
Abstract
Rotavirus (RV) is a leading pathogen causing diarrhea in children. In this study, a total of 51 fecal samples from children with RV enteritis, 29 post-treatment fecal samples, and 38 fecal samples from age-matched healthy controls were collected. Microbial DNA was isolated from the samples followed by high throughput Illumina sequencing targeting 16 S rRNA gene. Compared to the healthy group, the RV-infected group exhibited reduced microbial diversity. Both groups shared Firmicutes as the dominant phylum. Additionally, the abundance of Proteobacteria increased significantly in the RV-infected group. At the genus level, among the top 50 most abundant genera, 34 showed significant differences, with these differential genera correlating with certain clinical indicators such as dehydration levels and C-reactive protein (CRP). Notably, there were no significant differences in the microbiota before and after treatment in RV-infected children. Only 8.82% (3/34) of the differential genera in the post-treatment group showed a recovery trend towards the healthy state. This study enhances the understanding of how RV infection alters the gut microbiota structure in children and provides a scientific basis for improving clinical diagnosis and treatment strategies.
Similar content being viewed by others


Introduction
The human gut microbiome is highly complex and dynamic, with a population far exceeding the total number of human cells1,2,3,4. These microorganisms, predominantly bacteria, coexist symbiotically with their host, playing crucial roles in various physiological processes5,6. Through their structure and metabolic products, they regulate host metabolism and maintain homeostasis7,8,9. Changes in the gut microbiota are closely linked to a wide range of diseases10,11.
Rotavirus (RV) is a non-enveloped double-stranded RNA virus and the predominant pathogen causing diarrhea in children under 5 years old, transmitted via the fecal-oral route. Despite the World Health Organization (WHO) introducing vaccines over a decade ago to prevent human RV infections, the virus still causes over 200,000 deaths annually12,13. Viral infections, including poliovirus, norovirus, influenza virus, and murine rotavirus, can alter the structure and function of the gut microbiota14,15,16,17,18,19. Studies have shown that RV infection increases mucin secretion in the small intestine mucosa, altering the diversity and composition of the gut microbiota20. RV is influenced by mucin-degrading microbes; for instance, Bacteroides and Akkermansia species have mucin-degrading capabilities that reduce RV’s ability to bind to mucins21,22, thereby promoting RV infection. RV infection has been demonstrated to reduce the diversity of the gut microbiome, indicating a reciprocal relationship and mutual influence between RV and the gut microbiota. Post-RV infection, the gut microbiota is temporarily altered by reduced diversity and increased prevalence of opportunistic pathogens23.
Research on rotavirus enteritis and the gut microbiota, both domestically and internationally, has primarily focused on comparisons between diseased children and healthy controls. However, studies investigating changes in the gut microbiota of children with rotavirus enteritis before and after treatment are relatively scarce. Therefore, this study utilizes 16 S rRNA gene sequencing technology to explore the characteristics of the gut microbiota in children with rotavirus enteritis and its changes before and after treatment, aiming to provide clinical insights into the impact of rotavirus enteritis on the gut microbiota of infants and young children.
Result
Participants’ characteristics
There were no significant differences between the RV group and the healthy control group in terms of gender, delivery method, age, height, and weight (P > 0.05) (Supplementary Table 1).
Among the 51 children in the RV group, 29 (56.9%) had a fever and 22 (43.1%) had no fever, and 34 (66.7%) had a history of antibiotic exposure. The length of hospitalization was 3.44 ± 1.25 days. Of these 51 children, 32 had mild dehydration, 16 had moderate dehydration, and 3 had severe dehydration. The average WBC count was 13.07 ± 6.84 × 10⁹/L, and the average CRP level was 6.02 ± 3.47 mg/L. The clinical normal range for WBC is 6–15 × 10⁹/L, and for CRP is 0.17–2.2 mg/L. Detailed results for other clinical indicators, including AST, ALT, BUN, and Scr, are presented in Table 1.
Data quality control and amplicon sequence variant clustering
The RV and control groups obtained a total of 6,747,480 raw sequences. After removing low-quality sequences and denoising, 6,448,250 high-quality reads (95.6% of the total) were retained. There were 73,251.18 ± 14,864.17 valid sequences in RV group and 71,380.00 ± 13,833.51 in the control group (P = 0.547). No significant difference was observed in the number of denoised sequences between the two groups. Pan/core species analysis curves (Fig. 1A,B) and rarefaction curves (Supplementary Fig. 1) approaching a plateau or asymptote indicate that the sample size and sequencing depth were adequate.
The valid sequences from both groups were clustered into amplicon sequence variants at 100% similarity, and the amplicon sequence variant (ASV) abundance for each sample was counted. A total of 1009 ASVs were detected across all samples. In the RV group, unique ASVs accounted for 24.48% (247/1009) of the total ASVs, while in the control group, unique ASVs accounted for 18.53% (187/1009). Additionally, 56.99% (575/1009) of the ASVs were shared between both groups (Fig. 1C).

