
Abstract
Acinetobacter baumannii is an opportunistic Gram-negative pathogen responsible for various infections, such as those of the bloodstream and lungs, which often resist antibiotic therapy. In the course of an infection, the human innate immune system’s phagocytic cells are activated producing nitric oxide (NO) that cause bacterial injury. While the antimicrobial effects of nitrosative stress and the bacterial resistance mechanisms are well-characterized for several pathogens, the adaptations of Acinetobacter spp. to NO have not been studied. In this work, we define the transcriptional response of A. baumannii to nitrosative stress induced by NO donor exposure. A. baumannii triggers the expression of several transporters, including those involved in iron and siderophore synthesis. One of the most significantly NO-induced genes is a putative flavohemoglobin. The loss of function of this gene in the mutant strain led to decreased fitness of A. baumannii under NO stress. We also identified the A. baumannii nitric oxide sensor NsrR and demonstrated that NsrR regulates the hemoglobin gene. Combining biochemical, kinetic, and structural prediction studies we show that A. baumannii hemoglobin exhibits nitric oxide dioxygenase and reductase activities and has an atypical structural domain composition. Moreover, we reveal that Acinetobacter hemoglobins have evolved into an independent branch and are phylogenetically distant from other bacterial hemoglobins. Altogether, our findings demonstrate that A. baumannii hemoglobins represent a novel class of NO-detoxifying defense proteins that evolve from flavohemoglobin.
Similar content being viewed by others


Introduction
Acinetobacter baumannii infections can lead to various health problems, ranging from mild skin and soft tissue infections to pneumonia and severe bloodstream infections. It is often associated with the use of medical devices such as catheters and ventilators, including patients in intensive care units. A. baumannii can cause outbreaks, and mortality rates have increased with the emergence of multi-resistant and difficult-to-treat strains. This has resulted in A. baumannii inclusion in the World Health Organization (WHO) list of bacteria, for which new types of antibiotics are urgently needed1.
Pathogens thrive and spread successfully by adapting to the host environment and evading the innate immune system, the body’s primary defense mechanism. This also encompasses the cells that generate reactive nitrogen species (RNS) and reactive oxygen species (ROS) that have harmful effects. However, pathogens have specific systems to shield themselves from these challenges2. Among the most important nitric oxide (NO) detoxifiers are bacterial globins (Hb) that can be classified into three classes: flavohemoglobins (often designated as Hmp or FHb), truncated globins (TRHb), and single-domain globins (SDHb)3,4,5. Flavohemoglobins contain a globin domain, that binds a single heme molecule, and the NADPH reductase domain, which can be subdivided into FAD- and NADH-binding domains, and protect cells from NO6. Truncated globins are shorter versions that lack distinct structural elements found in conventional globins, and their functions are more variable, including a role in oxidative stress response7. Single-domain globins consist of only one globin domain and typically exhibit a compact tertiary structure. Some of the known functions include detoxification of ROS, NO metabolism, and sensing of environmental signals8. All these enzymes are found in various organisms including bacteria, archaea, and even some unicellular eukaryotes, with E. coli flavohemoglobin being particularly well-characterized9.
Nitrosative and oxidative stress are crucial in the immune system’s defense against bacterial pathogens. The response of Acinetobacter baumannii to these stresses has been well-documented. Host NADPH oxidase is important for clearing oxidative stress, and its deficiency impairs this process, increasing susceptibility to infection. However, A. baumannii possesses robust mechanisms to neutralize reactive oxygen species (ROS)10,11. UspA is essential for the bacterium’s ability to cope both oxidative and nutrient stress, as well as for its virulence. Moreover, catalase enzymes KatG and KatE serve a dual function, protecting the bacteria from oxidative stress while simultaneously making them more susceptible to immune-mediated killing12,13. A. baumannii also expresses OxyR, a key regulator of the H2O2 stress response14. Manganese (Mn) is critical for A. baumannii viability under oxidative stress, and the transcriptional regulator MumR contributes to survival by regulating specific metabolic pathways in response to H2O215. Additionally, disrupting the phenylacetate (PAA) pathway reduces the pathogen’s virulence, highlighting its role in adaptation to oxidative and antibiotic stress16. Finally, TrmB, a tRNA methyltransferase, is essential for A. baumannii survival during pulmonary infections17.
Although strategies leveraging nitric oxide (NO) as a bactericidal agent have been explored18, how A. baumannii strains counteract the deleterious effects of nitrosative stress remains essentially unclear.
In this work, we exposed A. baumannii to nitrosative stress and examined its response. The transcriptional pattern was affected with several genes having their expression modified, including a gene annotated as a putative flavohemoglobin. Phenotypic studies showed that this protein confers NO protection to the pathogen, and biochemical and structural studies revealed that this protein is a hemoglobin-like protein but with an unprecedent domain composition.
Results
Acinetobacter b aumannii response to nitrosative stress
Acinetobacter baumannii AB5075, recognized as a model strain due to its high capacity to induce disease and increased virulence in animal models19, was tested for its resistance to nitrosative stress. We exposed A. baumannii cells to a nitric oxide (NO) donor, specifically Spermine NONOate and observed that NO halted growth in a concentration dependent way (Fig. 1). The growth recovery occurred after ~ 2 h, indicating that A. baumannii has survival mechanisms for NO protection (Fig. 1).

Effect of nitric oxide on the growth of A. baumannii. A. baumannii AB5075-UW was grown aerobically in LB at 37 °C and 150 rpm, without NO treatment (triangle), and with the addition of Spermine NONOate at 50 µM (square), 100 µM (diamond), 150 µM (circle), and 200 µM (cross) after 1 h of growth. Error bars represent the standard deviation of at least three independent experiments.
We next examined the transcriptomic response of A. baumannii to Spermine NONOate by RNA sequencing (RNAseq), which analysis revealed that 339 genes exhibiting differential expression, comprising 191 upregulated and 148 downregulated genes (Fig. 2A-B and Supplementary Tables S4 and S5).
The RNAseq data was validated through quantitative real-time polymerase chain reaction (qRT-PCR), with the mean cycle quantification (Cq) values normalized relative to 16 S rRNA (see Methods). The genes were chosen based on fold change (FC) > 17 and ≤ 0.18 and compared with that observed in the RNAseq analysis. Figure 2C-D show that the gene expression levels exhibit a consistent correlation between the two assays (qRT-PCR and RNA-seq), confirming their differential expression after nitrosative stress exposure.
The functional categorization of genes with significant changes in expression levels (FC < 0.05) revealed that transporters comprise the largest group (Fig. 2A), with 21 genes upregulated and 16 genes downregulated. Notably, nitrosative stress influences cellular iron metabolism, as shown by the increase in the expression of genes encoding iron transporters and related with siderophore synthesis. In particular, NO induces the bauDCEBA polycistronic locus that is involved in the iron acquisition process mediated by acinetobactin, with acinetobactin being a catechol siderophore20. We also observed the upregulation of genes for Fe-S cluster biogenesis that suggests impairment of the Fe-S containing proteins or reduced iron availability. The induction of the transcriptional regulator responsible for activating copper tolerance gene cueR suggests the disruption of copper homeostasis upon NO treatment.
Among the highest upregulated genes is RS01970 that encodes a putative flavohemoglobin, and genes encoding nitrate and nitrite reductases. Furthermore, the induction of several genes encoding putative reductases and nitroreductases, such as RS09215, RS08715 and RS02215, suggests their potential role in detoxifying various nitrogen derived compounds.

A. baumannii transcriptional analysis in response to NO. (A) Functional classification of NO-responsive genes resulting from RNA-seq analysis. White and black bars indicate the number of genes with decreased and increased expression, respectively, in cells treated with 50 µM of Spermine NONOate for 15 min. (B) RNA-seq analysis. Statistically significant (FDR < 0.05) upregulated and downregulated genes are depicted in green and red, respectively. Horizontal dashed lines are placed at logFC = -1 and logFC = 1. The vertical dashed line is placed at average log counts per million = 3. (C-D) Quantitative real-time PCR (qRT-PCR) of selected upregulated genes (C) or downregulated genes (D) in A. baumannii AB5075-UW revealed by RNA-seq. Cells untreated (white dots) and exposed to Spermine NONOate (50 µM) (black dots) for 15 min exposure are represented. Relative gene expression was calculated by normalizing RS01970 Cq values with 16 S Cq values. Each value in the graph represents one independent sample. Statistical significance was determined using the Holm-Sidak method, with alpha = 0.1, without assuming a consistent SD. ***: p-value < 0.01; **: p-value < 0.05.
