
Abstract
Biochemical recurrence (BCR) of prostate cancer (PCa) negatively impacts patients’ post-surgery quality of life, and the traditional predictive models have shown limited accuracy. This study develops an AI-based prognostic model using deep learning that incorporates androgen receptor (AR) regional features from whole-slide images (WSIs). Data from 545 patients across two centres are used for training and validation. The model showed strong performances, with high accuracy in identifying regions with high AR expression and BCR prediction. This AI model may help identify high-risk patients, aiding in better treatment strategies, particularly in underdeveloped areas.
Similar content being viewed by others


Introduction
Prostate cancer (PCa) is one of the most common malignancies of the genitourinary system worldwide1,2. Currently, radical prostatectomy is considered one of the most effective treatments3. However, despite undergoing surgery or radiotherapy, and even standard androgen deprivation therapy (ADT), 27-53% of patients still BCR, which severely impacts their quality of life4. Clinical characteristics such as age, tumour grade, and tumour stage, are crucial for assessing patient survival5. However, relying solely on clinical information often fails to fully capture the complex features of the tumour, necessitating the integration of histopathological features to improve the accuracy of prognostic assessments6. Despite advances in treatment methods, significant challenges remain in personalized treatments and prognostic evaluations7.
The androgen receptor (AR) plays a crucial role in prostate cancer (PCa)8. AR is a nuclear receptor that is essential for regulating the proliferation, differentiation, and survival of prostate cells9. As early as the 1840s, researchers first demonstrated that castration could lead to the regression of prostate cancer, highlighting the importance of the androgen signalling pathway in PCa10. Consequently, AR has become a key target in the treatment of prostate cancer11. AR expression is traditionally assessed through immunohistochemistry (IHC) staining techniques. However, this method has several limitations, including being labour intensive and highly subjective and yielding inconsistent results12,13. Therefore, there is a significant demand for automated analysis systems in the field of pathology to greatly reduce workload, improve reproducibility, and enhance diagnostic accuracy14,15,16.
In recent years, with advancements in computational power and the widespread use of whole slide imaging (WSI), artificial intelligence (AI) has made significant strides across various fields17,18,19. Particularly in the automated analysis of histopathology, AI technology can extract subvisual features from digital images that are imperceptible to the naked eye, enabling disease diagnosis and prognostic predictions20,21,22. Numerous AI-based diagnostic and prognostic systems have already been successfully developed and have achieved promising results in clinical applications23,24,25,26,27.
In this study, we develop an artificial intelligence (AI) system capable of predicting AR expression from digital H&E slides with high accuracy. The system also employs visualization techniques to display the predicted AR regions, thereby expanding the scope of pathological recognition in prostate cancer. By establishing a neural network-based prognostic model, we effectively predict the postoperative prognosis of prostate cancer patients via prognostically relevant cellular features.
Methods
Clinical samples
We included data on prostate cancer patients over two time periods: Zhongda Hospital, Southeast University: 452 patients with prostate cancer who underwent radical prostatectomy at the hospital between January 2014 and December 2019 were selected. Huai’an First People’s Hospital: A total of 118 prostate cancer patients who underwent radical prostatectomy in the hospital from January 2017 to December 2019 were selected, as shown in Table 1. Overall, 55/341(16.1%), 10/99 (10.1%), and 11/105 (10.5%) patients experienced BCR in the training, internal validation, and external validation cohort, respectively. In the entire cohort, approximately 71% of biochemical recurrences occurred within the first three years post-surgery. The median follow-up time was 20.8 (95% confidence intervals [CIs], 17.4–25.3) months.
The cohort was screened to include only treatment-naive patients, resulting in a final sample size of 545 cases. We reviewed electronic medical records and pathology reports to extract baseline characteristics (e.g., age at surgery, preoperative PSA level, BMI, surgical approach), pathological variables (e.g., Gleason score, TNM stage, surgical margin status, seminal vesicle invasion, extracapsular extension, prostate volume). The Gleason score was determined according to the 2015 revised Gleason grading system28, TNM staging was based on the 8th edition of the AJCC(American Joint Committee on Cancer) standards29, and prostate volume was calculated using the “Terris formula”30.
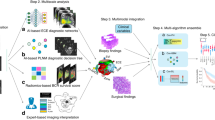
For the first two years after surgery, patients are followed up every three months as part of routine care; thereafter, follow-up occurs every six months until five years, after which patients are followed up annually according to institutional practice. The primary endpoint of the study is the biochemical recurrence-free survival (BCR-free survival). This survival period is calculated from the date of surgery to the date of BCR or censored at the last follow-up (up to five years). BCR is defined as a continuous increase in PSA levels greater than 0.1 ng/ml for at least three occasions, at least six weeks after surgery, with a final PSA level > 0.2 ng/ml or PSA ≥ 0.4 ng/ml, or the initiation of secondary treatment due to rising PSA levels. This definition is based on previous reports and is associated with the likelihood of future PSA progression31. Follow-up data were obtained via telephone interviews, and the study flowchart is shown in Fig. 1.

Process diagram of the pathological artificial intelligence model research. Study flowchart of the pathological artificial intelligence prognosis model.
