = 14, cases = 14, including 8 cats with HCM, and 6 cats with RCM).
Phenotyping
The cardiac phenotype was defined by echocardiography and/or gross pathology and histopathology, with owner consent. Echocardiography was performed by a board-certified veterinary cardiologist, or by a cardiology resident under the supervision of a board-certified veterinary cardiologist, using a Vivid E9 or Vivid I ultrasound machine (GE Systems, Hatfield, Hertfordshire, UK), with a 7.5 or 12 MHz phased-array transducer. Standard echocardiographic views were acquired, and video loops recorded9. All studies were measured off-line using dedicated echocardiographic software (EchoPac, GE Systems, Hatfield, Hertfordshire, UK).
On echocardiography, the thickness of the left ventricular free wall (LVFW) and interventricular septum (IVS) was measured by a leading edge to leading edge technique from a 2D right parasternal long-axis (RPLA) four- or five-chambered view, and a short-axis view at the papillary muscle level (RPSA). The thickest end-diastolic segment was averaged over three different cardiac cycles in each view (RPLA and RPSA). End-diastolic frames were defined as the first frame after mitral valve closure in RPLA and as the time point in the cardiac cycle of greatest left ventricular internal diameter in RPSA9. The greatest end-diastolic wall thickness of these measured views (RPLA septal, RPLA free wall, RPSA septal, RPSA free wall) was defined as LVWT and used for data analysis. Left atrial linear dimensions were measured as left atrial to aortic ratio (LA/Ao ratio) and left atrial diameter (LAD). The LA/Ao was measured as the ratio of the left atrium to aorta measured in 2D from an RPSA view at the heart base, in the frame after aortic valve closure55. The LAD was measured as the cranial-caudal LA dimension from a RPLA 4-chambered view, in the frame before mitral valve opening56. Left ventricular (LVFS%) fractional shortening was measured by M-mode from a right parasternal short-axis at the papillary muscle. Systolic anterior motion of the mitral valve (SAM) was assessed on colour Doppler and 2D echo from a right parasternal long-axis five-chamber view.
HCM was defined as LVWT ≥ 5.5 mm at end-diastole. Cases with concurrent disease that could contribute to LVH were excluded from the study. These conditions included systemic hypertension (systolic blood pressure > 160 mmHg)57, aortic stenosis or hyperthyroidism58. Healthy cats (control group) were defined as having a LVWT < 5.5 mm and aged ≥ 9 years old to minimise inclusion of cats with late onset HCM. Necropsy examinations were performed by a single trained observer (Lois Wilkie), and HCM was defined as a hypertrophied LV in the presence of myofiber disarray and interstitial/replacement fibrosis on histopathology59.
In the Birman cat cohort, in addition to HCM cases we also included RCM cases, defined as the presence of left or biatrial enlargement (left and/or right atrial diameter in RPLA view > 16 mm), LVWT ≤ 5.5 mm and normal left ventricular systolic function (LVFS% > 30%).
Post-Mortem Diagnosis of Hypertrophic Cardiomyopathy (HCM) and Restrictive Cardiomyopathy (RCM) in Cats
Macroscopic Evaluation
Gross pathology reports were reviewed for body weight, cardiac weight, and ventricular wall thicknesses. Measurements of right ventricular free wall (RVFW), interventricular septum (IVS), and left ventricular free wall (LVFW) were taken on a transverse cross-section, one-third from apex to base. The expected RVFW: IVS: LVFW ratio was 1:3:3, with relative cardiac weight reference range of 0.28–0.88% of body weight60. HCM was characterised by increased cardiac weight and LVH.
Histopathological Examination
Cardiac tissues were formalin-fixed, paraffin-embedded, and stained with haematoxylin-eosin and Masson’s trichrome60. A single observer blindly evaluated slides using the Olympus BX51TF microscope at magnifications ranging from x10 to x10060. A semiquantitative scoring system was followed assessing: fibrosis (interstitial, perivascular, replacement, subendocardial); intramural arteriolosclerosis; myocyte degeneration; inflammatory cell infiltrate and fat infiltration (absent/present); and myocyte hypertrophy and disarray (graded 0–3)60.