Within the downregulated genes, besides the ion transporters, putative amino acid permeases, outer membrane proteins, and ABC transporters, the genes associated with DNA metabolism, pilus, flagellum and secretion systems, chemotaxis, and heme biosynthesis were also repressed. Interestingly, the gene cgdC, which encodes coproporphyrinogen III decarboxylase involved in heme synthesis, was downregulated. This enzyme catalyzes the conversion of coproporphyrinogen III into protoporphyrinogen IX in the protoporphyrin-dependent pathway, suggesting repression of heme biosynthesis under NO stress conditions. Glutaredoxin and two distinct peroxiredoxins (putative thiol peroxidase and alkyl hydroperoxide reductase enzymes) were likewise repressed. These enzymes are linked with preserving a reducing cellular environment and are recognized for their pivotal role in defending against oxidative stress.
We focused our work on one of the highest induced genes RS01970 (FC ~ 32) to infer its importance in the protection of A. baumannii against NO.
RS01970 encodes a nitrosative stress protecting hemoglobin
Bacterial flavohemoglobins are typically linked with NO detoxification, employing NO dioxygenase activity to convert NO to nitrate under aerobic conditions21,22.
The high induction of RS01970 led us to investigate the phenotype of an A. baumannii strain with an insertion mutation in this gene (Table S1) when exposed to NO donors with different lifetimes. Figure 3A illustrates that the nitrosative stress caused growth impairment in both strains, but this effect was much more pronounced in the ΔRS01970 mutant. The utilization of a NO donor with a significantly extended half-life changed the growth pattern of the mutant in comparison to the wild-type. Under these conditions, the growth of the ΔRS01970 strain was halted completely in the presence of 500 µM of DETA NONOate, while that of the wild-type remained essentially unaffected (Fig. 3B).
To further ascertain the function of RS01970, we conducted complementation assays to test whether RS01970 could replace the major aerobic E. coli NO detoxifier flavohemoglobin. For that purpose, plasmids expressing RS01970 (254 amino acids) or a truncated version of RS019701–148 (that lacks the predicted FAD-binding-like domain, see below) were introduced in E. coli Δhmp and exposed to DETA-NONOate under aerobic conditions. Figure 3C shows that both strains remained as sensitive to the stress as the parental strain. Next, we investigated whether oxygen levels in the growth medium influenced the outcomes by conducting similar experiments under microaerobic conditions. In this case, the expression of RS01970 in Δhmp partially cancelled the sensitivity of this mutant strain to nitrosative stress, while the expression of RS019701–148 did not (Fig. 3D and Supplementary Figure S1). This indicates that RS01970 protects A. baumannii from nitrosative stress.

RS01970 protects A. baumannii from NO stress. (A-B) Growth of A. baumannii AB5075-UW (wt, circle) and its ΔRS01970 (triangle) derivative strain. Cells were grown aerobically in LB at 37 °C and 150 rpm. Both, Spermine NONOate (50 µM) (A) and DETA NONOate (500 µM) (B) were added after 1 h (red line) or not (black line). (C-D) Growth of E. coli BW25113 (wt, circle) and its Δhmp derivative strain carrying p0 (pFLAG-CTC empty plasmid, diamond), pHb (pFLAG-CTC expressing RS01970, triangle) or ptrHB (pFLAG-CTC expressing RS01970 truncated protein, square). Cells were grown in LB, under aerobic conditions (C) and microaerobic conditions (D), in the presence of DETA NONOate (500 µM), added at t0. Error bars represent the standard deviation of three independent biological samples.
A cinetobacter b aumannii RS01970 is regulated by a NsrR transcriptional regulator
Analysis of A. baumannii genome shows that RS01970 gene (FC ~ 32 in qRT-PCR assays) is adjacently located to a putative transcriptional regulator RS01965 (FC ~ 7.5) (Fig. 4A). Moreover, the upstream region of the RS01970 gene (located ~ 50 base pairs upstream of the ATG start codon) contains the NsrR-like binding sequence 5´-AAGATGTATTTTAAATACATCT-3´. This regulator exhibits 37% identity and 61% sequence similarity (89% sequence coverage) with E. coli NsrR (Fig. 4B). Bacterial NsrR is an iron-sulfur transcriptional regulator belonging to the Rrf2 family. It functions by binding to promoter regions and repressing the expression of genes related to nitrosative stress. Typically, high levels of nitrosative stress disrupt the iron-sulfur cofactor causing conformational changes in NsrR that prevent it from binding to promoter regions, thereby allowing the transcription of target genes to proceed23.
To gain insight into the regulatory NO mechanisms of A. baumannii, we tested RS01965 as a transcriptional regulator of the RS01970 protein. Firstly, we observed that the strain lacking the nsrR gene (ΔRS01965 strain) performed similarly in the presence and absence of the NO donor (Supplementary Figure S2). Next, the regulation of RS01970 by RS01965 was evaluated by qRT-PCR using total RNAs extracted from wild-type and ΔRS01965 strains that were harvested at the exponential growth phase. The relative expression of the RS01970 gene is approximately four orders of magnitude higher in the ΔRS01965 mutant strain compared to the wild-type strain (Fig. 4C), showing that in A. baumannii-NsrR is the regulator of RS01970.

RS01965 is a NsrR-like protein. (A) Schematic representation of genomic organization of A. baumannii AB5075-UWRS01970 locus. RS01950 (dicarboxylate/amino acid: cation symporter), RS01955 (metaldependent hydrolase), RS01960 (YihAfamily ribosome biogenesis GTP-binding protein), RS01965 (Rrf2 family transcriptional regulator), RS01975 (DUF853 domain-containing protein). (B) Multiple protein sequence alignment with selected bacterial NsrR: Pseudomonas aeruginosa (A0A069QAL4), Lactiplantibacillus plantarum (A0A0G9FFQ2), Streptomycescoelicolor (Q9L132), Bacillus subtilis (O07573), Moraxella catarrhalis (B7T0V2), Neisseria meningitidis (X5FBC9), Yersinia pestis (Q0WJT1), Escherichia coli (P0AF63), and Salmonella Typhimurium (A0A0F6BAN2) and RS01965 NsrR (Acinetobacter baumannii). Protein alignment was made using Clustal Omega with custom parameters and colored using Jalview (2.11.3.0). UniProt accession numbers are presented in parentheses. (C) Quantitative real-time PCR (qRT-PCR) of RS01970 gene in the wild-type strain (white circles) and ΔRS01965 strain (black circles). Relative gene expression was calculated by normalizing RS01970 Cq values with 16 S Cq values. Each value in the graph represents one independent sample. Statistical significance was determined using the Holm-Sidak method, with alpha = 0.1, without assuming a consistent SD. **: p-value < 0.05.
A. b aumannii RS01970 exhibits nitric oxide dioxygenase and reductase activity
In the A. baumannii genome, RS01970 is annotated as a flavohemoglobin. This type of bacterial proteins is characterized by containing a hemoglobin-like domain at the N-terminus that includes one b-type heme, and a ferredoxin-NADP + reductase-like domain at the C-terminus, which includes FAD- and NADPH-binding motifs. Although the amino acid sequence of A. baumannii RS01970 apparently contains the heme- and flavin-binding domains, it lacks the NADH reductase domain (Fig. 5). To confirm the presence of the cofactors, RS01970 was produced recombinantly in E. coli and characterized. The UV-visible spectrum of purified RS01970 confirmed the presence of a heme group as judged by absorbance maxima at 410, 540 and 575 nm of the oxidized form and their shift to 420, 525 and 560 nm upon reduction with sodium dithionite (Supplementary Figure S3). The heme content was estimated as 0.9 ± 0.3 heme molecules per protein. The presence of a flavin group was not detected in the as-isolated protein, as judged by the absence of the typical absorbance at 450 nm in the visible spectrum. To assess the capacity of RS01970 to bind FAD, we incubated the protein with an excess of FAD, removed the unbound flavin by passage in a desalting resin, and recorded the UV-visible spectrum. Despite several attempts, binding to FAD was never observed.
