
Abstract
Coral-associated microbes have essential roles in promoting and regulating host function and health. As climate change advances and other environmental perturbations increasingly impact corals, it is becoming ever more important that we understand the composition of the microbial communities hosted. Without this baseline it is impossible to assess the magnitude and direction of any future changes in microbial community structure. Here, we characterised both the bacterial and Symbiodiniaceae communities in four coral species (Diploastrea heliopora, Porites lutea, Pachyseris speciosa, and Pocillopora acuta) collected from Sabah, Malaysia. Our findings reveal distinct microbial communities associated with different coral species tending to reflect the varied life history strategies of their hosts. Microbial communities could be differentiated by collection site, with shifts in Symbiodiniaceae communities towards more stress tolerant types seen in samples collected on the shallow Sunda Shelf. Additionally, we identified a core microbiome within species and a more discrete core between all species. We show bacterial and Symbiodiniaceae communities are structured by host species and appear to be influenced by host life history characteristics. Furthermore, we identified a core microbiome for each species finding that several amplicon sequence variants were shared between hosts, this suggests a key role in coral health regardless of species identity. Given the paucity of work performed in megadiverse regions such as the Coral Triangle, this research takes on increased importance in our efforts to understand how the coral holobiont functions and how it could be altered as climate change advances.
Similar content being viewed by others


Introduction
Corals are keystone species in reef habitats, serving a multitude of roles in maintaining a healthy ecosystem1. In performing these roles, they are reliant on the symbioses that exist between the host and its microbial constituents, including, but not limited to bacteria and Symbiodiniaceae2,3. These symbiotic relationships are crucial for the healthy functioning of the coral holobiont, with bacteria involved in nutrient cycling, metabolism and immune system functioning4,5, while Symbiodiniaceae provide a significant proportion of their hosts nutritional requirements through photoautotrophy6. Importantly, these symbionts can also help promote adaptation to environmental change7,8,9,10,11. Given this, an understanding of the symbiotic microbial communities and how they may change is an important consideration in efforts to understand how corals are expected to adapt as climate change advances. However, without baseline work describing the current community structure and microbial compositions, determining the magnitude and direction of any changes is impossible.
Corals display a variety of life-history strategies that can influence microbial community composition12, with coral species, life-history and traits (e.g., competitive, weedy, stress-tolerant, opportunistic or generalist) all influencing microbial community assemblage13,14,15. Competitive species have fast growth rates and reproduce via broadcast spawning, whereas species classified as weedy tend to be brooders with smaller colony sizes and shorter generation times. Stress tolerant species have slow growth rates tending to reproduce by broadcast spawning and are highly fecund, with species described as generalist or opportunistic having moderate growth rates and a degree of overlap with competitive, weedy and stress-tolerant life histories, for a complete description of coral life history traits see Darling et al. 201216. Additionally, in terms of how corals shape their microbial community, they can be divided into two groups (i.e., microbial conformers or regulators)12. Conformers have a microbiome that changes in response to the environment, while regulators are able to maintain stable microbiomes across environments. Similarly, some microbial taxa show specialized versus generalist behaviors. Members of the Symbiodiniaceae genus, Durusdinium, specifically D. trenchii are considered more heat-tolerant and can confer an advantage to their host in elevated temperatures17,18,19, and some bacteria are implicated in the suppression of bacterial pathogens and play a role in maintaining a healthy microbiome, while others are involved in sulphur and nutrient cycling processes20. Here, we investigate the bacterial and Symbiombiodiniaceae communities of four coral species, Diploastrea heliopora, Porites lutea, Pachyseris speciosa and Pocillopora acuta, collected around the Malaysian state of Sabah (Borneo). Specifically, we determine whether (1) these communities show local spatial patterns and host-specificity, and (2) a ‘core microbiome’ is present within and amongst species. The concept of a ‘core microbiome’ was initially explored in humans where it was defined as a group of microbial taxa shared by all, or most humans21. The shared nature of these microbes led to suggestions of an important role in the maintenance of host functioning22, similar ideas have been applied in natural ecosystems where it is hypothesised that this core has roles in the maintenance of ecological functioning9,23,24. Here we apply this concept in an effort to determine whether a ‘core microbiome’ exists between the four coral host species collected in this work.
We hypothesise that the bacterial and Symbiodiniaceae communities will differ by host species, a consequence of their different life history strategies e.g., D. heliopora and P. lutea demonstrate stress tolerant strategies and traits, while P. speciosa is a generalist16,25 and P. acuta is classed as an opportunistic species26. Additionally, but to a lesser extent in comparison to host species, we expect that site specific environmental conditions will influence microbial community composition27.
This study seeks to characterise the microbial diversity associated with reef-building corals in an area that remains understudied but contains highly diverse and rich coral reef communities. Much of the world’s coral reef biodiversity resides in the global south, yet much of the research capacity is focused on the much less diverse and arguably less healthy reefs of the global north28. If coral reefs are to persist into the future it is critical that we work on understanding these megadiverse marine ecosystems and their essential microbial constituents, especially those at the epicenter of global marine biodiversity, for example the Coral Triangle and its surrounding regions. It is here that the evolutionary novelty required to adapt to a rapidly changing climate likely resides.