Blood and tissue collection and DNA extraction
Myocardial samples collected at necropsy were received from Birman breeders following death with suspicion of heart disease. For the Across-breeds cats, liver samples were obtained following routine necropsy examinations at the RVC. Residual blood (derived from clinical testing) was used for this project from blood collected by either a qualified veterinarian or veterinary nurse following echocardiography (using the same equipment and expert for each diagnosis) to exclude systemic diseases that could affect the heart and to measure cardiac biomarkers. DNA was extracted from whole blood/liver/myocardial samples using two commercial kits: DNeasy Blood and Tissue Kit (Qiagen®) and GeneJet Whole Blood Genomic DNA Purification Mini Kit (Thermo Scientific®) according to the manufacturers’ instructions. DNA quality and quantity were assessed using Denovix DS-11 Series spectrophotometer and Invitrogen Qubit 4 Fluorometer, respectively.
Feline HCM gene panel and targeted next-generation sequencing (tNGS)
We developed a gene panel for feline HCM and RCM based on candidate genes previously implicated in human cardiomyopathies (Table 3). This feline panel was equivalent to the Illumina TruSight Cardio Panel61 which is applied in suspected cases of human cardiomyopathy. In the first study (Across-breeds cohort) we included a panel of 18 candidate genes (Table 3). The same panel was used in the second study (Birman cohort) with the exclusion of two genes. These two genes were excluded due to limited variation, with no exonic variation being identified in these genes from the first study.
Targeted next-generation sequencing analysis
The raw sequencing data (FASTQ files) were assessed for quality control using FASTQC (v10.1)62 and trimmed to exclude adapter sequencing using Trimmomatic (v0.36)63 prior to mapping the reads on Felis Catus v9.0 genome assembly64 using the BWA aligner65. The matching variant file for the Felis Catus v9.0 genome assembly (Ensembl release version 95)66 was sorted against the reference dictionary to obtain known variant sites using Picard toolkit (v2.21.7)67. The reads were indexed, and duplicates removed using SAMTOOLS (v1.3)68. Base recalibration and variant calling to detect SNVs and indel variants were performed with the GATK (v.3.8) software69 using HaplotypeCaller70. Joint VCF files were created for cases and controls (for each study separately). Two separate VCF files for the Birman cases were created: one including both HCM and RCM cases and another only HCM. We ran a grouped analysis for cats with HCM and RCM phenotypes, as RCM has been suggested to be part of the HCM spectrum, i.e., these two phenotypes might represent diverse expressions of the same disease49,50,71,72. The SNV locations were obtained from Felis Catus v9.0 genome assembly using the Ensembl genome browser release version 9573. SNV annotation was performed using the Ensembl variant effect predictor (VEP) tool74.
The data from each study were analysed separately. Allelic and genotypic frequencies of genetic (SNV and indel) variants with a predicted high, moderate, or modifier functional impact according to VEP were compared between cases and controls to assess if there are statistically significant differences between the two groups. The Chi-squared test (χ2), with a significance level set at P ≤ 0.05 was used in this respect. A correction for multiple testing (0.05 divided by number of genes tested) was also applied.
To identify if any of the SNVs of interest in 3’UTR and other non-coding regions were located within a putative regulatory region we further interrogated these SNVs using Softberry software75. Specifically, to identify potential functional roles of our SNVs of interest we used BEDTools76 to extract SNV sequences 1500 bp either side of our SNV and ran comparisons against the corresponding 3000 bp sequence extracted from our sample containing the reference allele. These 3000 bp sequences were inputted into Softberry tools FPROM promoter predictor to look for predicted promoter regions in our significant 3’UTR SNVs and the NSITE tool to search for regulatory motifs in our 3’UTR and intronic regions77.
Genomic association studies
A bed format genotypic file was generated from the VCF file for cases and controls using the PLINK software (v1.90)78,79,80. Each of the datasets was subjected to quality control (qc) measures using the following thresholds: call rate < 90%, minor allele frequency < 0.05, and Hardy-Weinberg equilibrium P < 10− 6. A genomic relationship matrix was created for all animals using the GEMMA (v0.98.1) algorithm81. GEMMA was used to run the genomic association analyses for HCM susceptibility using a mixed model where the genomic relationship matrix was included as a random effect to account for possible population stratification and age, sex, and breed as fixed effects in the first study (Across-breeds cat cohort), and lambda correction applied to the P-values. The same model with the exclusion of breed as a fixed effect was used in the second study (Birman cat cohort). The significance level was set at P ≤ 0.05 and a Bonferroni correction for multiple testing was applied. Python382 in Jupyter notebook83 (for Mac OS) was used to create Manhattan plots to present the genomic analyses results.