The Pan/core curves and Venn diagram descriptions for the RV and control groups. (A) Pan/core curve of the RV group; (B) and control group; (C) Venn diagram illustrating unique and shared ASVs associated with RV group and control group intestinal microbiota.
Alpha diversity and beta diversity analysis of gut microbiota
In this study, alpha diversity analysis was based on ASV, Chao1, and Ace indices, which reflect species richness, as well as the Shannon and Simpson indices, which reflect diversity. The alpha diversity indices of the gut microbiota in the RV group and the control group were compared, as shown in Supplementary Tables 2 and Fig. 2, revealing significant differences between the two groups (P < 0.001). These results indicate that the diversity and richness of the gut microbiota in the RV group were lower than those in the control group.

Box plots of the alpha diversity indices of the gut microbiota in the two groups (***P < 0.001, ****P < 0.0001).
To analyze beta diversity, principal coordinate analysis (PCoA) was performed (Fig. 3A), revealing clustering trends within the gut microbiota of each group. Further analysis using analysis of similarities (ANOSIM) (Fig. 3B) showed significant differences in gut microbiota distribution between the two groups (P = 0.001, R = 0.186).

Beta diversity analysis between RV group and healthy control group. (A) show the two-dimensional PCoA plot of samples: circles represent 95% confidence intervals; different colors or shapes denote different sample groups, with closer proximity indicating greater similarity in species composition for each sample. (B) ANOSIM similarity analysis: the horizontal axis labeled “between” indicates the between-group differences, and “RV” and “control” represent the within-group differences for the corresponding groups.
Composition of gut microbiota
In this study, 15 phyla and 274 genera were identified in both the RV and control groups. At the phylum level, the top four phyla accounted for 99.38% of the total relative abundance: Firmicutes (50.16%), Actinobacteriota (21.05%), Bacteroidota (14.36%), and Proteobacteria (13.80%). In the RV group, the proportions of Firmicutes and Proteobacteria increased compared to the control group. The top four phyla in both groups were Firmicutes, Actinobacteria, Proteobacteria, and Bacteroidetes, with Firmicutes and Actinobacteria occupying the first and second positions, respectively (Fig. 4A).
At the genus level, the top 50 most abundant genera accounted for 97.73% of the total detected genera. The top 10 genera comprised 77.81% of the total genera, with the following abundances: Bifidobacterium (20.28%), Enterococcus (11.48%), Bacteroides (11.33%), Escherichia-Shigella (8.46%), Veillonella (6.99%), Streptococcus (5.67%), Faecalibacterium (5.49%), Blautia (3.18%), Megamonas (2.57%), and Enterobacter (2.36%). In the RV group, Enterococcus (19.75%) and Bifidobacterium (17.87%) were predominant, with Lactobacillus (2.86%) and Parabacteroides (2.25%) being unique to the RV group. In the control group, Bifidobacterium (23.51%) and Bacteroides (13.51%) were more prevalent, with Ruminococcus-gnavus-group (4.96%) and Anaerostipes (2.30%) being unique to the control group Fig. 4B).

The gut microbiota composition at the phylum and genus levels in the RV and control groups. (A) The two groups formed a histogram of intestinal microbiota at the phyla, and the fractions with an abundance of less than 1% were combined into the others. (B) Heatmap of the top 50 genera by relative abundance.
Analysis of gut microbiota differences
Species differences between the two groups were analyzed at both the phylum and genus levels, with a total of 1 phylum and 34 genera showing significant differences. At the phylum level, the relative abundance of Proteobacteria was significantly higher in the RV group.
At the genus level, the RV group showed a significant increase in the abundance of Enterococcus, Streptococcus, Escherichia-Shigella, Lactobacillus, Raoultella, Clostridioides, unclassified Lactobacillaceae, Phascolarctobacterium, Neisseria, Enterobacteriaceae-unclassified, and Rothia. In contrast, there was a significant decrease in the abundance of Clostridium-innocuum-group, Butyricicoccus, Hungatella, Lachnoclostridium, Monoglobus, Dialister, Lachnospiraceae-NK4A136-group, Dorea, Ruminococcus torques group, Romboutsia, Clostridium sensu stricto1, Prevotella, Collinsella, Eubacterium hallii group, Fusicatenibacter, Ruminococcus, unclassified Lachnospiraceae, Roseburia, Anaerostipes, Enterobacter, Ruminococcus gnavus group, Bacteroides and Blautia, in the RV group (Fig. 5).