Methods
Sample collection
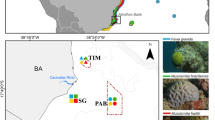
Corals from four sites in Sabah, Malaysia were collected between February and March 2020. Tissue samples measuring approximately 2 cm2 were collected from each of the following species Diploastrea heliopora, Pachyseris speciosa, Pocillopora acuta, and Porites lutea. Collections were made from reefs in Kota Kinabalu, Labuan, Lankayan, and Mataking, of these four sites Mataking is the only one located within the Coral Triangle biodiversity hotspot (Fig. 1), at each location 20 individual samples from each species were taken. All samples were collected from depths between 5 and 10 m and appeared visibly healthy with no apparent signs of bleaching or disease. Upon collection, samples were placed in individual, sterile Whirl–Pak bags and placed in 100% ethanol within 2 h, they were then stored at − 20 °C until transferred to − 80 °C prior to DNA extraction.

Location of sampling sites in Sabah, Malaysia. Figure was created using The Global Multi-Resolution Topography tool (https://gmrt.org/).
DNA extraction and library preparation
DNA extraction was performed with a Qiagen DNeasy Blood and Tissue Kit and followed all manufacturer’s instructions. PCR amplification of the bacterial 16S rRNA gene V4 region was performed using the 515F and 806R primers (515F: 5′- GTGYCAGCMGCCGCGGTAA -3′; 806R: 5′- GGACTACNVGGGTWTCTAAT-3′) and modified to include Illumina adaptors and unique barcodes following Caporaso et al.29. To reduce Plastid and mitochondrial amplification, peptide nucleic acids (PNAs) were added (mPNA: GGC AAG TGT TCT TCG GA; pPNA: GGC TCA ACC CTG GAC AG) (PNAGENE, Daejeon, South Korea). We followed the thermocycling conditions and protocol described in30. Briefly, PCR was performed in 25 µl volumes containing 1 μl of undiluted template, 0.1 μl of KAPA 3G Enzyme (Kapa Biosystems, Inc, Wilmington, MA, USA), 0.75 μl of each primer at 10 μM, 2.5 μl of mPNA and 2.5 μl pPNA at 50 μM, 1.5 μl of 1.5 mg/ml BSA, 12.5 μl KAPA PCR buffer and water to 25 µl. PCR cycling protocol was 94 °C for 180 s, followed by 35 cycles of 94 °C for 45 s, 75 °C for 10 s, 50 °C for 60 s and 72 °C for 90 s, with a final extension at 72 °C for 10 min. Negative controls were included to identify any potential contamination.
For Symbiodiniaceae, the nuclear ribosomal internal transcribed spacer 2 (ITS2) region was amplified following the two-step protocol described in31. Briefly, the ITS2 region was amplified using the SYM_VAR primer pair (SYM_VAR_5.8S2: 5′–GAA TTG CAG AAC TCC GTG AAC C–3′, SYM_VAR_REV: 5′–CGG GTT CWC TTG TYT GAC TTC ATG C–3′)32 in 12.5 µl reaction volumes containing 1 µl of sample DNA diluted 1:10 with PCR grade water, 0.5 µl of forward and reverse primers each at 10 μM, 6.25 µl of 2 × KAPA HiFi HotStart ReadyMix and water to make a final volume of 12.5 µl. PCR cycling conditions consisted of an initial step of 95 °C for 3 min followed by 25 cycles of 95 °C for 30 s, 55 °C for 30 s, and 72 °C for 30 s, with a final extension step at 72 °C for 5 min. PCR products were then cleaned using AMPure XP magnetic beads (Agencourt). A reduced cycle PCR was used to attach unique barcodes and Illumina adaptors 33, reactions were performed in 12.5 µl volumes containing 2 µL of amplicon from the previous round, 0.5 µl of each uniquely indexed primer at 10 μM, 6.25 µL of 2 × KAPA HiFi HotStart ReadyMix, and water to a total volume of 12.5 µl. PCR cycling conditions were 95 °C for 3 min for initial denaturation, followed by 8 cycles of 95 °C for 30 s, 55 °C for 30 s, and 72 °C for 30 s, with a final extension step at 72 °C for 5 min. PCR negatives were included again to identify any possible contamination.
Prior to pooling, PCR products were cleaned and normalized using SequalPrep™ Normalization Plates (Thermo Fisher Scientific, Waltham, MA, USA). Samples were pooled into two libraries (bacteria or Symbiodiniaceae) and each library was sequenced independently on the Illumina MiSeq platform (V3 chemistry, 300 bp paired-end reads, 30% Phi-X spike), with sequencing performed by Macrogen, Inc.
Sequencing and data processing
All bioinformatic steps, unless mentioned otherwise, were performed in R v4.3.134. For 16S rRNA gene amplicons, cutadapt v4.4 implemented in Python v3.11.4 was used to remove primers from demultiplexed sequences35,36. Quality profiles of reads were checked and reads filtered and trimmed using the DADA2 package v1.28.037. D. heliopora and P. speciosa forward and reverse reads were trimmed to 250 and 150 base pairs respectively. Filtering was performed with a maximum expected error setting of 2 for both forward and reverse reads. P. acuta and P. lutea forward and reverse reads were trimmed to 180 and 120 base pairs, respectively, with a maximum expected error setting of 2 and 5 for forward and reverse reads. All other settings remained at their defaults. Error rates were then estimated and paired dereplicated reads were used infer amplicon sequence variants (ASVs). Any chimeras were identified and removed along with contaminants using control “blank” samples with the prevalence method implemented in the decontam package v1.20.038. Taxonomic assignment was performed, to species level where possible, based on the SILVA v138.1 16S rRNA gene database39. ASVs that were assigned as “mitochondria” or “chloroplast” were removed. ASVs were further filtered to retain only those that were found in at least 5% of all samples40. Other than three P. speciosa samples (Kota Kinabalu: n = 2, Mataking: n = 1) all samples passed quality control and were used in downstream analysis.