Image preprocessing
All H&E- and AR-stained histopathological slides were digitized using a NanoZoomer S360 digital slide scanner at 20x magnification, achieving a maximum resolution of 20x. Low-quality images due to age or low resolution were excluded. The scanner produced whole-slide images (WSIs) in ndpi file format. The AR status for all the patients was determined through immunohistochemical (IHC) staining, with two genitourinary pathologists reviewing and annotating high-AR-expressing regions. Discrepancies were resolved by consensus with a third pathologist, ensuring consistent annotation across all cases.
For image pre-processing, the Otsu method was employed to segment the WSIs of H&E-stained slides, eliminating the background areas32. Within AR regions, QuPath v0.1.2 was used to meticulously outline polygons33, labelling areas with high AR expression. AR high-expression regions are defined on the basis of the commonly used clinical immunohistochemical (IHC) scoring system. The nuclear staining intensity was classified into four categories: no staining (score of 0), light yellow (score of 1), yellow (score of 2), and brown (score of 3). Tumour lesions with a staining intensity of 2 or 3 were defined as regions with high AR expression. Given the complexity and time-intensive nature of annotating entire slides, only the AR high-expression regions and other relevant areas within the tumour foci were annotated, significantly reducing the workload and enhancing the specificity of the image recognition process. To prevent confusion during model training, unmarked tissue areas and backgrounds were excluded from the training images, and the model was focused on learning from the annotated sections.
The annotated images were cropped into 512 × 512 pixel PNG tiles. After screening, approximately 250 AR high-expression tiles and 250 other region tiles were randomly selected per slide to construct the training and testing datasets. Specifically, the number of AR high-expression tiles per slide ranged from 213 to over 250, with an average of 250 tiles. Similarly, the number of tiles from other regions ranged from 183 to over 250 per slide, with an average of 250 tiles. The semantic segmentation models were trained at the pixel level, learning spatial, grayscale, and color information to classify each pixel and accurately integrate it into the output.
Deep learning models
UNet model for AR expression prediction
The UNet architecture is a widely utilized deep learning framework for image segmentation tasks, particularly in the medical imaging domain34. In this study, we developed an AI-AR model based on the UNet + + architecture to predict androgen receptor (AR) expression in H&E-stained images. The AI-AR model incorporates additional skip connections to merge feature maps at multiple scales, which improves segmentation accuracy and robustness.
To address the variability in H&E staining, we applied a colour normalization method to the input images, ensuring a consistent colour distribution across different slides. Specifically, we employed Z-Score Normalization, a statistical method that transforms the data by adjusting its mean to 0 and its standard deviation to 135. By applying Z-Score standardization to the input images, we reduced the impact of varying pixel intensity ranges across different tissue samples, allowing the AI model to more effectively learn the relevant features and improve its performance during training. The standardized images were then used to train the AI-AR model, with AR-labelled patches employed for fine-tuning the model weights. This process helped reduce the degree of staining variability, ensuring more accurate detection of regions with high AR expression. The hyperparameters were optimized using cross-validation, and the model performance was evaluated on an independent internal validation set comprising unseen patches. The output of the AI-AR model includes pixelwise predictions of the AR expression status for each input H&E image.
In medical image segmentation tasks, class imbalance, especially in tumour segmentation, often results in significant differences between the quantities of the normal and tumour tissues. To address this, we have used the following metrics:
1. Mean Pixel Accuracy (MPA): MPA calculates the average pixel accuracy for each class in image segmentation tasks. It measures how accurately each class’s predicted pixels match the true label pixels. MPA was calculated based on the following formula (1)
C is the total number of classes, TPi (True Positive) is the number of pixels correctly predicted as positive for class i, FPi (False Positive) is the number of pixels incorrectly predicted as positive for class i, FNi (False Negative) is the number of pixels incorrectly predicted as negative for class i, TNi (True Negative) is the number of pixels correctly predicted as negative for class i.
2. Mean Dice Coefficient (mDice): The mDice is a measure of the similarity between the predicted segmentation and the ground truth. It ranges from 0 (no overlap) to 1 (perfect overlap). The formula (2) for mDice is is presented as follows.
Ai is the predicted region for class i, Bi is the ground truth region for class i, |Ai∩Bi | is the number of overlapping pixels between the predicted and true regions for class i, |Ai |+|Bi | are the total number of pixels in the predicted and true regions, respectively.
3. Mean Recall (mRecall): mRecall, also known as mean sensitivity, measures the model’s ability to correctly identify the positive class (i.e., tumor or AR-high regions). It is calculated as the average of the recall values for each class. Mean Recall was calculated based on the following formula (3).
4. The mIoU (mean intersection and merger ratio) is a commonly used evaluation metric in image segmentation tasks, especially in semantic segmentation tasks. It measures the overlap between the model-predicted region and the true labelled region, and is usually used to evaluate the accuracy of model segmentation. The formula (4) for mIoU is presented as follows.
|Ai∪Bi |is the number of concatenated pixels in the predicted and real regions.