Allelic frequencies for variants of interest in the general cat population
The allelic frequencies of genetic variants associated with HCM and RCM were assessed in the general cat population using the 99Lives feline whole genome sequences (WGS) database, which includes data from 341 cats from random bred and pedigree breeds (https://cvm.missouri.edu/research/feline-genetics-and-comparative-medicine-laboratory/99-lives/)30. This comprehensive genomic resource constructed from diverse sampling of domestic cats provides robust estimates of variant prevalence in the broader feline population30. Notably, phenotyping for HCM or RCM is not part of the inclusion criteria for this database. Given the prevalence of these diseases in the cat population, it is plausible that several individuals within this dataset could be genetically predisposed to HCM or RCM.
Data availability
Sequence data that support the findings of this study have been deposited in the Sequence Read Archive (SRA) repository with the BioProject accession code PRJNA1083230. Upon publication, the data will be made fully accessible to the public without restrictions through the SRA repository. The data are currently available in read-only format via the following link: https://dataview.ncbi.nlm.nih.gov/object/PRJNA1083230?reviewer=c2989md3eb80b925ug7pmk265u.
Abbreviations
- ACTC1 :
-
Actin Alpha Cardiac Muscle 1
- ACTN2 :
-
Actinin Alpha 2
- ALMS1 :
-
Alstrom Syndrome 1
- ARVC:
-
Arrhythmogenic Right Ventricular Cardiomyopathy
- CSRP3 :
-
Cysteine and Glycine Rich Protein 3
- DCM:
-
Dilated Cardiomyopathy
- DSH:
-
Domestic Shorthair
- HCM:
-
Hypertrophic Cardiomyopathy
- LA/Ao:
-
Left Atrium to Aorta Ratio
- LAD:
-
Left Atrial Diameter
- LVH:
-
Left Ventricular Hypertrophy
- LVWT:
-
Left Ventricular Wall Thickness at end-diastole
- MYBPC3 :
-
Myosin-Binding Protein C
- MYH7 :
-
Beta-Myosin Heavy Chain 7
- MYL2 :
-
Regulatory Myosin Light Chain
- MYL3 :
-
Essential Myosin Light Chain
- NFC:
-
Norwegian Forest Cat
- RCM:
-
Restrictive Cardiomyopathy
- SNV:
-
Single Nucleotide Variant
- tNGS:
-
Targeted Next-Generation Sequencing
- TNNI3 :
-
Troponin I3, Cardiac Type
- TNNT2 :
-
Troponin T, Cardiac Type
- TPM1 :
-
Tropomyosin 1
References
Wexler, R. K., Elton, T., Pleister, A. & Feldman, D. Cardiomyopathy: An overview. Am. Fam. Phys. 79, 778–784 (2009).
Payne, J. R. et al. Prognostic indicators in cats with hypertrophic cardiomyopathy. J. Vet. Intern. Med. 27, 1427–1436 (2013).
Luis Fuentes, V. et al. ACVIM consensus statement guidelines for the classification, diagnosis, and management of cardiomyopathies in cats. J. Vet. Intern. Med. 34, 1062–1077 (2020).
Ommen, S. R. et al. 2020 AHA/ACC Guideline for the diagnosis and treatment of patients with hypertrophic cardiomyopathy. J. Am. Coll. Cardiol. 76, 159–240 (2020).
Maron, B. J. et al. Prevalence of hypertrophic cardiomyopathy in a general population of young adults. Circulation 92, 785–789 (1995).
Marian, A. J. & Roberts, R. The molecular genetic basis for hypertrophic cardiomyopathy. J. Mol. Cell. Cardiol. 33, 655–670 (2001).