Analysis of ITS2 Symbiodiniaceae samples was performed using SymPortal v0.3.2441. All steps were performed using SymPortal’s default settings. Sequence filtering and quality control were performed using MOTHUR, BLASTn was used identify Symbiodiniaceae, and minimum entropy decomposition (MED) nodes identified42,43. Defining intragenomic sequence variants (DIVs) and downstream type profiles of Symbiodiniaceae were then inferred within the SymPortal framework to assess community composition. Downstream analysis was performed for both DIVs and type profiles separately.
Community analysis
Symbiodiniaceae and bacterial communities were visualized on separate non-metric multidimensional scaling (nMDS) plots using the Bray–Curtis metric with ellipses indicating 95% confidence intervals. Permutational multivariate ANOVA (PERMANOVA) was performed with Bray–Curtis distances with host species and site as predictors using vegan v2.6.444. The Shannon diversity index for each sample was calculated and grouped by host species and site. Relative abundance bar plots by host species and site were constructed for samples to show distribution of bacterial phyla, and Symbiodiniaceae genera and type profiles. Only the top 20 ASVs and type profiles from each species were used for these plots. A plot of the majority sequence for type profiles was constructed to simplify the type profile relative abundance. This majority sequence is the sequence that makes up the largest abundance of each type profile, as identified by SymPortal. Individual sample relative abundance plots were also constructed and relative abundance was also plotted as heatmaps, grouped by host species and site.
The core microbiome was arbitrarily defined as bacterial ASVs present in at least 50% of individuals with a detection threshold of 1%45,46,47,48. The core_members function from the microbiome package v1.22.0 was used to extract these core microbes for all samples and for each host species49. These sequences were searched using BLASTn to infer their individual characteristics and presence in other studies.
Results
Bacteria
After quality control 3,171,579 bacterial reads were retained for analysis. Ordinations indicate that bacterial communities can be differentiated by host species, with clustering by species observed (Fig. 2a). This community differentiation is further supported by our PERMANOVA results which indicated significant differences between species (PREMANOVA R2 = 0.22, F = 32.55 p = 0.001). Plots of Shannon diversity indicate that the bacterial communities in P. speciosa and D. heliopora have the highest and second highest diversities in all but one location, Mataking. With bacterial diversity across all four species at Mataking appearing broadly similar (Fig. 3). In three of the four sites bacterial diversity is lowest in P. acuta (Fig. 3a). Our PERMANOVA tests indicate significant structuring in bacterial communities by site (PREMANOVA R2 = 0.08, F = 11.15 p = 0.001), with this structure supported by our ordinations (Fig. 4a). The bacterial phylum Proteobacteria is largely dominant across all host species, and is especially prevalent in P. lutea and P. acuta (SI Fig. 1–3). Bacteroidota has the highest abundance in P. speciosa (mean relative abundance 20.9 ± SD 0.10 and is largely absent in P. lutea and P. acuta (mean relative abundance 0.45 ± SD 0.002 and 3.41 ± SD 0.04 respectively) (SI Fig. 1).

Non-metric multidimensional scaling (nMDS) plots of Bray–Curtis distances for (a) bacterial ASVs and (b) Symbiodiniaceae sequences. Ellipses indicate 95% confidence intervals.

Shannon diversity plots of (a) bacterial communities and (b) Symbiodiniaceae profiles.

Non-metric multidimensional scaling (nMDS) plots of (a) bacterial ASVs and (b) Symbiodiniaceae sequences by each host species. Based upon the Bray–Curtis metric with ellipses indicating 95% confidence intervals.
Using the criteria defined above to identify a core microbiome (ASVs present in at least 50% of individuals with a detection threshold of 1%) D. heliopora had a core of 13 bacterial ASVs belonging to eight families and P. speciosa had 48 bacterial ASVs from 32 families. We found four bacterial ASVs from three bacterial families in the P. acuta samples, with the P. lutea core microbiome containing 16 bacterial ASVs from six families (Fig. 6, SI Fig. 4). Out of these ASVs, six were shared in the core microbiome of three host species while none were shared among all four host species’ core microbiome. The six ASVs shared between the three species belong to four families: Alteromonadaceae, Cyanobiaceae, Vibrionaceae, and Pseudoalteromonadaceae. Using the criteria described to define the core microbiome, no ASVs in the core microbiome are shared. However, 22 ASVs are found to be present across all four hosts.