HoverNet for feature extraction
Hover-Net is a state-of-the-art deep learning model specifically designed for semantic segmentation in histopathological images. Introduced by Lienkamp et al. in 2019, Hover-Net addresses several limitations of the traditional segmentation methods, such as inadequate precision in delineating object boundaries and recognizing small objects. The core innovations of Hover-Net include multiscale feature fusion, multitask learning, deep supervision, and a lightweight architecture for real-time inference23,24.
In this study, we utilized Hover-Net to extract features from histopathological images with evident AR characteristics. The model was trained to segment various types of nuclei, including nontumorous epithelial cells, tumorous epithelial cells, inflammatory cells, stromal cells, and necrotic cells. From the tumour regions, we manually extracted 262-dimensional nuclear features encompassing nuclear texture, morphology, spatial arrangement, and tumour microenvironment (TME) characteristics.
Survival analysis
The DeepSurv model architecture is a deep learning framework tailored for survival analysis tasks. It employs a neural network structure to process input data and predict survival outcomes. The model comprises input layers, hidden layers with nonlinear activation functions, and an output layer that produces predicted survival probabilities36.
Integrated Data: These procedures involve integrating heterogeneous data sources, including the features extracted from the pathology slides and clinical data, into the DeepSurv model. Optimization techniques such as stochastic gradient descent or Adam optimization are used to minimize a loss function, typically the negative log-partial likelihood.
For each survival model, performance metrics, including C-index and AUC, were calculated, and 95% confidence intervals (CIs) were generated using bootstrapping.
Statistical analysis
This study strictly adhered to all the relevant guidelines and regulations. Additionally, it also complied with privacy protection and information security regulations for medical data, and all collected data were strictly deidentified to ensure the security of patient privacy. Categorical variables were expressed as n (%) and were compared utilizing either the chi-square test or Fisher’s exact test. The mean values (standard deviations, SDs) of normally distributed continuous variables were reported and compared via Student’s t test. For nonnormally distributed continuous variables, the median values along with the interquartile range (IQR) were presented, and comparisons were conducted using the Mann‒Whitney U test. All the statistical analyses were two-tailed, and statistical significance was defined as a p value less than 0.05. The statistical analyses were performed using R version 4.2.1.
Software and tools
Python Version: The OpenSlide library (v3.4.1) was utilized within Python (3.10.1, Python Software Foundation) for image tile extraction.
Deep Learning Framework: PyTorch, a Python-based deep learning framework, was employed for model development and testing.
Computational Resources: Model development and testing were conducted on a computer equipped with an NVIDIA RTX 3090 GPU, featuring 30 GB of memory and operating at 2.4 GHz.
Results
The predictive ability of the AR segmentation models
In this study, we utilized datasets from Zhongda Hospital Southeast University and Huai’an First People’s Hospital, comprising H&E images from 570 (452 + 118) patients, to train and test AI-AR. Our objective was to determine whether AR expression could be identified solely from digital H&E images of prostate cancer patients. After removing images with excessive markings or large missing areas, we divided the dataset into a training set (352 patients), a testing set (88 patients), and a validation set (105 patients). All 545 H&E-stained slides were annotated at the pixel level by professional pathologists. Following a fivefold cross-validation on the training set, we trained AI-AR using all the WSIs from the training set and evaluated it on the testing set WSIs. The results are illustrated in Fig. 2.

Working diagram of the segmentation model. Diagram of the development and validation of an artificial intelligence prognostic model for pathology. Two pathologists labelled all the HE images based on IHC images. In the training stage, a CNN model (Unet++) was trained using the Zhongda Hospital Affiliated to Southeast University (purple 1) training set, and the AR segmentation model (AR-Net) was trained. In the validation stage, the internal validation set (purple 2) of Zhongda Hospital affiliated to Southeast University and the external validation set (green) of the First People’s Hospital of Huai’an City were input into the convolutional neural network to obtain the average pixel accuracy of each slide, and the output of the image patch blocks was stitched together to obtain a heatmap, which intuitively depicted the AR high expression prediction interval. In the predicted prognostic stage, the predicted AR regions were extracted from the topological and nuclear texture features using Hover-Net, and the predicted ROC curves were obtained by combining clinical data. CNN convolutional neural network, WSI whole slide image, H&E haematoxylin-eosin, IHC Immunohistochemistry, AR Androgen Receptor.
The AR segmentation model achieves an MPA of 0.86, an mIoU of 0.68, an mDice of 0.82, and an mRecall of 0.78 on the internal validation set. These results exceeded expectations, reaffirming the importance of increasing the dataset size to enhance predictive efficacy.
While the predictive performance on the external validation set was relatively less satisfactory, it still yielded promising outcomes, with an MPA of 0.80, mIoU of 0.65, mDice of 0.82, and mRecall of 0.80, as detailed in Table 2. Compared with the validation set, the model demonstrated a more accurate and stable MPA and mDice on the training set, further validating the predictive efficacy of the segmentation model. The performance on the external validation set underscores the necessity of incorporating data from diverse centres to improve the model’s generalizability.