The nitric oxide dioxygenase (NOD) activity is catalyzed by canonical flavohemoglobins that typically possess FAD-binding and NADH reductase domains. Given that A. baumannii RS01970 lacks the NADH reductase domain, we utilized the E. coli NADH flavorubredoxin reductase (NorW) as the electron partner24. First, we analyzed the capacity of NorW to reduce A. baumannii RS01970. To do so, we incubated NorW with RS01970 under anaerobic conditions and followed the reduction of heme (A560nm) after addition of NADH. Our results indicated that reduction was not efficient, however in the presence of an excess of FAD in solution, RS01970’s heme reduction occurs more rapidly (Supplementary Figure S4A). Therefore, we proceeded to determine the NOD activity of A. baumannii RS01970 in the presence of NorW and FAD. We set up a reaction mixture with RS01970 (0.5 µM), E. coli NorW (1 µM), NADH (1 mM) and FAD (2 µM) and monitored the NO consumption rate. Under these conditions, a NO dioxygenase activity of 0.31 ± 0.09 heme− 1·s− 1 was determined (Supplementary Figure S4B). We also tested the role of the last 97 amino acid residues corresponding to the putative FAD-binding domain in the enzyme activity. The NOD activity of the truncated RS019701–148 protein, which was produced containing 1.1 ± 0.2 heme/protein, exhibited a NO consumption rate of 0.20 ± 0.06 heme− 1·s− 1.
In the subsequent assays, we evaluated the NO reductase activity, i.e., the conversion of NO into N2O under oxygen-free conditions of the RS01970 and RS019701–148 enzymes also using the E. coli NorW reductase. Values of 0.61 ± 0.09 heme− 1·s− 1 and 0.37 ± 0.09 heme− 1·s− 1 were obtained for RS01970 and truncated RS019701–148 protein, respectively.
Overall, our findings indicate that A. baumannii RS01970 is a NO-protecting hemoglobin (Hb), but not a flavohemoglobin and although the enzyme lacks an NADH reductase domain, it can receive electrons from a heterologous reductase. A comprehensive analysis of the A. baumannii genome indicates RS01970 is the sole hemoglobin of the pathogen.
The lack of a NADH reductase domain in A. baumannii hemoglobin suggests that it may contain an as-yet-unidentified reductase for this hemoglobin. The RNA-seq analysis showed that several reductases were overexpressed under nitrosative stress, notably RS09215, a FAD-dependent oxidoreductase that exhibits a fold change of ~ 139. Therefore, we hypothesized RS09215 as a potential reductase, which is further supported by the amino acid sequence similarity with E. coli NorW reductase (24% identity, 43% similarity and 74% coverage). The superimposition of the predicted model structure of A. baumannii RS09215 and E. coli NorW resulted in an RMSD between 123 pruned atom pairs is 1.25 angstroms; (across all 342 pairs: 8.232) (Supplementary Figure S5). Hence, we assessed the functionality of RS09215 as physiological reductase of A. baumannii hemoglobin. We constructed a plasmid containing the RS09215 gene sequence fused with an N-terminal His-tag, expressed and purified the protein. To ascertain the capacity of RS09215 to reduce A. baumannii hemoglobin, we incubated under anaerobic conditions A. baumannii hemoglobin (3 µM) with RS09215 (6 µM) in the presence of an excess of FAD (20 µM) and NADH (1 mM) and monitored the absorbance at 555 nm to follow the heme reduction. However, all assays showed that RS09215 is not able to reduce A. baumannii hemoglobin.
Acinetobacter b aumannii hemoglobin is structurally different from truncated and single-domain hemoglobins
Bacterial flavohemoglobins are formed by approximately 400 amino acid residues. Besides the heme located at the N-terminal, they typically feature binding sites for NADH and FAD in their C-terminal domain that are localized between amino acid residues 151–251 (FAD-binding domain) and 259–396 (NADH-binding domain) (E. coli flavohemoglobin numbering). The A. baumannii Hb has 254 amino acid residues corresponding to the length of the globin and an extra domain, but lacking the C-terminus region that in the flavohemoglobins corresponds to the NADH reductase domain (Fig. 5).

Amino acid sequence alignment of RS01970 hemoglobin with bacterial flavohemoglobins. Multiple protein sequence alignment of RS01970 hemoglobin (Acinetobacter baumannii) with and flavohemoglobins from Mycobacterium tuberculosis (Q7ARS9), Pseudomonas aeruginosa (Q9I0H4), Vibrio cholerae (Q9KMY3), Salmonella Typhimurium (A0A0F6B4X5), Escherichia coli (P24232), Aspergillus oryzae (Q2U124), Cupriavidus necator (Alcaligenes eutrophus, P39662), Staphylococcus aureus (Q2G2P2) and Saccharomices cerevisiae (P39676). The different domains are indicated with coloured lines: globin domain (red), FAD-binding domain (orange) and NADH reductase domain (blue). Conserved residues important for globin structure and FAD-binding are marked with red and orange asterisks, respectively. Trx-like FAD-binding domain is shown in an orange box. Protein alignment was made using Clustal Omega with custom parameters and colored using Jalview (2.11.3.0). UniProt accession numbers are presented in parentheses.
In flavohemoglobins, the FAD-binding motif is prevalent and consists of the residues Arg204, Gln205, Tyr206, and Ser207 (E. coli flavohemoglobin numbering). Also, Tyr188 is pointed as relevant for the orientation of the isoalloxazine ring3. In M. tuberculosis flavohemoglobin, a different FAD-binding motif is present, characterized by the Asp(x)8GlyxxPro sequence (starting from residue 250 in M. tuberculosis numbering) and resembling that of thioredoxin reductase3,25. However, none of these motifs are present in A. baumannii hemoglobin (Fig. 5). Moreover, when compared to known bacterial single-domain and truncated hemoglobins, A. baumannii hemoglobin is also unique in that it contains an extra stretch of approximately 120 amino acid residues. We also compared the primary sequence of A. baumannii hemoglobin with those of representative bacterial truncated and single-domain globins that contain around 130 amino acid residues. The set of highly conserved residues, which are essential for structural stability or heme interaction in canonical flavohemoglobins, is also present in A. baumannii hemoglobin (Fig. 6).

Amino acid sequence alignment of RS01970 hemoglobin with bacterial truncated and single-domain globins. Multiple protein sequence alignment with RS01970 hemoglobin (Acinetobacter baumannii) and truncated globins from Campylobacter jejuni (Q0PB48), Brazyrhizobium sp. (A0A4Y9ULR3), Staphylococcus aureus (A0A0D6HX52), Bacillus subtilis (O31607), Corynebacterium diphteriae (A0A0D6GTN5), Mycobacterium tuberculosis (P9WN23 TrHbO, P9WN25 TrHbN), Pseudomonas syringae (A0A2K4WQV2), Caulobacter crescentus (A0A290N176), Synechocystis sp. (P73925) and single-domain globins from Pseudomonas aeruginosa (Q9HX49), Vibrio parahaemolyticus (Q87H58), Aquifex aeolicus (O66586), Vireoscilla stercoraria (P04252), Campylobacter jejuni (Q0P842), Campylobacter coli (Q7WYM5), Gloebacter violaceus (Q7NKA6), Shewanella amazonensis (A1SBC4), Bradyrhizobium diazoefficiens (Q89RG5), Rhodopirellula baltica (Q7UWN2). Protein alignment was made using Clustal Omega with custom parameters and colored using Jalview (2.11.3.0) with only strictly conserved residues marked in black. UniProt accession numbers are presented in parentheses.
To investigate whether variances in the primary sequence of A. baumannii hemoglobin result in distinct structural conformations, we used Alphafold 3 to predict its structure26. The A. baumannii hemoglobin model exhibits a typical 3/3 helix fold, characteristic of flavohemoglobins, presenting a six-stranded β-barrel consisting of two domains, one of which is the globin domain (Fig. 7A). On the contrary, single-domain and truncated heme-containing globins are usually assembled into a 2/2 alpha-helical sandwich27.
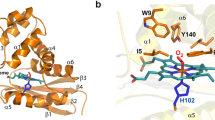
A. baumannii Hb globin domain exhibits the classical architecture in the proximal region of the heme pocket with the hydrogen-bonding network triad (His84, Tyr94, and Glu136), along with Tyr28 and Gln52 (Fig. 7A), which play roles in stabilizing heme-bound dioxygen3. The fold of the globin domain is very similar to that observed in the Vitreoscilla stercoraria single-domain globin (RMSD of 0.8 Å between the 128 C pairs, Fig. 7B) and to the E. coli flavohemoglobin28 (RMSD of 1.1 Å between the 198 C pairs, Fig. 7C). Interestingly, the structural model predicts an additional α-helix spanning residues Leu44 to Ser49, which is not present in the Vitreoscilla stercoraria structure.