Symbiodiniaceae
After quality control, 9,375,672 Symbiodiniaceae reads were retained for analysis. Ordinations indicate that Symbiodiniaceae communities can be differentiated by host species, (Fig. 2b). This community differentiation is further supported by our PERMANOVA results which indicated significant differences between species (PREMANOVA R2 = 0.12, F = 15.04 p = 0.001). For Symbiodiniaceae communities, P. speciosa shows variable diversity across all sites, of the four species it has the highest diversity at Kota Kinabalu and Labuan, while it shows the lowest diversity at Lankayan and Mataking. Symbiodiniaceae diversity in P. acuta is similar across Kota Kinabalu, Labuan and Lankayan, but of all four species it has the highest diversity in Mataking. While Symbiodiniaceae diversity in P. lutea and D. heliopora is broadly comparable across all four sites, and generally lower in comparison to the other species examined (Fig. 3b). However, it should be noted that rDNA copy number in Cladocopium can be five times higher than Durusdinium, therefore Symbiodiniaceae diversity metrics should be treated with a degree of caution, especially in mixed endosymbiont communities where both Cladocopium and Durusdinium are present. Nevertheless, these metrics can still give a broad over view of diversity provided these limitations are considered50. Symbiodiniaceae communities can be significantly differentiated by site (PREMANOVA R2 = 0.05, F = 6.30 p = 0.001). This is further supported by the ordinations, while there is evidence of clustering, the separation of communities is less well defined in comparison to microbial communities (Fig. 4a, b). Across all four species we identified 20 Symbiodiniaceae type profiles with all Symbiodiniaceae belonging to the genera, Cladocopium or Durusdinium (Fig. 5b, SI Fig. 5, 6). Each of the four host species are dominated by different type profiles, in D. heliopora, three of the four sites are dominated by type profile C3u/C3/C115, while Mataking is dominated by C3 and has C40, and type C40/C3 that is not found in the other locations. P. speciosa samples are largely composed of type C27 and D1, whereas Mataking has a reduced abundance of C27, a high abundance of C21 and C27/C15h types. P. acuta samples from three of the four sample sites are primarily composed of type profiles D1, or D1b, with Mataking differing markedly from other sites again, P. acuta from Mataking is dominated by C1d/C1, and C1d, both of which are absent in the other locations. In comparison to the other three host species, P. lutea, has a much more uniform Symbiodiniaceae type profile that is dominated by type C15 across all sites.

Bar plots showing relative abundance of (a) family of top 20 bacterial taxa and (b) the majority sequence of the top 20 Symbiodiniaceae profiles for each host species.

Heatmaps (a) showing relative abundance of all amplicon sequence variants (ASVs) found in the core microbiomes of each individual coral host species (b) ASVs identified as belonging to the core microbiome using the predefined criteria (occurring in 50% of all individuals with a detection threshold of 1%). KK Kota Kinabalu, LA Labuan, LY Lankayan, MT Mataking.
Discussion
This is the first examination and characterization of coral associated microbes to be performed around the Malaysian state of Sabah, on the island of Borneo. Collections spanned a distance of nearly 1000 km and include part of the Coral Triangle diversity hotspot as well as the South China Sea. Unsurprisingly, and similar to other studies performed throughout the Southeast Asian region26,27,51 we show that bacterial and Symbiodiniaceae communities from the four collected host species of coral are structured by, and can be differentiated by geographic location and species.
Bacterial community patterns
Life history characteristics of the coral host can be useful when describing the relationship between host and its associated microbial communities, for example, Diploastrea heliopora and Porites lutea are considered stress tolerant species52, Pocillopora acuta is described as an opportunistic colonizer, while Pachyseris speciosa is considered a generalist53,54. Broadly in line with these characteristics, and as previously reported26 we find that the more stress tolerant species tend to have less diverse microbial communities, whereas the more opportunistic and generalist species have a more diverse microbial communities which should allow them to persist in a wider range of conditions. This pattern is evident in bacterial communities with diversity highest in the generalist, P. speciosa. But, contrary to expectations the opportunistic colonizer P. acuta has the lowest bacterial diversity. Further work is needed to confirm this, but it is possible that P. acuta is a microbiome regulator and as such is able to select its microbial constituents. Adding weight to this suggestion, the low microbial diversity is indicative of a microbiome that has been tailored to host specific requirements through microbial winnowing55 with similar, low bacterial diversity reported in this species previously56,57. These species-specific differences are supported by our ordinations that show well defined clusters indicating differences in the hosted bacterial communities between coral species. Diploastrea heliopora shows the lowest degree of differentiation in bacterial community structure, this is consistent with work performed in Peninsular Malaysia48 and could be a consequence of stress tolerant species requiring fewer symbiotic partners for survival26,58. A similar, but less pronounced pattern is seen in P. lutea, another species described as stress tolerant. Bacterial community structure is more defined in Pocillopora acuta and Pachyseris speciosa, described as opportunistic or as a generalist respectively. It is likely that the life history characteristics of P. acuta and P. speciosa allows these species to take advantage of disturbance or exist in a wide range of habitats, to do this, it is probable they acquire bacteria that are prevalent in the local environment, hence we see clear differentiation in community structure between sampling locations. In fact, a number of studies exploring the microbiome of P. acuta and how it responds to new environments when transplanted indicate that it is strongly influenced by and quickly conforms to local conditions59,60,61.
The bacterial communities in all four coral species are dominated by Proteobacteria, and to a lesser extent Bacteroidota, Cyanobacteria, and Firmicutes. These general associations are well documented in corals throughout the world11,14,62,63,64,65. The bacterial family Colwelliaceae observed in P. speciosa and P. lutea has been implicated in buffering the negative effects of photooxidative stress linked to bleaching66, this family would be a good candidate for future research attempting to determine how microbes can contribute to coral stress adaptation. This association fits well with the generalist and stress tolerant life histories these corals have, and may in part be responsible for their ability to persist in stressful conditions.