Lopes, L. R., Rahman, M. S. & Elliott, P. M. A systematic review and meta-analysis of genotype-phenotype associations in patients with hypertrophic cardiomyopathy caused by sarcomeric protein mutations. Heart 99, 1800–1811 (2013).
Semsarian, C., Ingles, J., Maron, M. S. & Maron, B. J. New perspectives on the prevalence of hypertrophic cardiomyopathy. J. Am. Coll. Cardiol. 65, 1249–1254 (2015).
Payne, J. R., Brodbelt, D. C. & Luis Fuentes, V. Cardiomyopathy prevalence in 780 apparently healthy cats in rehoming centres (the CatScan study). J. Vet. Cardiol. 17, 244–257 (2015).
Gersh, B. J. et al. 2011 ACCF/AHA guideline for the diagnosis and treatment of hypertrophic cardiomyopathy: Executive summary. J. Thorac. Cardiovasc. Surg. 124, 2761–2796 (2011).
Zamorano, J. L. et al. 2014 ESC Guidelines on diagnosis and management of hypertrophic cardiomyopathy. Eur. Heart J. 35, 2733–2779 (2014).
Maron, B. J. et al. Hypertrophic cardiomyopathy. J. Am. Coll. Cardiol. 64, 83–99 (2014).
Ho, C. Y. et al. Genetic advances in sarcomeric cardiomyopathies: State of the art. Cardiovasc. Res. 105, 397–408 (2015).
Meurs, K. M. et al. A cardiac myosin binding protein C mutation in the Maine Coon cat with familial hypertrophic cardiomyopathy. Hum. Mol. Genet. 14, 3587–3593 (2005).
Meurs, K. M., Norgard, M. M., Ederer, M. M., Hendrix, K. P. & Kittleson, M. D. A substitution mutation in the myosin binding protein C gene in ragdoll hypertrophic cardiomyopathy. Genomics 90, 261–264 (2007).
Schipper, T. et al. A feline orthologue of the human MYH7 c.5647G>A (p.(Glu1883Lys)) variant causes hypertrophic cardiomyopathy in a Domestic Shorthair cat. Eur. J. Hum. Genet. 27, 1724–1730 (2019).
Meurs, K. M. et al. A deleterious mutation in the ALMS1 gene in a naturally occurring model of hypertrophic cardiomyopathy in the Sphynx cat. Orphanet. J. Rare Dis. 16, 108 (2021).
Shenje, L. T. et al. Mutations in Alström protein impair terminal differentiation of cardiomyocytes. Nat. Commun. 5, 3416 (2014).
McNamara, J. W., Schuckman, M., Becker, R. C. & Sadayappan, S. A novel homozygous intronic variant in TNNT2 associates with feline cardiomyopathy. Front. Physiol. 11, 66 (2020).
Schipper, T. et al. The TNNT2:c.95–108G>A variant is common in Maine Coons and shows no association with hypertrophic cardiomyopathy. Anim. Genet. 53, 526–529 (2022).
Boeykens, F. et al. Classification of feline hypertrophic cardiomyopathy-associated gene variants according to the American College of Medical Genetics and Genomics guidelines. Front. Vet. Sci. 11, 66 (2024).
Bottillo, I. et al. Molecular analysis of sarcomeric and non-sarcomeric genes in patients with hypertrophic cardiomyopathy. Gene 577, 227–235 (2016).
Wu, W. et al. Novel phenotype-genotype correlations of restrictive cardiomyopathy with myosin-binding protein C (MYBPC3) gene mutations tested by next-generation sequencing. J. Am. Heart. Assoc. 4, 66 (2015).
Kubo, T. et al. Prevalence, clinical significance, and genetic basis of hypertrophic cardiomyopathy with restrictive phenotype. J. Am. Coll. Cardiol. 49, 2419–2426 (2007).
Olivotto, I. et al. Obesity and its association to phenotype and clinical course in hypertrophic cardiomyopathy. J. Am. Coll. Cardiol. 62, 449–457 (2013).
Lesurf, R. et al. Whole genome sequencing delineates regulatory, copy number, and cryptic splice variants in early onset cardiomyopathy. NPJ Genom. Med. 7, 18 (2022).
Vadgama, N. et al. De novo and inherited variants in coding and regulatory regions in genetic cardiomyopathies. Hum. Genomics 16, 1–20 (2022).