We employed the Grad-CAM technique to visualize the segmentation model’s predictions further using heatmaps37. Grad-CAM (Gradient-weighted Class Activation Mapping) is a powerful method that highlights the important regions of an image that contribute most to the model’s prediction. To enhance the interpretability of the heatmap, we overlaid boundary contours on the predicted regions to clearly display the areas identified by the model. This process generates probability prediction values, which were compared against a predefined segmentation threshold of 0.5 to determine the class of segmented image patches and generate probability prediction heatmaps. In the probability prediction maps, brighter red and blue colours indicate higher probabilities of the pixels belonging to specific classifications, whereas mixed colours indicate a greater uncertainty in classification. Figure 3 illustrates the segmentation model’s prediction outcomes, revealing that the model performs well in distinguishing high-AR-expressing regions from other types of areas in WSI images of prostate cancer. The model effectively predicts most target areas, with the probability prediction heatmaps closely overlapping with the manually delineated images. This consistency between the AI-predicted and manually annotated regions indicates the interpretability of the segmentation model’s predictions.

Segmentation model for predicting heatmaps. Examples of manual annotation and automatic tissue segmentation.
Target audience for the AR segmentation model
To further investigate which population is more suitable for our AI-AR model, we standardized the prediction results on the basis of the predicted MPA of the AR regions, selecting a median cut-off value of 0.878 to distinguish between high-accuracy and low-accuracy groups. We conducted single-factor and multifactor logistic regression analyses, incorporating collected clinical factors (age, BMI, TPSA, prostate volume, surgical margin, seminal vesicle invasion, extracapsular extension, TNM stage, Gleason score, AR expression, Her2, Ki67, ERG). Factors with a single-factor P-value < 0.1 were included in the multifactor logistic regression.
Ultimately, through single-factor and multifactor selection, we identified the independent factors influencing the high and low accuracies:
TPSA = 10–20 ng/ml (OR = 0.547, 95% CI 0.319–0.937, P = 0.028).
Negative surgical margin (OR = 0.600, 95% CI 0.379–0.950, P = 0.029).
Seminal vesicle invasion was positive (OR = 1.932, 95% CI 1.091–3.421, P = 0.024).
AR1 positive (OR = 2.120, 95% CI 1.044–3.368, P = 0.010).
AR2 positive (OR = 2.120, 95% CI 1.225–3.872, P = 0.010).
P53 mutation (OR = 1.606, 95% CI 1.049–2.461, P = 0.029).
The logistic regression analysis suggested that individuals with seminal vesicle invasion, weak positive AR expression, and P53 mutation were better suited for our model. A TPSA concentration of 10–20 ng/ml and negative surgical margins indicate poorer predictive capabilities. These findings provide valuable insights for the application of our AI-AR model (Table 3).
Feature extraction of Hover-Net for the AR area
In this study, images showing notable high AR expression features (MPA > 0.878) were selected for segmenting the characteristics of the cell nucleus to distinguish various types, including nontumorous epithelial cells, tumorous epithelial cells, inflammatory cells, stromal cells, and necrotic cells. We manually extracted nuclear features (texture, morphology, spatial arrangement) from the AR high-expression regions on the HE slides and analysed the tumour microenvironment spectra (relationships between tumour nuclei, stromal nuclei/tumour nuclei, and immune cell nuclei/immune cell nuclei with stromal nuclei), totalling 262 dimensions. These features, including the nuclear texture, shape, spatial arrangement, and TME-related features, are relevant for predicting patient prognosis.
The morphology/texture features of the nuclei (46 descriptors) were quantified using metrics such as the area, cell eccentricity, roundness, and elongation ratio. Spatial arrangement features (216 descriptors) captured the topological differences and spatial structures among the tumour nuclei, utilizing parameters such as n-subgraphs (Nsubgraph), degrees (degrees), coreness (core), and closeness (close). Using the random forest feature_importance function, 20 features were selected to manage dimensionality and avoid model overfitting.
The final set of twenty features included graph characteristics such as Coreness (tumour-stroma), meanEdgeLength (tumour-stroma), Eccentricity (inflammation-stroma), and nuclear texture attributes such as Entropy (stroma) and IntensityMax (inflammation), as detailed in Fig. 4. These cellular and clinical data features were then integrated to predict patient prognosis.

Cellular characteristics of regions highly expressed in AR.
The impact of clinical data on the prognosis of PCa
In our study, single-factor Cox regression analysis was conducted on the various clinical variables associated with BCR in patients. The results (Table 4) indicated that age, TPSA, surgical margin status, N stage, Gleason score, Ki67, and ERG were potential risk factors for BCR after RP (radical prostatectomy). When these risk factors were included in a multifactor Cox regression model, surgical margin status (HR 3.024, 95% CI 1.268–7.213, P = 0.013), N stage (HR 21.171, 95% CI 1.361-329.405, P = 0.029), and ERG expression (HR 0.275, 95% CI 0.124–0.613, P = 0.002) emerged as independent prognostic factors for predicting BCR in prostate cancer patients post-RP. Conversely, age, the TPSA, the Gleason score, and Ki67 expression did not significantly differ in this model.