Symbiodiniaceae community patterns
Coral-associated symbidiniaceae communities typically vary by species and geographic location26,67. The Porites lutea colonies examined here are dominated by Symbiodiniaceae type C15 and its variants, this C15 association is found throughout Southeast Asia52,67,68,69 and more generally throughout the world70. Similar to other work performed in the South China Sea71 we find the Diploastrea heliopora host is dominated by type C3u/C3/C115 at three of the four sites, whereas this type is absent from Mataking and of the four sites type C40/C3 is only present here. At all sites, and as previously reported from the Malay Peninsula26,58, type C27 is most prevalent in Pachyseris speciosa, with type C21 only found at Mataking. Pocillopora acuta is dominated by types D1 and D1b in three of the sites, whereas Mataking is dominated by types C1d and C1d/C1. The Symbiodiniaceae community composition of P. lutea shows little variation across all four sites, while the other three species have similar Symbiodiniaceae communities at Kota Kinabalu, Labuan and Lankayan, whereas Symbiodiniaceae communities differ considerably in P. speciosa, D. heliopora and P. acuta collected from Mataking in comparison to the other sample sites (Fig. 5b). This difference is particularly striking in P. acuta where a large shift from Durusdinium to Cladocopium is seen. Further work is needed to confirm this and with the data we have, we can only speculate why the samples collected from Mataking are frequently different. Unlike the other collection sites, Mataking is in close proximity to the deep waters of the Celebes Sea, whereas the other three sites are in shallower coastal water associated with the Sunda Shelf. Shallower water does heat up faster and corals found here could experience warmer waters and have shifted their Symbiodiniaceae communities in response to the more stress tolerant Durusdinium, members of this genus, particularly D. trenchii have been shown to confer an advantage in warmer waters17,18,71. Similar phenomena has previously been reported in Pocillopora spp from the eastern tropical pacific and has been suggested as a mechanism that may confer increased resilience to ocean warming11. Whereas the deeper water of the Celebes Sea could buffer and prevent heat stress in corals from Mataking, consequently we see a different Symbiodiniaceae community, one that is adapted to the different conditions experienced here. This site is also the only one within the Coral Triangle biodiversity hotspot, an area where coral biodiversity reaches its maxima72. Work shows that the composition of the coral community in the immediate area can influence the endosymbionts found in a host73. Given the elevated coral diversity of the Coral Triangle region, this could, in part also be responsible for the different Symbiodiniaceae communities seen at Mataking and could play a role in structuring Symbiodiniaceae communities in hosts that acquire their symbionts from the environment, rather than through vertical transmission. However, it should be noted that despite having significantly lower coral host diversity, previous work suggests the Caribbean actually has more Symbiodiniaceae species than the Indo-Pacific74. Although, this may change as high throughput DNA sequencing approaches are increasingly applied to examine Symbiodiniaceae communities and diversity throughout the Coral Triangle and other hotspots of marine biodiversity.
Both D. heliopora and P. speciosa have more mixed Symbiodiniaceae profiles at Kota Kinabalu in comparison to P. acuata and P. lutea, mixed communities have been implicated as advantageous in certain environmental conditions such as elevated temperatures or low light environments75,76. As the capital of Sabah, it is not unreasonable to suggest that the corals found at Kota Kinabalu are subject to the increased anthropogenic stresses associated with a human population that is larger than that at other locations in Sabah (e.g., terrestrial runoff). Further work is needed to confirm this, but, this mixed profile maybe a response to the increased stresses unique to Kota Kinabulu.
Core microbiome
Six core microbial ASVs are shared between three of the four corals—ASV2, ASV67, ASV1, ASV3, ASV6, and ASV7. These ASVs have been identified in other work examining coral microbial communities, this ubiquity suggests a role in promoting host health. ASV2 (Alteromonas spp) and ASV67 (Synechococcus CC9902 spp) are shared between P. speciosa, P. acuta, and P. lutea, while the remaining four are shared among D. heliopora, P. speciosa, and P. lutea.
Pseudoalteromonas (ASV 7) and Vibrio (ASVs 1 & 3) have been observed associated with numerous coral hosts, suggesting important roles of these bacteria in maintaining microbiome functioning48,77,78. ASVs 6 (Algicola bacteriolytica) & 7 (Pseudoalteromonas spp) are involved in dimethylsulfoniopropionate (DMSP) metabolism, DMSP metabolizers are part of the Beneficial Microorganisms for Corals (BMC) group and have been identified as having important roles in nutrient cycling and preventing bacterial pathogen outbreak, helping to maintain the hosts microbiome10. ASV2 has been implicated as having potential roles in nitrogen fixation and could offer pathways of alternative fixation during periods of stress (e.g., thermal bleaching) and thus increase coral host resilience79. ASV67 has been recovered from coral tissue where it is thought to have roles in nutrient uptake80,81,82.
Studies such as this are increasingly common place throughout the world, however, they remain comparatively rare in the global epicenter of marine biodiversity. Given this, studies such as the one performed here are valuable as they provide an initial foundation upon which we can build and develop more hypothesis driven research. For example, future work will more specifically examine differences in environmental conditions and coral community composition between sampling sites around the island of Borneo to determine why differences in microbial community composition exist. Additionally, as coral reef restoration activities become increasingly popular, it will become more important that we understand the diversity contained within an ecosystem, for example, if we do not know what is there already, how will we determine if restoration is successful? Additionally, a more complete understanding of the microbial constituents that are present in marine ecosystems could help restoration practitioners determine and prioritize the most suitable candidate sites for restoration.
Data availability
All raw sequences obtained in this study have been deposited in the National Center for Biotechnology Information under BioProject record PRJNA1028667.
References
Paoli, C., Montefalcone, M., Morri, C., Vassallo, P. & Bianchi, C. N. Ecosystem Functions and Services of the Marine Animal Forests 1271–1312 (Springer, 2017).
Blackall, L. L., Wilson, B. & Van Oppen, M. J. Coral—the world’s most diverse symbiotic ecosystem. Mol. Ecol. 24, 5330–5347 (2015).
Rabbani, G., Huang, D. & Wainwright, B. J. The mycobiome of Pocillopora acuta in Singapore. Coral Reefs 40, 1419–1427 (2021).