Another structural feature that distinguishes the truncated and single-domain hemoglobin fold from the conventional globin fold lies in the vicinity of the heme proximal side27,29. However, A. baumannii hemoglobin shows considerable structural deviations in this region, which is reflected, for example, in the high RMSD value of the superimposition of the M. tuberculosis truncated hemoglobin with A. baumannii hemoglobin (RMSD of 8.3 Å considering 117 C pairs (Fig. 7D).

A. baumannii hemoglobin structural model and its comparison with flavohemoglobin, single domain and truncated globins. (A) AlphaFold 3 model structure for A. baumannii hemoglobin (green) showing the two domains and the relative position of the b-type heme. Highlight on the b-type heme and the conserved residues in its vicinity. (B-D) Structural superposition of AlphaFold 3 model structure for A. baumannii hemoglobin (green) with the Vitreoscilla stercoraria single domain globin (PDB1VHB, pink) (B); the E. coli flavohemoglobin (PDB1GVH, yellow) (C); and Mycobacterium tuberculosis truncated globin (PDB1IDR, gray) (D).
The second domain of the A. baumannii hemoglobin shares some structural similarity with the FAD domain of E. coli flavohemoglobin. However, when the structure of A. baumannii hemoglobin is modeled with an included FAD molecule, the cofactor assumes a different orientation compared to its position in E. coli flavohemoglobin (Fig. 8A). The electrostatic surface of E. coli flavohemoglobin shows a positively charged region that accommodates and orientates the phosphate groups of FAD molecule (Fig. 8B-C). This region is mainly due to the presence of Arg204, which despite being replaced by another positively charged residue in A. baumannii, Lys204, in this case its surroundings have a much more negative character due to the presence of many negatively charged amino acids (Asp45, Asp46, Glu224, Glu225, Asp226 and Glu229), which are not present in the E. coli flavohemoglobin. This results in a prediction of a different orientation of the FAD molecule in A. baumannii Hb model, where its phosphate groups are pointing to an alternative positively charged region (Arg51, Arg54 and Arg79), that in the E. coli protein is negatively charged (Asp52) (Fig. 8D). In addition to these factors, the presence of the NADH-binding domain in the E. coli flavohemoglobin structure most likely helps in protecting the FAD molecule from being completely exposed to the solvent. On the contrary, the absence of this domain in A. baumannii Hb would leave the FAD molecule exposed, potentially facilitating its labile behavior. These predicted structural features combined with the absence of the amino acids required for binding the FAD molecule may explain why A. baumannii Hb could not be isolated with this cofactor.

In silico prediction of the FAD position in the A. baumannii hemoglobin structural model. (A) Relative position of the model structure of A. baumannii hemoglobin’s FAD-binding domain predicted by AlphaFold 3 (green) superimposed with the E. coli flavohemoglobin’s FAD-binding and reductase domains (PDB1GVH, yellow). The relative position of the FAD molecule predicted for A. baumannii hemoglobin and the FAD molecule of the E. coli flavohemoglobin are shown. The b-type hemes of the two models are also shown for position clarification. Coulombic electrostatic potential calculated for E. coli flavohemoglobin (B) and A. baumannii hemoglobin (C) showing the electrostatic patterns in the vicinity of the FAD molecules. The Coulombic electrostatic potential surfaces were calculated and displayed in ChimeraX using ANTECHAMBER for charge calculation and AMBER Protein Force Fields54,55 and the results are presented in units of kcal/(mol.e) at 298 K (red, -20; blue, 30). (D) Highlight of the positively and negatively charged amino acids that contribute to the different Coulombic electrostatic potential pattern predicted for both models.
Phylogenetic analysis
We sought to explore the distribution of this novel hemoglobin type in bacteria and understand its evolutionary relationships by performing a phylogenetic analysis using our genomic dataset that comprises 33,959 genomes from 163 bacterial phyla and 1,350 genomes from 25 archaeal phyla30. We conducted a DIAMOND BLAST search using the A. baumannii hemoglobin protein sequence RS01970 as a query that was blasted against the DIAMOND database comprising the 35,309 genomes using cut-offs of 25% identity and an E-value lower than 10− 10. From these results we performed two different analyses. First, we filtered the results with a cut-off of 25% identity and an E-value lower than 10− 10. Second, since A. baumannii Hb consists of only 254 amino acid residues, we created a second set of sequences by using an additional filter for proteins between 200 and 300 amino acid residues in length. In this set and upon closer inspection of the alignment, different domain extensions were identified in several sequences. This is the case for instance of the cyanobacterial sequences, where, although having residues characteristic for the 3/3 alpha helical globin fold structure conserved, an additional well-conserved N-terminal domain is present that sets them apart from other globins (Fig. 9).
Maximum likelihood phylogenies were computed for both sets of data (Figs. 9 and 10) with the ultrafast bootstrap support values presented in Supplementary Datafiles. It is worth to mention that in this analysis, truncated globins as well as some of the single-domain globins or flavohemoglobins did not pass the cutoff of sequence identity (namely, Vibrio cholerae single-domain globin and Mycobacterium tuberculosis flavohemoglobin), and therefore were not included in this analysis.
In the phylogenetic reconstruction using globin sequences with lengths between 200 and 300, we observed that the root separates six long branched sequences, belonging to diverse taxonomic affiliations from the remaining globin sequences. Notably, in four out of those six sequences an additional N-terminal domain extension of around 39–65 amino acids is present, suggesting that they might perform a different function than the globins on the other side of the root (Fig. 9). Since those sequences are found in metagenomic assemblies, it also cannot be excluded that these N-terminus extensions are derived from sequencing artifacts. Thus, to determine the actual function of these proteins in vivo, further experimental studies are required.

Phylogenetic reconstruction of A. baumannii hemoglobin with proteins containing 200–300 amino acids. Maximum-likelihood reconstruction of hemoglobin (Using Q.pfam + I + G general Q matrix model). BLASTp DIAMOND results were filtered by protein length (200–300 aa), identity (> 25%), and E-value (< 10− 10). Black circles indicate ultrafast bootstrap support above 90. Clades are labelled with their respective taxon. Only bootstrap values for major split nodes are indicated. Green and blue zones, depict subclades from Clade 1, the branch containing Acinetobacter new hemoglobin type is colored in magenta as discussed in text and Fig. 10. The scale bar represents the number of substitutions per site.
The other side of the root comprises most of the globin sequences, including A. baumannii hemoglobin sequence, and it can be divided into several clades having five actinobacteria sequences as basal clade. The sequences from Acinetobacter species form a well-supported later branching clade (circled in Fig. 9) along with two sequences from Prolinoborus and Serratia, likely resulting from horizontal gene transfer. This clade is part of a larger green clade, indicating that A. baumannii hemoglobin orthologues are conserved in the Acinetobacter genus. This suggests that this protein derived from a single fusion event, specific to the Acinetobacter genus.
Having established that A. baumannii hemoglobin RS01970 orthologues are present in several Acinetobacter species and form a well-supported monophyletic clade, to further inspect the history of A. baumannii hemoglobin-like proteins in the context of globin evolution, a second phylogeny using all hits without the protein length filtering was performed (Fig. 10). The topologies of both phylogenies are similar and, as before, the root separates long-branch sequences from the remaining ones, where also two clades can be observed. Within the long-branch clade, both short-sequences and single-domain globins such as the ones from Bradyrhizobium and Rhodopirellula are found. Since sequences belonging to the same phylum are present on both sides of the root, this could indicate that the root separates different types of globins which may have a different function. However, since none of the globins within the long-branch clade is characterized, further experimental characterizations are needed to assign the in vivo function. On the other side of the root, the basal clade contains sequences from four different phyla (Actinobacteria, Nitrospirae, Proteobacteria and Verrucomicrobia), which might also represent a distinctive globin type. This subclade is also present in the small phylogenetic distribution (Fig. 9) but only represented by Actinobacteria sequences, since the others did not pass the sequence length cutoff. As in this clade both short and long sequences are found, this might indicate that either independent events of fission of the last domain, followed by a functional specialization, have occurred or instead the shorter proteins kept the original function as the longer ones, and the loss of the third domain, although possibly leading to lower activity, might be, at least partially, compensated by the saving in ATP for amino acid synthesis.