Ripoll Vera, T. et al. The R820W mutation in the MYBPC3 gene, associated with hypertrophic cardiomyopathy in cats, causes hypertrophic cardiomyopathy and left ventricular non-compaction in humans. Int. J. Cardiol. 145, 405–407 (2010).
Trehiou-Sechi, E. et al. Comparative echocardiographic and clinical features of hypertrophic cardiomyopathy in 5 breeds of cats: A retrospective analysis of 344 cases (2001–2011). J. Vet. Intern. Med. 26, 532–541 (2012).
Lyons, L. A. et al. Whole genome sequencing in cats, identifies new models for blindness in AIPL1 and somite segmentation in HES7. BMC Genomics 17, 1–11 (2016).
Satoh, M., Minami, Y., Takahashi, Y., Tabuchi, T. & Nakamura, M. Expression of microRNA-208 is associated with adverse clinical outcomes in human dilated cardiomyopathy. J. Card. Fail. 16, 404–410 (2010).
Schiaffino, S. & Reggiani, C. Fiber types in mammalian skeletal muscles. Physiol. Rev. 91, 1447–1531 (2011).
Malizia, A. P. & Wang, D. Z. miRNA in cardiomyocyte development. Wiley Interdiscip. Rev. Syst. Biol. Med. 3, 183–190 (2011).
Chiti, E., Di Paolo, M., Turillazzi, E. & Rocchi, A. MicroRNAs in hypertrophic, arrhythmogenic and dilated cardiomyopathy. Diagnostics 11, 66 (2021).
Paul, P. et al. Interplay between miRNAs and human diseases. J. Cell Physiol. 233, 2007–2018 (2018).
Hajjari, M., Mowla, S. J. & Faghihi, M. A. Editorial: molecular function and regulation of non-coding RNAs in multifactorial diseases. Front. Genet 7, 66 (2016).
Cammaerts, S., Strazisar, M., Rijk, P. . De. & Del Favero, J. Genetic variants in microRNA genes: impact on microRNA expression, function, and disease. Front. Genet. 6, 66 (2015).
Corsten, M. F. et al. Circulating MicroRNA-208b and MicroRNA-499 reflect myocardial damage in cardiovascular disease. Circ. Cardiovasc. Genet. 3, 499–506 (2010).
Agiannitopoulos, K. et al. Expression of miR-208b and miR-499 in Greek patients with acute myocardial infarction. In Vivo 32, 313–318 (2018).
Wronska, A., Kurkowska-Jastrzebska, I. & Santulli, G. Application of microRNAs in diagnosis and treatment of cardiovascular disease. Acta Physiologica 213, 60–83 (2015).
Fang, L. et al. Circulating microRNAs as biomarkers for diffuse myocardial fibrosis in patients with hypertrophic cardiomyopathy. J. Transl. Med. 13, 314 (2015).
Palacín, M. et al. Profile of MicroRNAs differentially produced in hearts from patients with hypertrophic cardiomyopathy and sarcomeric mutations. Clin. Chem. 57, 1614–1616 (2011).
Baulina, N. et al. Circulating miR-499a-5p is a potential biomarker of MYH7-associated hypertrophic cardiomyopathy. Int. J. Mol. Sci. 23, 3791 (2022).
Weber, K., Rostert, N., Bauersachs, S. & Wess, G. Serum microRNA profiles in cats with hypertrophic cardiomyopathy. Mol. Cell. Biochem. 402, 171–180 (2015).
Mendes de Almeida, R. et al. Whole gene sequencing identifies deep-intronic variants with potential functional impact in patients with hypertrophic cardiomyopathy. PLoS ONE 12, 66 (2017).
Vaz-Drago, R., Custódio, N. & Carmo-Fonseca, M. Deep intronic mutations and human disease. Hum. Genet. 136, 1093–1111 (2017).
Komamura, K. et al. The role of a common TNNT2 polymorphism in cardiac hypertrophy. J. Hum. Genet. 49, 129–133 (2004).
Gomes, A. V., Potter, J. D. & Szczesna-Cordary, D. The role of Troponins in muscle contraction. IUBMB Life 54, 323–333 (2002).