The DeepSurv model predicts the prognosis of prostate cancer patients after RP
When the discriminative ability of the D-S model was compared via Harrell’s C-index and the time-dependent AUC (5-year), the D-S model demonstrated robust performance across the training, internal validation, and external validation cohorts. Specifically, the C-index values for the D-S model were 0.80, 0.83, and 0.79 in the respective cohorts, whereas the time-dependent AUC (5-year) values were 0.99, 0.79, and 0.85. These metrics consistently indicated the superior predictive performance of the D-S model for early recurrence compared with the other models (Table 5). Additionally, the ROC curve results of the D-S model exceeded those of the CAPRA-S, Cox, and TNM models across all three cohorts (Fig. 5).

Comparison of the prognostic ROC curves among the different models.
Discussion
IHC has become the primary method for detecting tumour biomarkers and is widely regarded as a reliable alternative to expensive molecular tests. However, IHC staining has several limitations, including cost inefficiency, a lack of expediency, and susceptibility to significant errors due to various factors. For example, in breast cancer, IHC testing for oestrogen and progesterone receptors may be inaccurate in up to 20% of cases. Validating the IHC-derived biomarker status using HE staining could reduce the variability and lower the cost of pathology workflows, saving time in clinical decision-making. To address these challenges, we developed a predictive model using deep neural networks trained on pixel-level annotated HE images. This model autonomously identifies regions of heightened AR receptor expression from unlabelled HE stains, achieving performances on internal validation data comparable to that of pathologists (MPA = 0.86).
Recent advancements highlight the potential of integrating pathology with AI technologies, with the goal of attaining diagnostic efficiencies equivalent to that of the actual pathologists38,39. Significant progress has been made in developing AI models for surgical clinical management that autonomously detects malignancies and lymph node micrometastases, and predict receptor statuses such as oestrogen, progesterone, and ERBB2 in breast cancer patients via HE-stained tissue microarrays (TMAs) and whole-slide images (WSIs). Similar advancements have been made to predict microsatellite instability and BRAF mutation statuses in colorectal cancer, Gleason scores in prostate cancer biopsy tissues, and tumour mutation burden and gene mutations in lung adenocarcinoma40,41,42,43,44.
Our study innovates by leveraging pathologist-annotated H&E slides to enhance AI training, significantly improving the model’s ability to learn and predict, despite increased labour intensity. The use of H&E-stained slides in AI training also addresses one of the key limitations of IHC, making the diagnostic process more accessible, especially in resource-limited settings. Our AI model, which is based on a semantic segmentation architecture (augmented from the UNet model with multiple skip connections), enhances segmentation accuracy and robustness by focusing on semantic relationships among pixels rather than relying on graphical features. This methodological approach improves the detection of nuanced features in tissue sections, which may be missed by the conventional methods.
We trained our model using 80% WSIs and validated it using fivefold cross-validation to optimize the parameters, aiming for a maximum accuracy with the existing data. Logistic regression analyses revealed varying accuracies across conditions such as TPSA levels and surgical margins but consistently higher accuracy in cases of seminal vesicle invasion and AR1+, AR2+, and P53 mutation positivity. These results suggest that the AI model could be particularly beneficial for predicting outcomes in certain high-risk patient groups, which aligns with recent research showing that AR expression is a critical factor in prostate cancer prognosis45,46.
While clinical pathologists can estimate tumour malignancies from H&E slides, accurately determining biomarker expression, AR at the protein level, in particular, remains challenging owing to the undefined relationships with prostate cancer tissue morphology. Although IHC remains the gold standard for protein-level biomarker detection, the use of H&E slides in conjunction with AI offers significant practical advantages. Our AI model predicts AR levels with similar accuracies to the pathologists, offering a feasible solution for underserved areas where IHC resources may be limited. Moreover, the ability to adapt the model to predict other molecular markers using extensive H&E slide databases opens new possibilities for AI-assisted diagnostics. This approach may even surpass the bioinformatics-based methods in terms of sensitivity.
The incorporation of the tumour microenvironment (TME) into our AI model could further increase its predictive accuracy, as recent studies suggest that TME features, such as immune cell infiltration and extracellular matrix remodelling, play a significant role in cancer prognosis47. Integrating such features could provide a more comprehensive understanding of cancer progression and patient outcomes48. Our study lays the groundwork for future research that combines AI with TME data, such as sequencing and spatial transcriptomics, to further improve prognostic accuracy.
The application of AI in clinical pathology is not without challenges. While our model has demonstrated a strong performance in predicting AR expression, further validation on external datasets and multicentre studies is essential to ensure its generalizability across diverse populations. Additionally, deploying deep learning in clinical settings requires rigorous validation, as well as addressing the potential concerns related to data privacy and model interpretability. Future work should focus on securing robust AI applications in clinical settings, including real-time integration with pathology workflows.
In conclusion, deep learning systems show great promise in routine pathology tasks, such as AR prediction and prostate cancer prognosis. Our model’s ability to identify distinct features in regions with high AR expression could aid in the development of more accurate diagnostic tools and improve patient management strategies. The role of AI in enhancing cancer prognosis and treatment planning is increasingly evident, and our work serves as a foundational step in integrating AI into prostate cancer diagnostics.