Ainsworth, T. D., Fordyce, A. J. & Camp, E. F. The other microeukaryotes of the coral reef microbiome. Trends Microbiol. 25, 980–991 (2017).
Dubinsky, Z. & Stambler, N. Coral Reefs: An Ecosystem in Transition (Springer Science & Business Media, 2010).
Muscatine, L. & Porter, J. W. Reef corals: Mutualistic symbioses adapted to nutrient-poor environments. BioScience 27, 454–460 (1977).
Rosenberg, E., Koren, O., Reshef, L., Efrony, R. & Zilber-Rosenberg, I. The role of microorganisms in coral health, disease and evolution. Nat. Rev. Microbiol. 5, 355–362 (2007).
Thompson, J. R., Rivera, H. E., Closek, C. J. & Medina, M. Microbes in the coral holobiont: partners through evolution, development, and ecological interactions. Front. Cell. Infect. Microbiol. 4, 176 (2015).
Bourne, D. G., Morrow, K. M. & Webster, N. S. Insights into the coral microbiome: underpinning the health and resilience of reef ecosystems. Annu. Rev. Microbiol. 70, 317–340 (2016).
Peixoto, R. S., Rosado, P. M., Leite, D. C. A., Rosado, A. S. & Bourne, D. G. Beneficial microorganisms for corals (BMC): proposed mechanisms for coral health and resilience. Front. Microbiol. 8, 341 (2017).
Palacio-Castro, A. M. et al. Increased dominance of heat-tolerant symbionts creates resilient coral reefs in near-term ocean warming. Proc. Natl. Acad. Sci. 120, e2202388120 (2023).
Ziegler, M. et al. Coral bacterial community structure responds to environmental change in a host-specific manner. Nat. Commun. 10, 3092 (2019).
Hernandez-Agreda, A., Leggat, W., Bongaerts, P., Herrera, C. & Ainsworth, T. D. Rethinking the coral microbiome: simplicity exists within a diverse microbial biosphere. mBio https://doi.org/10.1128/mBio.00812-18 (2018).
Liang, J. et al. Distinct bacterial communities associated with massive and branching scleractinian corals and potential linkages to coral susceptibility to thermal or cold stress. Front. Microbiol. 8, 979 (2017).
Sharp, K. H., Distel, D. & Paul, V. J. Diversity and dynamics of bacterial communities in early life stages of the Caribbean coral Porites astreoides. ISME J. 6, 790–801 (2012).
Darling, E. S., Alvarez-Filip, L., Oliver, T. A., McClanahan, T. R. & Côté, I. M. Evaluating life-history strategies of reef corals from species traits. Ecol. Lett. 15, 1378–1386 (2012).
Cunning, R. & Baker, A. C. Thermotolerant coral symbionts modulate heat stress-responsive genes in their hosts. Mol. Ecol. 29, 2940–2950 (2020).
Howells, E. J. et al. Corals in the hottest reefs in the world exhibit symbiont fidelity not flexibility. Mol. Ecol. 29, 899–911 (2020).
Qin, Z. et al. Diversity of Symbiodiniaceae in 15 coral species from the Southern South China Sea: potential relationship with coral thermal adaptability. Front. Microbiol. 10, 2343 (2019).
Peixoto, R. S., Rosado, P. M., Leite, D. C. A., Rosado, A. S. & Bourne, D. G. Beneficial microorganisms for corals (BMC): Proposed mechanisms for coral health and resilience. Front. Microbiol. https://doi.org/10.3389/fmicb.2017.00341 (2017).
Turnbaugh, P. J. et al. The human microbiome project. Nature 449, 804–810 (2007).
Risely, A. Applying the core microbiome to understand host–microbe systems. J. Anim. Ecol. 89, 1549–1558 (2020).
Trevathan-Tackett, S. M. et al. A horizon scan of priorities for coastal marine microbiome research. Nat. Ecol. Evol. 3, 1509–1520 (2019).
Wainwright, B. J. et al. The core mangrove microbiome reveals shared taxa potentially involved in nutrient cycling and promoting host survival. Environ. Microbiome 18, 47 (2023).
Tanzil, J. T. I. et al. A preliminary characterisation of Symbiodinium diversity in some common corals from Singapore. COSMOS 12, 15–27 (2016).
Ong, J. H. et al. Species and spatio-environmental effects on coral endosymbiont communities in Southeast Asia. Coral Reefs 41, 1131–1145 (2022).
Ng, M. S. et al. Highly diverse symbiodiniaceae types hosted by corals in a global hotspot of marine biodiversity. Microb. Ecol. 87, 92 (2024).
Fisher, R. et al. Global mismatch between research effort and conservation needs of tropical coral reefs. Conserv. Lett. 4, 64–72 (2011).
Caporaso, J. G. et al. Global patterns of 16S rRNA diversity at a depth of millions of sequences per sample. Proc. Natl. Acad. Sci. 108, 4516–4522 (2011).
Wainwright, B. J., Zahn, G. L., Afiq-Rosli, L., Tanzil, J. T. & Huang, D. Host age is not a consistent predictor of microbial diversity in the coral Porites lutea. Sci. Rep. 10, 14376 (2020).
Jain, S. S. et al. Homogenization of endosymbiont communities hosted by equatorial corals during the 2016 mass bleaching event. Microorganisms 8, 1370 (2020).
Hume, B. C. et al. An improved primer set and amplification protocol with increased specificity and sensitivity targeting the Symbiodinium ITS2 region. PeerJ. 6, e4816 (2018).