Vio, R. et al. Hypertrophic cardiomyopathy and primary restrictive cardiomyopathy: Similarities, differences and phenocopies. J. Clin. Med. 10, 66 (2021).
Fox, P. R., Basso, C., Thiene, G. & Maron, B. J. Spontaneously occurring restrictive nonhypertrophied cardiomyopathy in domestic cats: A new animal model of human disease. Cardiovasc. Pathol. 23, 28–34 (2014).
Geier, C. et al. Beyond the sarcomere: CSRP3 mutations cause hypertrophic cardiomyopathy. Hum. Mol. Genet. 17, 2753–2765 (2008).
Ehsan, M. et al. Mutant muscle LIM Protein C58G causes cardiomyopathy through protein depletion. J. Mol. Cell. Cardiol. 121, 287–296 (2018).
Fokstuen, S. et al. A DNA resequencing array for pathogenic mutation detection in hypertrophic cardiomyopathy. Hum. Mutat. 29, 879–885 (2008).
Ingles, J. et al. Evaluating the clinical validity of hypertrophic cardiomyopathy genes. Circ. Genom. Precis. Med. 12, 57–64 (2019).
Abbott, J. A. & MacLean, H. N. Two-dimensional echocardiographic assessment of the feline left atrium. J. Vet. Intern. Med. 20, 111–119 (2006).
Schober, K. E., Maerz, I., Ludewig, E. & Stern, J. A. Diagnostic accuracy of electrocardiography and thoracic radiography in the assessment of left atrial size in cats: Comparison with transthoracic 2-dimensional echocardiography. J. Vet. Intern. Med. 21, 709–718 (2007).
Taylor, S. S. et al. ISFM consensus guidelines on the diagnosis and management of hypertension in cats. J. Feline Med. Surg. 19, 288–303 (2017).
Bond, B. R., Fox, P. R., Peterson, M. E. & Skavaril, R. V. Echocardiographic findings in 103 cats with hyperthyroidism. J. Am. Vet. Med. Assoc. 192, 1546–1549 (1988).
Novo Matos, J. et al. Micro-computed tomography (micro-CT) for the assessment of myocardial disarray, fibrosis and ventricular mass in a feline model of hypertrophic cardiomyopathy. Sci. Rep. 10, 66 (2020).
Wilkie, L. J., Smith, K. & Luis Fuentes, V. Cardiac pathology findings in 252 cats presented for necropsy: A comparison of cats with unexpected death versus other deaths. J. Vet. Cardiol. 17, 329–340 (2015).
Pua, C. J. et al. Development of a comprehensive sequencing assay for inherited cardiac condition genes. J. Cardiovasc. Transl. Res. 9, 3–11 (2016).
Andrews, S. FastQC: A quality control tool for high throughput sequence data. Babraham Bioinformatics. https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (2010).
Bolger, A. M., Lohse, M. & Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120 (2014).
Washington University Genome Sequencing Center (GSC). Felis_catus_9.0 (GCA_000181335.4). Ensembl ftp://ftp.ensembl.org/pub/release-98/fasta/felis_catus/dna/Felis_catus.Felis_catus_9.0.dna.toplevel.fa.gz (2017).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 25, 1754–1760 (2009).
Ensembl. Felis_catus_variation_vcf. Ensembl http://ensembl.org/pub/release-95/variation/vcf/felis_catus/felis_catus.vcf.gz (2018).
Broad Institute. Picard Tools. Broad Institute. http://broadinstitute.github.io/picard/ (2019).
Li, H. et al. The sequence alignment/map format and SAMtools. Bioinformatics 25, 2078–2079 (2009).
Van der Auwera, G. A. & O’Connor, B. D. Genomics in the Cloud (O’Reilly, 2020).
Van der Auwera, G. A. et al. From FastQ data to high-confidence variant calls: The genome analysis toolkit best practices pipeline. Curr. Protoc. Bioinfor. 43, 66 (2013).
Angelini, A. et al. Morphologic spectrum of primary restrictive cardiomyopathy. Am. J. Cardiol. 80, 1046–1050 (1997).
Hirota, Y. et al. Spectrum of restrictive cardiomyopathy: Report of the national survey in Japan. Am. Heart J. 120, 188–194 (1990).