Limitations of the study
First, it is retrospective and requires additional prospective validation. Second, our data originate from two centres, and although the predictive performance on internal test datasets remains adequate, multicentre studies are necessary for broader validation. Consequently, we have initiated multicentre research initiatives. Third, despite efforts to mitigate heterogeneity stemming from variations in tissue sampling sites, factors such as tissue loss, inherent tumour diversity, and tumour size remain inevitable sources of error. Fourth, the current opacity in AI learning and decision-making processes, particularly in medical contexts, represents an inherent limitation. While integrating additional learning models can mitigate this issue, it often adds complexity to the study. Our study enhances interpretability by manually annotating high-expression regions, and rigorously selecting these areas to predict AR expression levels. Fifth, our study did not perform extensive comparisons with widely recognized international prognostic models, as many risk factors did not reach statistical significance in our Cox regression analysis. Hence, the predictive outcomes may be somewhat skewed. Sixth, the study did not compare the results with expert subjective evaluations, which could restrict the broader applicability of the diagnostic performance assessed by our model. Finally, our study did not delve into deeper cellular mechanistic explanations, necessitating the inclusion of clinical sample sequencing data for further investigation.
Data availability
Further information and requests for resources should be directed to and will be fulfilled by the lead contact, Yifeng Xue (494809908@qq.com).
References
Bray, F. et al. Global cancer statistics 2022: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 74, 229–263. https://doi.org/10.3322/caac.21834 (2024).
Zhang, M. et al. Near-infrared dye-labeled anti-prostate stem cell antigen minibody enables real-time fluorescence imaging and targeted surgery in translational mouse models. Clin. Cancer Res. 25, 188–200. https://doi.org/10.1158/1078-0432.CCR-18-1382 (2019).
Guillaumier, S. et al. A Multicentre study of 5-year outcomes following focal therapy in treating clinically significant nonmetastatic prostate cancer. Eur. Urol. 74, 422–429. https://doi.org/10.1016/j.eururo.2018.06.006 (2018).
Heidenreich, A. et al. EAU guidelines on prostate cancer. Part 1: screening, diagnosis, and local treatment with curative intent-update 2013. Eur. Urol. 65, 124–137. https://doi.org/10.1016/j.eururo.2013.09.046 (2014).
Pandiaraja, M. et al. Utilisation and impact of predict prostate on decision-making among clinicians and patients in a specialist tertiary referral centre: a retrospective cohort study. BJUI Compass. 5, 489–496. https://doi.org/10.1002/bco2.311 (2024).
Wang, S. et al. Features of tumor-microenvironment images predict targeted therapy survival benefit in patients with EGFR-mutant lung cancer. J. Clin. Invest. https://doi.org/10.1172/JCI160330 (2023).
Roubaud, G., Liaw, B. C., Oh, W. K. & Mulholland, D. J. Strategies to avoid treatment-induced lineage crisis in advanced prostate cancer. Nat. Rev. Clin. Oncol. 14, 269–283. https://doi.org/10.1038/nrclinonc.2016.181 (2017).
Bainbridge, A. et al. IKBKE activity enhances AR levels in advanced prostate cancer via modulation of the Hippo pathway. Nucleic Acids Res. 48, 5366–5382. https://doi.org/10.1093/nar/gkaa271 (2020).
Shorning, B. Y., Dass, M. S., Smalley, M. J. & Pearson, H. B. The PI3K-AKT-mTOR pathway and prostate cancer: at the crossroads of AR, MAPK, and WNT signaling. Int. J. Mol. Sci. https://doi.org/10.3390/ijms21124507 (2020).
Avila, D. M., Zoppi, S. & McPhaul, M. J. The androgen receptor (AR) in syndromes of androgen insensitivity and in prostate cancer. J. Steroid Biochem. Mol. Biol. 76, 135–142. https://doi.org/10.1016/s0960-0760(00)00158-8 (2001).
Valdes-Mora, F. et al. Acetylated histone variant H2A.Z is involved in the activation of neo-enhancers in prostate cancer. Nat. Commun. 8, 1346. https://doi.org/10.1038/s41467-017-01393-8 (2017).
Naik, N. et al. Deep learning-enabled breast cancer hormonal receptor status determination from base-level H&E stains. Nat. Commun. 11, 5727. https://doi.org/10.1038/s41467-020-19334-3 (2020).
Shamai, G. et al. Deep learning-based image analysis predicts PD-L1 status from H&E-stained histopathology images in breast cancer. Nat. Commun. 13, 6753. https://doi.org/10.1038/s41467-022-34275-9 (2022).
Goecks, J., Jalili, V., Heiser, L. M. & Gray, J. W. How machine learning will transform Biomedicine. Cell 181, 92–101. https://doi.org/10.1016/j.cell.2020.03.022 (2020).
Wu, S. et al. Artificial intelligence-based model for lymph node metastases detection on whole slide images in bladder cancer: a retrospective, multicentre, diagnostic study. Lancet Oncol. 24, 360–370. https://doi.org/10.1016/S1470-2045(23)00061-X (2023).