Soon, N., Quek, Z. B. R., Pohl, S. & Wainwright, B. J. More than meets the eye: characterizing the cryptic species complex and Symbiodiniaceae communities in the reef-dwelling nudibranch Pteraeolidia ‘semperi’ (Nudibranchia: Aeolidioidea) from Singapore. J. Molluscan Stud. 89, eyad011 (2023).
R Core Team. R: a language and environment for statistical computing. Vienna: R Foundation for Statistical Computing. (2021).
Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 17, 10–12 (2011).
Van Rossum G. Python Programming Language. USENIX Annu Tech Conf. Santa Clara, CA, 1–36. (2007).
Callahan, B. J. et al. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 13, 581–583 (2016).
Davis, N. M., Proctor, D. M., Holmes, S. P., Relman, D. A. & Callahan, B. J. Simple statistical identification and removal of contaminant sequences in marker-gene and metagenomics data. Microbiome 6, 1–14 (2018).
Quast, C. et al. The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res. 41, D590–D596 (2012).
Wainwright, B. J., Afiq-Rosli, L., Zahn, G. L. & Huang, D. Characterisation of coral-associated bacterial communities in an urbanised marine environment shows strong divergence over small geographic scales. Coral Reefs 38, 1097–1106 (2019).
Hume, B. C. et al. SymPortal: A novel analytical framework and platform for coral algal symbiont next-generation sequencing ITS2 profiling. Mol. Ecol. Resour. 19, 1063–1080 (2019).
Eren, A. M. et al. Minimum entropy decomposition: unsupervised oligotyping for sensitive partitioning of high-throughput marker gene sequences. ISME J. 9, 968–979 (2015).
Schloss, P. D. et al. Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol. 75, 7537–7541 (2009).
Oksanen, J. et al. The vegan package. Community Ecol. Package 10, 719 (2007).
Ainsworth, T. et al. The coral core microbiome identifies rare bacterial taxa as ubiquitous endosymbionts. ISME J. 9, 2261–2274 (2015).
Hernandez-Agreda, A., Leggat, W., Bongaerts, P., Herrera, C. & Ainsworth, T. D. Rethinking the coral microbiome: Simplicity exists within a diverse microbial biosphere. MBio 9, e00812 (2018).
Hernandez-Agreda, A., Leggat, W. & Ainsworth, T. D. A comparative analysis of microbial DNA preparation methods for use with massive and branching coral growth forms. Front. Microbiol. https://doi.org/10.3389/fmicb.2018.02146 (2018).
Kanisan, D. et al. Diversity and distribution of microbial communities associated with reef corals of the Malay Peninsula. Microb. Ecol. 85, 37–48 (2022).
Lahti L, Shetty S. microbiome R package. http://microbiome.github.io. Accessed 03 Feb 2025 (2012).
Davies, S. W. et al. Building consensus around the assessment and interpretation of Symbiodiniaceae diversity. PeerJ. 11, e15023 (2023).
Kanisan, D. P. et al. Diversity and distribution of microbial communities associated with reef corals of the Malay Peninsula. Microb. Ecol. 85, 37–48 (2023).
Thuaibah Isa Tanzil, J. et al. A preliminary characterisation of symbiodinium diversity in some common corals from Singapore. Cosmos 12, 1–13 (2017).
Hernandez-Agreda, A., Leggat, W., Bongaerts, P. & Ainsworth, T. D. The microbial signature provides insight into the mechanistic basis of coral success across reef habitats. mBio https://doi.org/10.1128/mbio.00560-16 (2016).
Fong, J. & Todd, P. A. Spatio-temporal dynamics of coral–macroalgal interactions and their impacts on coral growth on urbanised reefs. Mar. Pollut. Bull. 172, 112849 (2021).
Bernasconi, R. et al. Establishment of coral-bacteria symbioses reveal changes in the core bacterial community with host ontogeny. Front. Microbiol. https://doi.org/10.3389/fmicb.2019.01529 (2019).
Bergman, J. L., Shaw, T., Egan, S. & Ainsworth, T. D. Assessing the coral microbiome at the scale of tissue-specific habitats within the coral meta-organism. Front. Mar. Sci. https://doi.org/10.3389/fmars.2022.985496 (2022).
Morrow, K. M., Pankey, M. S. & Lesser, M. P. Community structure of coral microbiomes is dependent on host morphology. Microbiome. 10, 113 (2022).
Smith, E. G. et al. Low symbiodiniaceae diversity in a turbid marginal reef environment. Coral Reefs 39, 545–553 (2020).
Botté, E. S. et al. Reef location has a greater impact than coral bleaching severity on the microbiome of Pocillopora acuta. Coral Reefs 41, 63–79 (2022).
Deignan, L. K. & McDougald, D. Differential response of the microbiome of Pocillopora acuta to reciprocal transplantation within Singapore. Microb. Ecol. 83, 608–618 (2022).
Haydon, T. D. et al. Rapid shifts in bacterial communities and homogeneity of Symbiodiniaceae in colonies of Pocillopora acuta transplanted between reef and mangrove environments. Front. Microbiol. 12, 756091 (2021).
Cai, L. et al. Exploring coral microbiome assemblages in the South China Sea. Sci. Rep. 8, 2428 (2018).
Alvarez-Yela, A. C., Mosquera-Rendón, J., Noreña-P, A., Cristancho, M. & López-Alvarez, D. Microbial diversity exploration of marine hosts at Serrana Bank, a coral atoll of the seaflower biosphere reserve. Front. Mar. Sci. https://doi.org/10.3389/fmars.2019.00338 (2019).