The remaining sequences in this clade are mostly classic flavohemoglobins, and those can be further split into two additional subclades, mimicking the ones found in Fig. 9. The remaining sequences within the green region can be divided into specific clusters of different types of flavohemoglobins. Within this subclade, we identified a large clade containing classical flavohemoglobin sequences exclusively from Actinobacteria. Furthermore, basal to this special clade are the classic flavohemoglobins protein sequences from Proteobacteria and Bacteroidetes, where one sequence belongs to the well-characterized Cupriavidus necator enzyme31 (Fig. 10). In the reduced phylogenetic representation (filtered by sequence length, Fig. 9) we found that Acinetobacter new type hemoglobin forms a specific subclade. The overall topology of the maximum-likelihood phylogeny containing all hits for the query not filtered by sequence length mirrors that of the phylogeny containing a subset of proteins with lengths ranging between 200 and 300 amino acids, as we observed that Acinetobacter sequences still cluster together forming a specific subclade (magenta branch in Fig. 9). However, in this analysis we also identified protein sequences from other Acinetobacter species that belong to classic flavohemoglobins and are basal to the A. baumannii Hb type subclade, which means that the novel Acinetobacter hemoglobin evolved from the classic flavohemoglobin within the same genus (Fig. 10). In the blue region, the characterized flavohemoglobin sequences from E. coli, S. enterica, and V. cholerae (Gammaproteobacteria) are found in early branches. Subsequently, another flavohemoglobin subclade, containing S. aureus and B. subtilis flavohemoglobins, is present. Within this subclade, we also found Vitreoscilla stercoraria and Campylobacter jejuni single-domain globins. These proteins co-cluster mostly with Epsilonproteobacteria and Clostridia, suggesting that Vitreoscilla acquired its single-domain globin from Clostridia via lateral gene transfer (LGT). In addition, a characterized single-domain globin from Aquifex aeolicus32,33, different from the other single-domain globins is also present in this clade. Interestingly, one single-domain globin from an archaeon of the Acidianus genus is also present in the blue clade, which is most likely due to an LGT event from Aquificae. Furthermore, this is the only archaeal sequence found in our analysis, and is found in a complete genome, thus indicating that it is likely to be a bona fide single-domain globin present in this organism. Since single-domain globins are found in two separate clades that stem from classic flavohemoglobins, this suggests that A. baumannii hemoglobin was formed by at least two events of single-domain globins evolving from classic flavohemoglobins.

Phylogenetic reconstruction of A. baumannii hemoglobin. Maximum-likelihood reconstruction of RS01970 (Using Q.pfam + R10). BLASTp DIAMOND results were filtered by identity (> 25%), and E-value (< 10− 10). The phylogeny is divided into Clades 1 and 2 by the root. Within Clade 2, major subclades are highlighted in green and blue. Specific clades containing only Actinobacteria or Acinetobacter are encircled with dotted lines. The branch containing the newly identified hemoglobin type is colored in magenta and marked with an asterisk. Organisms possessing experimentally identified flavohemoglobins and single-domain globins are depicted in blue and cyan, respectively. Red color are the phyla of the protein sequences present in Fig. 9. Clade containing Cyanobacterial sequences is also depicted in yellow. The scale bar represents the number of substitutions per site.
Discussion
In this work we determined the physiological and transcriptomic responses of A. baumannii to nitrosative stress and identified a nitric oxide-detoxifying enzyme acting in this human pathogen.
The limited role of nitric oxide synthase in host defense against A. baumannii18 aligns with our observation of the bacterium’s capacity to tolerate nitric oxide stress. We found that A. baumannii can withstand high concentrations of nitric oxide, whether generated by short-lived or long-lived NO donors. Moreover, the transcriptomic study, the first using NO stress conducted for an Acinetobacter species, revealed that the pathogen triggers a strong transcriptional response that supports its ability to overcome the deleterious effects of nitric oxide. A general stress response occurs, leading to the upregulation of genes involved in chemotaxis, chaperones, and DNA repair. Interestingly, genes encoding transporters and siderophores are among the two most upregulated groups, along with hemoglobin, nitrate and nitrite reductases, and nitroreductases. NO downregulated many genes, especially those involved in DNA metabolism, pilus and flagellum formation, secretion systems, chemotaxis, heme biosynthesis, as well as glutaredoxin and peroxiredoxins, which typically maintain a reducing intracellular environment and usually observed to be induced under NO stress conditions.
Previous studies on A. baumannii have only addressed the enzymes involved in ROS defense, such as superoxide dismutase and catalase, which are regulated by the ferric uptake regulator Fur. This regulation was reported to contribute to the reduced sensitivity of A. baumannii AB5075 to antibiotics, such as colistin, gentamicin, and rifampicin34,35. Moreover, hydrogen peroxide has been identified as a stressor produced in the lung during A. baumannii infection, and when A. baumannii is exposed to hydrogen peroxide differentially transcribes several genes that are under the control of the transcriptional regulator OxyR related with hydrogen peroxide detoxification14.
In this study we addressed the nitrosative stress response and its regulators, such as the RS01965 gene, which expresses an NsrR-type regulator. NsrR functions as a transcriptional repressor of the hemoglobin NO detoxifier gene, one of the highest NO induced genes. This hemoglobin, which displays nitric oxide dioxygenase activity, is the only globin-encoding protein found in this pathogen’s genome. Furthermore, among the genes highly upregulated by nitrosative stress are those that encode nitrate and nitrite reductases (Supplementary Table S4). This finding suggests their involvement in detoxifying the nitrate and nitrite by-products produced by the activities of hemoglobin and nitrate reductase, respectively.
We observed that the genetic inactivation of hemoglobin in A. baumannii decreases the resistance of the strain to NO which is consistent with the NO dioxygenase and NO reductase activities of the recombinant hemoglobin. The A. baumannii hemoglobin exhibits a specific NOD activity of 0.31 ± 0.09 heme− 1·s− 1. This value is 100-fold lower when compared to the E. coli flavohemoglobin (Hmp) NOD activity (90 heme− 1·s− 1) as reported by (Gardner et al., 2000). However, the NOD activity determined so far are very different amongst globins, from 0.05 heme− 1·s− 1 of the truncated hemoglobin of Mycobacterium tuberculosis36 to the 3.9 and 7.4 heme− 1·s− 1 determined by Gardner and coworkers for the Saccharomyces cerevisiae and Ralstonia eutropha (formerly Alcaligenes eutrophus), respectively37. Pathania and colleagues measured the NOD activity of recombinantly expressed E. coli Hmp, Vitreoscilla VHb, and M. tuberculosis HbN in E. coli cell extracts. Cells expressing Hmp showed an NO consumption rate ~ 10 and 100-fold higher than the cell extracts containing VHb and HbN, indicating a difference between Hmp and the truncated and single-domain globins. Although our results suggest that A. baumannii Hb has a turnover level comparable to that of truncated hemoglobin of Mycobacterium tuberculosis36, we cannot underestimate that the use of the non-physiological E. coli NorW, despite its capacity to transfer electrons to A. baumannii Hb, may result in a reduced enzyme’s activity due to a less effective electron transfer, as was previously observed with other enzymes38. Also, it is noteworthy to state that cell extracts containing a truncated version of Ralstonia eutropha, without the reductase domain, presented a ca. 4-fold reduction in its NOD activity than similar extracts containing the wild-type enzyme39, which indicates a relevant role of this domain in the enzyme’s activity.
The electron transfer experiments between the electron donor, NADH and A. baumannii Hb also showed that the reaction is faster in the presence of an excess of FAD. However, both the full-length A. baumannii Hb and the enzyme containing only the globin domain, i.e., the truncated version A. baumannii RS019701–148, exhibited a NOD activity in the same range (0.20 ± 0.06 µM NO·heme− 1·s− 1). These findings suggest that FAD functions not as the physiological mediator but as a simple mediator. In fact, most of the published NOD activity assays with the different globins are performed in the presence of an excess of FAD37,40. Despite our attempts, at this stage, it was not possible to identify the physiological electron donor which will require further studies. On the other hand, the NO reductase activity determined for A. baumannii hemoglobin and its truncated version, 0.61 ± 0.09 heme− 1·s− 1 and 0.37 ± 0.09 heme− 1·s− 1, respectively, are in line with the previously obtained for the flavohemoglobin of Staphylococcus aureus (0.7 s− 1)41 and ca. 10-fold higher than the determined for Saccharomyces cerevisiae, Ralstonia eutropha and Escherichia coli (0.02, 0.03 and 0.12 heme− 1·s− 1)37.