Zhou, J. et al. Integrative deep learning analysis improves colon adenocarcinoma patient stratification at risk for mortality. EBioMedicine 94, 104726. https://doi.org/10.1016/j.ebiom.2023.104726 (2023).
John, R. A. et al. Optogenetics inspired transition metal dichalcogenide neuristors for in-memory deep recurrent neural networks. Nat. Commun. 11, 3211. https://doi.org/10.1038/s41467-020-16985-0 (2020).
Liu, X., Li, Y. I. & Pritchard, J. K. Trans effects on gene expression can drive omnigenic inheritance. Cell 177, 1022–1034. https://doi.org/10.1016/j.cell.2019.04.014 (2019).
Levine, A. B. et al. Rise of the machines: advances in deep learning for cancer diagnosis. Trends Cancer. 5, 157–169. https://doi.org/10.1016/j.trecan.2019.02.002 (2019).
Aboutalib, S. S. et al. Deep learning to distinguish recalled but benign mammography images in breast cancer screening. Clin. Cancer Res. 24, 5902–5909. https://doi.org/10.1158/1078-0432.CCR-18-1115 (2018).
Hosny, A., Parmar, C., Quackenbush, J., Schwartz, L. H. & Aerts, H. Artificial intelligence in radiology. Nat. Rev. Cancer. 18, 500–510. https://doi.org/10.1038/s41568-018-0016-5 (2018).
Lin, H. et al. Fast ScanNet: fast and dense analysis of multi-gigapixel whole-slide images for cancer metastasis detection. IEEE Trans. Med. Imaging. 38, 1948–1958. https://doi.org/10.1109/TMI.2019.2891305 (2019).
Graham, S. et al. Hover-Net: simultaneous segmentation and classification of nuclei in multi-tissue histology images. Med. Image Anal. 58, 101563. https://doi.org/10.1016/j.media.2019.101563 (2019).
Zhao, S. et al. Single-cell morphological and topological atlas reveals the ecosystem diversity of human breast cancer. Nat. Commun. 14, 6796. https://doi.org/10.1038/s41467-023-42504-y (2023).
Tsai, P. C. et al. Histopathology images predict multi-omics aberrations and prognoses in colorectal cancer patients. Nat. Commun. 14, 2102. https://doi.org/10.1038/s41467-023-37179-4 (2023).
Gerke, S., Shachar, C., Chai, P. R. & Cohen, I. G. Regulatory, safety, and privacy concerns of home monitoring technologies during COVID-19. Nat. Med. 26, 1176–1182. https://doi.org/10.1038/s41591-020-0994-1 (2020).
Campanella, G. et al. Clinical-grade computational pathology using weakly supervised deep learning on whole slide images. Nat. Med. 25, 1301. https://doi.org/10.1038/s41591-019-0508-1 (2019).
Netto, G. J. et al. The 2022 World Health Organization classification of tumors of the urinary system and male genital organs-part B: prostate and urinary tract tumors. Eur. Urol. 82, 469–482. https://doi.org/10.1016/j.eururo.2022.07.002 (2022).
Paner, G. P. et al. Updates in the eighth edition of the tumor-node-metastasis staging classification for urologic cancers. Eur. Urol. 73, 560–569. https://doi.org/10.1016/j.eururo.2017.12.018 (2018).
Baohong, J. et al. ERG tumor type is less frequent in high grade and high stage prostate cancers of Chinese men. J. Cancer. 10, 1991–1996. https://doi.org/10.7150/jca.30025 (2019).
Brockman, J. A. et al. Nomogram predicting prostate cancer-specific mortality for men with biochemical recurrence after radical prostatectomy. Eur. Urol. 67, 1160–1167. https://doi.org/10.1016/j.eururo.2014.09.019 (2015).
Liu, Y., Liang, G. & Saha, P. K. A new multi-object image thresholding method based on correlation between object class uncertainty and intensity gradient. Med. Phys. 39, 514–532. https://doi.org/10.1118/1.3668058 (2012).
Bankhead, P. et al. QuPath: open source software for digital pathology image analysis. Sci. Rep. 7, 16878. https://doi.org/10.1038/s41598-017-17204-5 (2017).
Dey, G. et al. Closed mitosis requires local disassembly of the nuclear envelope. Nature 585, 119–123. https://doi.org/10.1038/s41586-020-2648-3 (2020).
Phillip, J. M. et al. Biophysical and biomolecular determination of cellular age in humans. Nat. Biomed. Eng. https://doi.org/10.1038/s41551-017-0093 (2017).
Katzman, J. L. et al. DeepSurv: personalized treatment recommender system using a Cox proportional hazards deep neural network. BMC Med. Res. Methodol. 18, 24. https://doi.org/10.1186/s12874-018-0482-1 (2018).
Jin, C. et al. Development and evaluation of an artificial intelligence system for COVID-19 diagnosis. Nat. Commun. 11, 5088. https://doi.org/10.1038/s41467-020-18685-1 (2020).