Bibi, F., Naseer, M. I. & Azhar, E. I. Assessing the diversity of bacterial communities from marine sponges and their bioactive compounds. Saudi J. Biol. Sci. 28, 2747–2754 (2021).
Qi, Z. et al. Spatial and interspecific differences in coral-associated bacterial diversity in Hainan, China. Mar. Pollut. Bull. 175, 113321 (2022).
Rachmawati R. Differential responses of coral-associated microbiomes to elevated temperatures across the Indonesian Archipelago at species, local, and regional scales. UCLA. (2018).
Tan, Y. T. R. et al. Endosymbiont diversity and community structure in Porites lutea from Southeast Asia are driven by a suite of environmental variables. Symbiosis https://doi.org/10.1007/s13199-020-00671-2 (2020).
Quek, Z. B. R. et al. Limited influence of seasonality on coral microbiomes and endosymbionts in an equatorial reef. Ecol. Indic. 146, 109878 (2023).
Chankong, A., Kongjandtre, N., Senanan, W. & Manthachitra, V. Community composition of Symbiodiniaceae among four scleractinian corals in the eastern Gulf of Thailand. Reg. Stud. Mar. Sci. 33, 100918 (2020).
D’Angelo, C. et al. Local adaptation constrains the distribution potential of heat-tolerant Symbiodinium from the Persian/Arabian Gulf. ISME J. 9, 2551–2560 (2015).
Qin, Z. et al. Diversity of symbiodiniaceae in 15 coral species from the Southern South China Sea: Potential relationship with coral thermal adaptability. Front. Microbiol. https://doi.org/10.3389/fmicb.2019.02343 (2019).
Hoeksema, B. W. Delineation of the Indo-Malayan centre of maximum marine biodiversity: The coral triangle. In Biogeography, Time, and Place: Distributions, Barriers, and Islands (ed. Renema, W.) 117–118 (Springer, 2007).
Williamson, O. M. et al. Neighboring colonies influence uptake of thermotolerant endosymbionts in threatened Caribbean coral recruits. Coral Reefs 40, 867–879 (2021).
LaJeunesse, T. C. et al. Low symbiont diversity in southern Great Barrier Reef corals, relative to those of the Caribbean. Limnol. Oceanogr. 48, 2046–2054 (2003).
Souza, M. R. et al. Community composition of coral associated Symbiodiniaceae is driven by fine scale environmental gradients. Ecology https://doi.org/10.1101/2021.11.10.468165 (2021).
Bhattacharya, D., Stephens, T. G., Chille, E. E., Benites, L. F. & Chan, C. X. Facultative lifestyle drives diversity of coral algal symbionts. Trends Ecol. Evol. 39, 239–247 (2024).
Kuek, F. W. I. et al. The potential roles of bacterial communities in coral defence: A case study at Talang-talang reef. Ocean Sci. J. 50, 269–282 (2015).
Pootakham, W. et al. High resolution profiling of coral-associated bacterial communities using full-length 16S rRNA sequence data from PacBio SMRT sequencing system. Sci. Rep. 7, 2774 (2017).
Benavides, M., Bednarz, V. N. & Ferrier-Pagès, C. Diazotrophs: overlooked key players within the coral symbiosis and tropical reef ecosystems?. Front. Mar. Sci. https://doi.org/10.3389/fmars.2017.00010 (2017).
Lesser, M. P., Mazel, C. H., Gorbunov, M. Y. & Falkowski, P. G. Discovery of symbiotic nitrogen-fixing cyanobacteria in corals. Science 305, 997–1000 (2004).
Neulinger, S. C., Järnegren, J., Ludvigsen, M., Lochte, K. & Dullo, W.-C. Phenotype-specific bacterial communities in the cold-water coral Lophelia pertusa (Scleractinia) and their implications for the coral’s nutrition, health, and distribution. Appl. Environ. Microbiol. 74, 7272–7285 (2008).
Tong, F., Zhang, P., Zhang, X. & Chen, P. Impact of oyster culture on coral reef bacterioplankton community composition and function in Daya Bay, China. Aquac. Environ. Interact. 13, 489–503 (2021).
Acknowledgements
We gratefully acknowledge field assistance from Sam Shu Qin, the marine biologists at Reef Guardian and Mataking Reef Resort as well as support staff and postgraduate students from Universiti Malaysia Terengganu (UMT) and Universiti Malaysia Sabah (UMS) for their assistance during sample collection.
Funding
This study was funded by Yale-NUS grants A-8001384-00-00, A-0007214-00-00, A-0007210-00-00.
Author information
Authors and Affiliations
Contributions
GR Data analysis, lab work, wrote first draft. LAR field work, logistics, read and improved first draft. JNL field work, logistics, permitting, read and improved first draft. ZW field work, logistics, permitting, read and improved first draft, BW responsible for experimental concept and design, supervised all work, read and edited each version.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Research and export permits
Research permits and licenses were obtained from the Malaysian Department of Marine Park (Permit No: JTLM 630-7 Jld. 9 (9)) and Sabah Biodiversity Council (Access License: JKM/MBS.1000-2/2 JLD.10 (65-69); Export License: JKM/MBS.1000-2/3 JLD.4 (51)). Coral tissue samples were shipped to Singapore according to CITES regulations (Export Permit No: 3275 (2020 6 ECTS1); Import Permit No: 20SG0078l2Cl).
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Rabbani, G., Afiq-Rosli, L., Lee, J.N. et al. Effects of life history strategy on the diversity and composition of the coral holobiont communities of Sabah, Malaysia. Sci Rep 15, 4459 (2025). https://doi.org/10.1038/s41598-025-88231-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-025-88231-w