The primary sequence and structural model of A. baumannii hemoglobin, when compared with other studied flavohemoglobins, single-domain, and truncated hemoglobins of bacteria, exhibit several differences. A. baumannii hemoglobin is formed by two domains, the canonical globin domain and a C-terminal sequence. In relation to flavohemoglobins, it does not have the residues commonly forming the FAD-binding motif. Moreover, it displays different electrostatic charge surface in the region that usually harbors the FAD molecule in flavohemoglobins. Simulation of the binding of FAD predicts a position of the molecule in a more solvent-exposed conformation (Fig. 8). The absence of FAD in the heterologously produced A. baumannii Hb also supports that the cofactor is either very labile or does not bind at all to the polypeptide chain as a recognizable binding motif is absent.
The hemoglobin of A. baumannii is smaller than the canonical flavohemoglobin as it does not have the reductase domain. It is also distinct from single-domain and truncated hemoglobins in that it has an extra C-terminal sequence of ~ 120 amino acid residues (Fig. 11). Although the model structure of A. baumannii hemoglobin folds like the classic 3/3 alpha-helical sandwich flavohemoglobins, the electrostatic surfaces of the A. baumannii hemoglobin and E. coli flavohemoglobin are different. This is probably because the E. coli enzyme functions as a single entity involving intramolecular electron transfer, whereas A. baumannii hemoglobin requires intermolecular electron transfer from another protein. In the latter, the C-terminal region is predicted to be required for the binding of the electronic mediator and/or the correct coupling with the physiological reductase. Furthermore, this feature makes A. baumannii hemoglobin also unique compared to truncated and single-domain hemoglobins, which do not have this extra protein sequence that in A. baumannii is functionally relevant.

Different types of globins. Graphical representation of the protein domains constituting the different globin types identified: flavohemoglobin, truncated globin and single-domain globin. A. baumannii bacterial hemoglobin contains a different C-terminal domain, of unknown function, similar to the FAD-binding domain of the classical flavohemoglobin but lacking the ability to bind FAD.
We did a throughout phylogenetic analysis using a very large database of 33,959 microbial genomes that allowed us to find around 900 sequences in the phylogeny, which is a much larger study with a different taxonomic distribution than the one recently published42 where A. baumannii hemoglobins were not identified as a separated branch. The generation of a maximum-likelihood phylogenetic trees highlights the uniqueness of A. baumannii hemoglobin as a phylogenetic different type of hemoglobin. Our data indicates that this new type of domain organization (Fig. 11) originated once within the genus Acinetobacter, as seen by the monophyletic organization of the clades in both phylogenetic reconstructions performed. Additionally, the ancestor of A. baumannii hemoglobin likely evolved from classical Acinetobacter flavohemoglobins, potentially due to a random mutation that resulted in the loss of the terminal domain. Future work will aim to clarify how this new type of hemoglobin constitutes an advantage for A. baumannii spp. fitness when compared to other bacterial hemoglobins.
Material & methods
Bacterial growth and exposure to nitrosative stress
The strains and plasmids used in this work are listed in Supplementary Tables S1 and S2. A. baumannii AB5075-UW and E. coli strains were grown under aerobic conditions in Luria-Bertani (LB) broth at 37 °C with agitation (150 rpm) and supplemented with antibiotics when indicated (50 mg mL− 1 of ampicillin or 50 mg mL− 1 of kanamycin). Cells were treated with two NO donors using freshly prepared stock solutions of Spermine NONOate (half-life of 39 min at 37 °C) and DETA-NONOate (half-life of 20 h at 37 °C) at 50 mM in 0.01 M NaOH using the indicated concentrations.
For the complementation assays, E. coli BW25113 wild-type (wt) and Δhmp strain were transformed, separately, with pFLAG-CTC empty plasmid and plasmids expressing A. baumannii RS01970, or a truncated form that lacks the last 106 amino acid residues (RS019701–148). For the aerobic complementation, cells were grown in Erlenmeyer flasks with agitation. Microaerobic conditions were attained by employing sealable flasks (50 mL), which were fully filled with culture and tightly sealed. DETA-NONOate (500 µM) was added at t0, and OD600 was monitored over time up to 6 h.
RNA extraction and quantitative real-time PCR (qRT-PCR)
Overnight cultures of A. baumannii AB5075-UW were diluted 1:100 in 50 mL of fresh LB medium, grown for 1 h, and treated with Spermine NONOate (50 µM). Following exposure for 15 min, cells were harvested by centrifugation (11,000 g, 10 min, 4 °C), resuspended in RNAlater (Sigma), and incubated at RT for ~ 4 h. Cells were centrifuged (11,000 g, 10 min, 4 °C), frozen in liquid nitrogen and stored at -20 °C. Cells manipulated in a similar way but not treated with the NO donor were used as controls. For each condition, we analyzed three independent biological samples.
RNA extraction was done using the Aurum Total RNA Mini Kit DNA-free RNA isolation kit (Bio-Rad), and DNA contaminants were removed using turbo DNA-free kit (Invitrogen). cDNA was synthesized from 1 µg of total RNA using Transcriptor High Fidelity cDNA synthesis kit (Roche Applied Science). qRT-PCRs were done in a LightCycler® 96 instrument using the LightCycler FastStart DNA Master SYBR green I kit (Roche Applied Science). All amplification reactions were carried out with the same amount of cDNA (10 ng) as the initial template, using specific pairs of oligonucleotides that were designed to amplify an internal region of 100 to 300 bp for each target gene (Supplementary Table S3). Assays were done for three biologically independent experiments, each with two replicates. Statistical analysis was performed using GraphPad Prism 8.0.2.
RNA sequencing analysis
Total RNA was depleted from rRNA using the species-specific (Bacteria) ribosomal RNA depletion Kit and examined in an Illumina Novaseq platform (150 bp, 20 M reads) (provided by StabVida, Portugal). The quality of the RNA-Seq raw data was analyzed using the FastQC tool, and reads were mapped into A. baumannii AB5075-UW genome (https://www.ncbi.nlm.nih.gov/datasets/genome/GCF_000963815.1/ downloaded from NCBI genome database) using Bowtie2 program43. The mapping files were sorted by genomic position using Samtools, and the quantification of transcript’s expression was done using FeatureCounts software44,45. Differential expression analyses were done with the R package edgeR46. All transcripts were considered as significant with a false discovery rate (FDR) correction of the p-value lower than 0.05. A further filtering of the results was performed by establishing cut-offs of expression (log2CPM – counts per million) higher than 3 and fold changes higher than 2. The functional annotation was performed with DAVID47,48.
Protein purification and heme quantification
Plasmids pET28a-His-RS01970 and pET28a-His-RS019701–148, pET28a-His-RS09215 expressing RS01970, truncated RS019701–148, and the reductase RS09215, respectively, were transformed into E. coli BL21DE3 cells. Overnight cultures were diluted 1:100 in fresh LB/kanamycin, and a ratio 2/5 of liquid medium/flask volume was used. For RS01970 and RS019701–148 expression, aminolevulinic acid (50 µM), and ammonium iron (II) sulfate (50 µM) were added. For expression of RS09215, the medium was supplemented with riboflavin (50 µM). Cells were grown to an OD600 ~ 0.4, at 37 °C and 150 rpm. At this stage, the expression of the protein was induced by the addition of isopropyl β-D‐1‐thiogalactopyranoside (IPTG) (500 µM), and growth proceed for 16–20 h at 20 °C and 90 rpm. Cells were pelleted by centrifugation (11,000 g, 10 min, 4 °C), resuspended in 20 mM Tris-HCl pH 8 (Buffer A), disrupted in a French Press at 1,000 psi, and centrifuged (42,000 g, 40 min, 4 °C). The supernatant was applied onto a chelating Sepharose fast flow column (Cytiva) charged with Ni2+, fractions eluted with Buffer A with 50 mM imidazole, dialysed iusing membranes Spectra/Por®, and further purified in a Hitrap QFF column (Cytiva). The fractions containing wild-type and truncated RS01970 were eluted with Buffer A with 200 mM of NaCl, concentrated, and buffer exchanged in PD-10 column using 20 mM Tris 10% glycerol (Buffer B). The ratio heme/protein was quantified by measuring the absorption spectra of pyridine hemochrome49. NADH: flavorubredoxin oxidoreductase (NorW) from E. coli was produced and purified as previously described24.