Pantanowitz, L. et al. An artificial intelligence algorithm for prostate cancer diagnosis in whole slide images of core needle biopsies: a blinded clinical validation and deployment study. Lancet Digit. Health 2, e407–e416. https://doi.org/10.1016/S2589-7500(20)30159-X (2020).
Acs, B., Rantalainen, M. & Hartman, J. Artificial intelligence as the next step towards precision pathology. J. Intern. Med. 288, 62–81. https://doi.org/10.1111/joim.13030 (2020).
Bulten, W. et al. Artificial intelligence for diagnosis and Gleason grading of prostate cancer: the PANDA challenge. Nat. Med. 28, 154–163. https://doi.org/10.1038/s41591-021-01620-2 (2022).
Blessin, N. C. et al. Automated Ki-67 labeling index assessment in prostate cancer using artificial intelligence and multiplex fluorescence immunohistochemistry. J. Pathol. 260, 5–16. https://doi.org/10.1002/path.6057 (2023).
Chen, Y. et al. Convolutional neural network quantification of gleason pattern 4 and association with biochemical recurrence in intermediate-grade prostate tumors. Mod. Pathol. 36, 100157. https://doi.org/10.1016/j.modpat.2023.100157 (2023).
Barisoni, L., Lafata, K. J., Hewitt, S. M., Madabhushi, A. & Balis, U. G. J. Digital pathology and computational image analysis in nephropathology. Nat. Rev. Nephrol. 16, 669–685. https://doi.org/10.1038/s41581-020-0321-6 (2020).
Zhang, J. et al. Nondestructive tissue analysis for ex vivo and in vivo cancer diagnosis using a handheld mass spectrometry system. Sci. Transl. Med. https://doi.org/10.1126/scitranslmed.aan3968 (2017).
Shin, S. B., Woo, S. U. & Yim, H. Cotargeting Plk1 and androgen receptor enhances the therapeutic sensitivity of paclitaxel-resistant prostate cancer. Ther. Adv. Med. Oncol. 11, 1758835919846375. https://doi.org/10.1177/1758835919846375 (2019).
Matuszczak, M., Schalken, J. A. & Salagierski, M. Prostate cancer liquid biopsy biomarkers’ clinical utility in diagnosis and prognosis. Cancers https://doi.org/10.3390/cancers13133373 (2021).
Pietrzak, S. et al. Correlation between selenium and zinc levels and survival among prostate cancer patients. Nutrients https://doi.org/10.3390/nu16040527 (2024).
Matuszczak, M. & Salagierski, M. Oligometastatic disease in prostate cancer. Evolving paradigm: current knowledge, diagnostic techniques and treatment strategies. Arch. Med. Sci. https://doi.org/10.5114/aoms/156170 (2022).
Acknowledgements
We thank all authors for their assistance in the preparation of this manuscript. This study was supported by the National Natural Science Foundation of China (82203151-Y.G), the Start-up Fund of Zhongda Hospital of Southeast University (CZXM-GSP-RC56-Y.G), the National Natural Science Foundation of China(82102799), Natural Science Foundation of Jiangsu Province(BK20210230), Zhongda Hospital Affiliated to Southeast University, Jiangsu Province High-Level Hospital Pairing Assistance Construction Funds(zdyyxy33), Open Research Fund of State Key Laboratory of Digital Medical Engineering.
Author information
Authors and Affiliations
Contributions
Jiawei Zhang, Feng Ding, Yitian Guo, Xiaoying Wei, Jibo Jing, Feng Xu, and Huixing Chen contributed equally to this study.J.Z., F.D., Y.G., X.W. Wei, J.J., F.X., and H.C. contributed to conceiving and designing the study. J.Z., X.N., B.L., and F.D. played roles in data collection. X.W. Wei and Z.G. curated the pathological examinations. X.W. Wang and F.D. trained and developed the artificial intelligence model. J.Z., Y.G., J.J., and F.D. were responsible for data analysis, data interpretation, and writing the original draft. Z.Y., M.C., F.X., and H.C. were responsible for writing–reviewing and editing, investigating. Y.X., D.J. and X.W. Wang provided study supervision. All authors read and approved the final manuscript.Correspondence to Yifeng Xue, Xiaobing Niu, Xiangxue Wang, Dongfang Jiang.
Corresponding authors
Ethics declarations
Competing interests
A patent related to this work is currently under review. The patent application was filed by Southeast University, with the following inventors: Xu Bin, Wang Xiangxue, Zhang Jiawei, Ding Feng, Yang Yifan, and Chen Ming. The application, filed under the number CN202311480504.3, is titled “A Method and System for Predicting AR Staining Image Classification Using H & E Image Features” and is currently in the application process.
Ethics approval and consent to participate
This retrospective study was approved by the institutional review board of Zhongda Hospital, Southeast University, and Huai’an First People’s Hospital(No. 2021ZDSYLL353-P01), and the requirement for informed consent was waived. The study was enrolled in a clinical trial(No. ChiCTR2200065689).
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Zhang, J., Ding, F., Guo, Y. et al. AI-based prediction of androgen receptor expression and its prognostic significance in prostate cancer. Sci Rep 15, 3985 (2025). https://doi.org/10.1038/s41598-025-88199-7
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-88199-7