Howe, K. L. et al. Ensembl 2021. Nucleic Acids Res. 49, 884–891 (2021).
McLaren, W. et al. The ensembl variant effect predictor. Genome Biol. 17, 122 (2016).
Softberry Inc. Softberry: Search for promoters/functional motifs. Softberry Inc. http://www.softberry.com/berry.phtml?topic=index&group=programs&subgroup=promoter (2010).
Quinlan, A. R. & Hall, I. M. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842 (2010).
Solovyev, V. V., Shahmuradov, I. A. & Salamov, A. A. Identification of promoter regions and regulatory sites. Methods Mol. Biol. 674, 57–83 (2010).
Purcell, S. & Chang, C. PLINK version 1.90. COG-genomics. www.cog-genomics.org/plink/1.9/ (2023).
Chang, C. C. et al. Second-generation PLINK: Rising to the challenge of larger and richer datasets. Gigascience 4, 7 (2015).
Purcell, S. et al. PLINK: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 81, 559–575 (2007).
Zhou, X. & Stephens, M. Efficient multivariate linear mixed model algorithms for genome-wide association studies. Nat. Methods 11, 407–409 (2014).
McKinney, W. Data structures for statistical computing in python. In Proceedings of the 9th Python in Science Conference 56–61 (Austin, 2010). https://doi.org/10.25080/Majora-92bf1922-00a.
Kluyver, T. et al. Jupyter Notebooks—A publishing format for reproducible computational workflows. In International Conference on Electronic Publishing 87–90 (2016). https://doi.org/10.3233/978-1-61499-649-1-87.
Acknowledgements
The authors acknowledge the financial support provided by the Biotechnology and Biological Sciences Research Council (BBSRC) studentship. Funding was provided via the UK Research and Innovation (UKRI) funder award reference 1903448. The authors acknowledge the Petplan Charitable Trust for making the research possible through funding provided to projects S18-6930731 and S190735-774. The authors acknowledge the Winn Feline Foundation and the Birman Cat Club for their continued support of the research. The authors express gratitude to the team at the Queen Mother Hospital for Animals at the Royal Veterinary College. The authors thank Petros Syrris (University College London) for their guidance on human hypertrophic cardiomyopathy. The authors acknowledge Oliver Foreman (Animal Health Trust) for their help with devising the original candidate gene panel and co-ordinates.
Author information
Authors and Affiliations
Contributions
Jade Raffle (JR): Collected clinical data on study participants, performed the bioinformatic and statistical analysis and interpretation of the sequencing datasets, and prepared the manuscript. Jose Novo Matos (JNM): Provided expertise on feline cardiology, recruited study participants, performed sample DNA extractions for submission to outsourced sequencing, contributed to data collection and study design, and assisted in revising the manuscript. Androniki Psifidi (AP): Provided expertise on clinical genetics, guidance on the research design and data interpretation, assisted in revising the manuscript, and contributed to conceiving and designing the genetic study of HCM resistance and securing funding. Virginia Luis Fuentes (VLF): Provided expertise on feline cardiology, conducted echocardiographic assessments, assisted in revising the manuscript, and contributed to conceiving and designing the genetic study of HCM resistance and securing funding. David J Connolly (DJC): Provided expertise on feline cardiology, conducted echocardiographic assessments, assisted in revising the manuscript, and contributed to conceiving and designing the genetic study of HCM resistance and securing funding. Perry Elliott (PE): Provided guidance on the human cardiology genomic background and the development of the feline targeted HCM panel. Richard Piercy (RP): Assisted in revising the manuscript and contributed to conceiving and designing the genetic study of HCM resistance and securing funding. Marsha Wallace (MW): Provided access to the 99Lives feline whole genome sequencing database and calculated the allelic frequencies of the variants of interest in the general cat population. Lois Wilkie (LW): Provided comprehensive pedigree information for the Birman cat cohort and conducted post-mortem examinations on cats included in the study.All authors read and approved the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Raffle, J., Novo Matos, J., Wallace, M. et al. Identification of novel genetic variants associated with feline cardiomyopathy using targeted next-generation sequencing. Sci Rep 15, 3871 (2025). https://doi.org/10.1038/s41598-025-87852-5
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-87852-5