Enzymatic assays
In the nitric oxide dioxygenase (NOD) and nitric oxide reductase (NOR) assays, the NO consumption was measured amperometrically with a modified Clark-type electrode, NO selective (ISO-NOP, World Precision Instruments, Sarasota, FL). Stock solutions of 1.9 mM NO were prepared by saturating the buffer with NO gas as previously described50. As the physiological electron donor of hemoglobin RS01970 is not known, we used E. coli NorW as an alternative reductase system. The NOD activity was measured at room temperature in a reaction mix containing buffer B, 1mM NADH, 2 µM FAD, 1 µM NorW and 6 µM NO, using the indicated enzyme concentrations. The NOR activity was determined under anaerobic conditions in buffer B and in the presence of an O2-scavenging system (10 mM glucose, 375 nM glucose oxidase and 750 nM catalase). Sequential additions of NO (up to 12 µM) were followed by the addition of 2 µM NorW and 1 mM NADH. For both NOD and NOR assays, the reactions were initiated by adding the enzyme (0.5 µM).
Amino acid sequence comparisons, structural and phylogenetic analyses
Alignments were done in the ClustalOmega website with predefined parameters51,52 and colored using Jalview (2.11.3.0) (https://www.jalview.org/).
The three-dimensional structure model of A. baumannii hemoglobin was predicted using AlphaFold 3 and considering the presence of a b-type heme and an FAD moiety26,53. Structural analysis, visualization and images were performed using UCSF ChimeraX 1.8 54,55,56. Electrostatic surfaces were calculated and displayed in ChimeraX using ANTECHAMBER for charge calculation and AMBER Protein Force Fields57,58.
For the phylogenetic analysis, we created a subset of our internal dataset, which originally comprises over 190,000 metagenomic assemblies obtained from NCBI in November 2019, with two Acidianus ambivalens assemblies added later30. This subset was generated by filtering genomic records based on previously mapped NCBI taxonomic information and genomic quality criteria, i.e., completeness and contamination calculated with the Rinke method59. All NCBI reference and representative genomes were retained. Additional genomic records were included, prioritizing complete genomes and higher quality assemblies. The final genomic dataset comprised 35,309 (meta)genomic assemblies, comprising 33,959 genomes from 163 bacterial phyla and 1,350 genomes from 25 archaeal phyla. Similarity searches were carried out using DIAMOND BLASTp (v2.0.15)60,61 in “ultra-sensitive” mode using “-k 0”. The protein sequence RS01970 was used as a query and was blasted against the DIAMOND database comprising the 35,309 genomes using cut-offs of 25% identity, and an E-value lower than 10− 10.
A maximum likelihood phylogenetic reconstruction was carried out using IQ-TREE considering 1000 ultrafast bootstraps, 1000 max iterations, 0.99 minimum correlation coefficient, 0.5 perturbation strength, 100 IQ-tree stopping rule, bnni (Optimize UFBoot trees by NNI on bootstrap alignment) and safe (Safe likelihood kernel to avoid numerical underflow) options activated62. The phylogenetic reconstruction was rooted by using the minimal ancestor deviation (MAD) method (version 2.2263), with a modified script to keep bootstrap values, kindly provided by Giddy Landan. FigTree was used for annotations and analysis (v1.4.4, http://tree.bio.ed.ac.uk/software/figtree/).
Data availability
The datasets generated and/or analysed during the current study are available in the Figshare repository, 10.6084/m9.figshare.27194004. The data will be made available upon request to Prof. Lígia Saraiva.
References
WHO publishes list of bacteria for which new antibiotics are urgently needed. https://www.who.int/news/item/27-02-2017-who-publishes-list-of-bacteria-for-which-new-antibiotics-are-urgently-needed
Bogdan, C. Nitric oxide and the immune response. Nat. Immunol. 2, 907–916 (2001).
Frey, A. D. & Kallio, P. T. Bacterial hemoglobins and flavohemoglobins: versatile proteins and their impact on microbiology and biotechnology. FEMS Microbiol. Rev. 27, 525–545 (2003).
Gell, D. A. Structure and function of haemoglobins. Blood Cells Mol. Dis. 70, 13–42 (2018).
Hausladen, A., Gow, A. J. & Stamler, J. S. Nitrosative stress: metabolic pathway involving the flavohemoglobin. Proc. Natl. Acad. Sci. U S A. 95, 14100–14105 (1998).
Forrester, M. T. & Foster, M. W. Protection from nitrosative stress: a central role for microbial flavohemoglobin. Free Radic Biol. Med. 52, 1620–1633 (2012).
Nardini, M., Pesce, A., Bolognesi, M. & Truncated 2/2) hemoglobin: unconventional structures and functional roles in vivo and in human pathogenesis. Mol. Asp Med. 84, 101049 (2022).
Vinogradov, S. N., Tinajero-Trejo, M., Poole, R. K. & Hoogewijs, D. Bacterial and archaeal globins - a revised perspective. Biochim. Biophys. Acta. 1834, 1789–1800 (2013).
Vinogradov, S. N. et al. Microbial eukaryote globins. Adv. Microb. Physiol. 63, 391–446 (2013).
García-Quintanilla, M., Pulido, M. R., López-Rojas, R., Pachón, J. & McConnell, M. J. Emerging therapies for multidrug resistant Acinetobacter baumannii. Trends Microbiol. 21, 157–163 (2013).
Sato, Y., Unno, Y., Miyazaki, C., Ubagai, T. & Ono, Y. Multidrug-resistant Acinetobacter baumannii resists reactive oxygen species and survives in macrophages. Sci. Rep. 9, 17462 (2019).
Elhosseiny, N. M., Amin, M. A., Yassin, A. S. & Attia, A. S. Acinetobacter baumannii universal stress protein A plays a pivotal role in stress response and is essential for pneumonia and sepsis pathogenesis. IJMM 305, 114–123 (2015).
Sun, D. et al. KatG and KatE confer Acinetobacter resistance to hydrogen peroxide but sensitize bacteria to killing by phagocytic respiratory burst. Life Sci. 148, 31–40 (2016).
Juttukonda, L. J. et al. Acinetobacter baumannii OxyR regulates the transcriptional response to hydrogen peroxide. Infect. Immun. 87, e00413–e00418 (2018).
Green, E. R., Juttukonda, L. J. & Skaar, E. P. The manganese-responsive transcriptional regulator MumR protects Acinetobacter baumannii from oxidative stress. Infect. Immun. 88, e00762–e00719 (2020).
Hooppaw, A. J. et al. The phenylacetic acid catabolic pathway regulates antibiotic and oxidative stress responses in Acinetobacter. mBio 13, e0186321 (2022).
McGuffey, J. C. et al. The tRNA methyltransferase TrmB is critical for Acinetobacter baumannii stress responses and pulmonary infection. mBio 14, e0141623 (2023).
Qiu, H., Kuolee, R., Harris, G. & Chen, W. Role of NADPH phagocyte oxidase in host defense against acute respiratory Acinetobacter baumannii infection in mice. Infect. Immun. 77, 1015–1021 (2009).
Jacobs, A. C. et al. AB5075, a highly virulent isolate of Acinetobacter baumannii, as a model strain for the evaluation of pathogenesis and antimicrobial treatments. mBio 5 https://doi.org/10.1128/mbio.01076-14 (2014).
Dorsey, C. W. et al. The siderophore-mediated iron acquisition systems of Acinetobacter baumannii ATCC 19606 and Vibrio anguillarum 775 are structurally and functionally related. Microbiol. (Reading Engl). 150, 3657–3667 (2004).
Gardner, P. R. et al. Hemoglobins dioxygenate nitric oxide with high fidelity. J. Inorg. Biochem. 100, 542–550 (2006).
Poole, R. K. Flavohaemoglobin: the pre-eminent nitric oxide–detoxifying machine of microorganisms. F1000Res (2020). 9, F1000 Faculty Rev-7.
Partridge, J. D., Bodenmiller, D. M., Humphrys, M. S. & Spiro, S. NsrR targets in the Escherichia coli genome: new insights into DNA sequence requirements for binding and a role for NsrR in the regulation of motility. Mol. Microbiol. 73, 680–694 (2009).
Gomes, C. M., Vicente, J. B., Wasserfallen, A. & Teixeira, M. Spectroscopic studies and characterization of a novel electron-transfer chain from Escherichia coli involving a flavorubredoxin and its flavoprotein reductase partner. Biochem 39, 16230–16237 (2000).
Thakur, N. et al. New insights into the function of flavohemoglobin in Mycobacterium tuberculosis: role as a NADPH-dependent disulfide reductase and D-lactate-dependent mycothione reductase. Front. Cell. Infect. Microbiol. 11, 796727 (2022).
Abramson, J. et al. Accurate structure prediction of biomolecular interactions with AlphaFold 3. Nature 1–8. https://doi.org/10.1038/s41586-024-07487-w (2024).
Milani, M. et al. Mycobacterium tuberculosis hemoglobin N displays a protein tunnel suited for O2 diffusion to the heme. EMBO J. 20, 3902–3909 (2001).
Gardner, A. M. & Gardner, P. R. Flavohemoglobin detoxifies nitric oxide in aerobic, but not anaerobic, Escherichia coli. Evidence for a novel inducible anaerobic nitric oxide-scavenging activity. J. Biol. Chem. 277, 8166–8171 (2002).
Pesce, A. et al. The N-terminal pre-A region of Mycobacterium tuberculosis 2/2HbN promotes NO-dioxygenase activity. FEBS J. 283, 305–322 (2016).
Neukirchen, S. & Sousa, F. L. DiSCo: a sequence-based type-specific predictor of dsr-dependent dissimilatory sulphur metabolism in microbial data. Microb. Genom. 7, 000603 (2021).
Ermler, U., Siddiqui, R. A., Cramm, R. & Friedrich, B. Crystal structure of the flavohemoglobin from Alcaligenes eutrophus at 1.75 a resolution. EMBO J. 14, 6067–6077 (1995).
Miranda, J. L., Maillett, D. H., Soman, J. & Olson, J. S. Thermoglobin, oxygen-avid hemoglobin in a bacterial hyperthermophile. JBC 280, 36754–36761 (2005).
Muraki, N., Takeda, K., Nam, D., Muraki, M. & Aono, S. Structural characterization of Y29F mutant of thermoglobin from a hyperthermophilic bacterium Aquifex Aeolicus. Chem. Lett. 50, 603–606 (2021).
Ajiboye, T. O., Skiebe, E. & Wilharm, G. Contributions of ferric uptake regulator Fur to the sensitivity and oxidative response of Acinetobacter baumannii to antibiotics. Microb. Pathog. 119, 35–41 (2018).
Fiester, S. E. & Actis, L. A. Stress responses in the opportunistic pathogen Acinetobacter baumannii. Future Microbiol. 8, 353–365 (2013).
Lama, A. et al. Role of pre-A motif in nitric oxide scavenging by truncated hemoglobin, HbN, of Mycobacterium tuberculosis. J. Biol. Chem. 284, 14457–14468 (2009).
Gardner, P. R. et al. Nitric-oxide dioxygenase activity and function of flavohemoglobins. Sensitivity to nitric oxide and carbon monoxide inhibition. J. Biol. Chem. 275, 31581–31587 (2000).
Kint, N. et al. How the anaerobic enteropathogen Clostridioides difficile tolerates low O2 tensions. mBio 11 https://doi.org/10.1128/mbio.01559-20 (2020).
Frey, A. D., Farrés, J., Bollinger, C. J. T. & Kallio, P. T. Bacterial hemoglobins and flavohemoglobins for alleviation of nitrosative stress in Escherichia coli. Appl. Environ. Microbiol. 68, 4835–4840 (2002).
Hill, S., Decorso, I., Nezamololama, N., Babaei, Z. & Rafferty, S. P. Catalytic differences between flavohemoglobins of Giardia intestinalis and E. coli. J. Pathog. 13, 480 (2024).
Nobre, L. S., Gonçalves, V. L. & Saraiva, L. M. Flavohemoglobin of Staphylococcus aureus. in Methods in Enzymology vol. 436 203–216 (Elsevier, (2008).
Jaspreet Kaur. In silico analysis of phylogeny, structure, and function of flavohemoproteins from metagenomic data. Biol. Bull Russ. Acad. Sci. 51, 1024–1038. (2024).
Langmead, B. & Salzberg, S. L. Fast gapped-read alignment with Bowtie 2. Nat. Methods. 9, 357–359 (2012).
Li, H. et al. The sequence Alignment/Map format and SAMtools. Bioinform 25, 2078–2079 (2009).
Liao, Y., Smyth, G. K. & Shi, W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinform 30, 923–930 (2014).
Robinson, M. D., McCarthy, D. J. & Smyth, G. K. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinform 26, 139–140 (2010).
Huang, D. W., Sherman, B. T. & Lempicki, R. A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 4, 44–57 (2009).
Sherman, B. T. et al. DAVID: a web server for functional enrichment analysis and functional annotation of gene lists (2021 update). Nucleic Acids Res. 50, W216–W221 (2022).
Berry, E. A. & Trumpower, B. L. Simultaneous determination of hemes a, b, and c from pyridine hemochrome spectra. Anal. Biochem. 161, 1–15 (1987).
Folgosa, F., Martins, M. C. & Teixeira, M. The multidomain flavodiiron protein from Clostridium difficile 630 is an NADH:oxygen oxidoreductase. Sci. Rep. 8, 10164 (2018).
Madeira, F. et al. Search and sequence analysis tools services from EMBL-EBI in 2022. Nucleic Acids Res. 50, W276–W279 (2022).
Sievers, F. et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 7, 539 (2011).
Mirdita, M. et al. ColabFold: making protein folding accessible to all. Nat. Methods. 19, 679–682 (2022).
Goddard, T. D. et al. UCSF ChimeraX: Meeting modern challenges in visualization and analysis. Protein Sci. 27, 14–25 (2018).
Meng, E. C. et al. UCSF ChimeraX: tools for structure building and analysis. Protein Sci. 32, e4792 (2023).
Pettersen, E. F. et al. UCSF ChimeraX: structure visualization for researchers, educators, and developers. Protein Sci. 30, 70–82 (2021).
Wang, J., Wang, W., Kollman, P. A. & Case, D. A. Automatic atom type and bond type perception in molecular mechanical calculations. J. Mol. Graphics Modell. 25, 247–260 (2006).
Wang, J., Wolf, R. M., Caldwell, J. W., Kollman, P. A. & Case, D. A. Development and testing of a general amber force field. J. Comput. Chem. 25, 1157–1174 (2004).
Rinke, C. et al. Insights into the phylogeny and coding potential of microbial dark matter. Nature 499, 431–437 (2013).
Buchfink, B., Reuter, K. & Drost, H. G. Sensitive protein alignments at tree-of-life scale using DIAMOND. Nat. Methods. 18, 366–368 (2021).
Buchfink, B., Xie, C. & Huson, D. H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods. 12, 59–60 (2015).
Trifinopoulos, J., Nguyen, L. T., von Haeseler, A. & Minh, B. Q. W-IQ-TREE: a fast online phylogenetic tool for maximum likelihood analysis. Nucleic Acids Res. 44, W232–W235 (2016).
Tria, F. D. K., Landan, G. & Dagan, T. Phylogenetic rooting using minimal ancestor deviation. Nat. Ecol. Evol. 1, 1–7 (2017).
Acknowledgements
This work has received funding from FCT through MOSTMICRO-ITQB R&D Unit (UIDB/04612/2020, UIDP/04612/2020) and LS4FUTURE Associated Laboratory (LA/P/0087/2020), the European Research Council (ERC) under the European Union’s Horizon 2020 research and innovation program (grant agreement 810856 and grant agreement 803768). JB is recipient of the MSCAIF- 2019 Individual Fellowship H2020-WF-02-2019, 101003441. For funding, F.L.S. thanks the Wiener Wissenschafts-, Forschungs- und Technologiefonds (grant agreement VRG15-007). The computational results of this work have been achieved using the Life Science Compute Cluster (LiSC) of the University of Vienna.
Author information
Authors and Affiliations
Contributions
JZ conducted all the experiments and took the lead in writing the manuscript. LMS conceived the project and gave critical feedback on all the steps.FF contributed to the NO activity assays and in the structural modelling analysis.VK and FS conceived and supervised the phylogenetic analysis.All authors contributed to the writing of the manuscript, shaping the research and analysis.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Beas, J.Z., Folgosa, F., Karavaeva, V. et al. A novel type of hemoglobin confers host-derived nitric oxide resistance to the opportunistic pathogen Acinetobacter baumannii. Sci Rep 15, 5969 (2025). https://doi.org/10.1038/s41598-025-88123-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-025-88123-z