Linear discriminant analysis (LDA) Effect Size Histogram for Marker Species: The vertical axis represents the taxa with significant differences between the groups, and the horizontal axis shows the logarithmic LDA scores for each taxon. Taxa are ranked according to their scores, indicating their specificity in sample grouping. The longer the bar, the more significant the difference.
Analysis of differential taxa and clinical correlations
A Spearman correlation analysis was conducted between the relative abundance of 34 differential genera and clinical data (WBC, CRP, ALT, AST, BUN, Scr, fever status, dehydration degree, antibiotic exposure history) (Fig. 6). The results indicate that there were 10 positive correlations and 16 negative correlations between the differential genera and clinical indicators. Specifically, Rothia and Enterococcus were positively correlated with CRP, while Anaerostipes, Lachnospiraceae-unclassified, and Lachnoclostridium were negatively correlated with CRP. Streptococcus, Rothia, and Lactobacillales-unclassified were positively correlated with the degree of dehydration, Blautia, Butyricicoccus, Phascolarctobacterium, Dorea, Escherichia-Shigella and Ruminococcus torgues were negatively correlated with the degree of dehydration. Bacteroides was negatively correlated with ALT. Hungatella, Clostridium-innocuum-group, Enterobacter, and Raoultella were positively correlated with BUN. Eubacterium-hallii-group, Phascolarctobacterium, Clostridium sensu stricto 1, and Lachnoclostridium were negatively correlated with fever. Antibiotics administration was positively correlated with Enterococcus and negatively correlated with Lactobacillus and Anaerostipes.

Distribution plot of correlation analysis between differential genera relative abundance and clinical data (*P < 0.05, **P < 0.01).
Differential changes in gut microbiota abundance before and after treatment in children with rotavirus enteritis
There were 51 cases before treatment (RV group) and 29 cases after treatment. Data assessment was conducted by calculating the sequence number, sequence length, and other parameters at each stage, with the preprocessing results summarized in Supplementary Table 3. There were no significant differences in ASV, Chao1 index, Ace index, Shannon index, or Simpson index between the pre-treatment and post-treatment groups (P > 0.05) (Supplementary Tables 4 and Supplementary Fig. 2). PCoA analysis showed that the gut microbiota clusters before and after treatment were similar, with no significant distinction (ANOSIM, R = 0.004, P = 0.443) (Supplementary Fig. 3). Both alpha diversity and beta diversity analyses indicated no significant changes in species diversity, richness, or microbiota clustering trends before and after treatment in children with RV enteritis.
An analysis of the relative abundance changes of the differential genera between the RV and healthy control groups was performed before and after treatment. Out of the 34 differential genera, three showed significant changes in relative abundance: an unclassified genus of Enterobacteriaceae (P = 0.0382), the genus Neisseria (P = 0.0056), and the genus Prevotella (P = 0.0489) (Supplementary Table 5). The relative abundance trends of these three differential genera indicated that post-treatment, the relative abundance of the unclassified Enterobacteriaceae and Neisseria decreased compared to pre-treatment levels, while the relative abundance of Prevotella increased. All three genera’s relative abundance trends post-treatment moved towards the levels observed in the healthy control group (Fig. 7).

Trend of relative abundance changes in differential bacterial genera.
Discussion
The gut microbiota is a complex ecosystem formed by multiple interacting factors, characterized by a wide variety of bacterial species and high genetic richness. The interaction between rotavirus and the gut microbiota is bidirectional23,24,25. Previous studies have primarily focused on comparing RV-infected children with healthy children26,27,28. However, research on the microbiome analysis and differential bacterial genera before and after RV enteritis treatment is relatively limited. This study demonstrates significant differences in microbiome diversity and taxonomic composition between RV-infected children and healthy controls. Similarly, differences in the microbiome were also observed before and after treatment in RV-infected children.
We sequenced 118 fecal samples to describe the composition of their gut microbiota and the dynamic trends before and after treatment. Compared with the healthy control group, alpha diversity analysis indicates that microbial diversity and richness are reduced in children with RV infection, and the microbiota was in a dysbiotic state, consistent with previous studies23,29,30,31,32. Beta diversity analysis and ANOSIM similarity analysis revealed clustering trends and structural changes in the gut microbiota of children with RV enteritis. Compared to pre-treatment, both alpha diversity and beta diversity analyses showed no significant difference. After treatment, only 8.82% of the differential genera (3/34) showed a recovery trend towards the healthy state, indicating no significant recovery of the gut microbiota during hospitalization. This result suggests that the complete recovery of the gut microbiota may require a longer period.
At the phylum level, we observed that Firmicutes dominated both the RV group and the healthy control group. Additionally, the abundance of Proteobacteria was significantly higher in the RV group compared to the control group. This finding is similar to that of Li et al.31, who observed an increased abundance of Proteobacteria in children with rotavirus infection compared to the control group. As reported by Engevik et al.20, RV infectivity and pathogenicity depend on changes in the microbiota, particularly the increase in Proteobacteria and decrease in Firmicutes. Sohail et al.27 also indicated that RV infection is positively correlated with an increased proportion of Proteobacteria. In a recent study28, the RV group was divided into RV-infected diarrheal and non-diarrheal groups. It was found that the Firmicutes phylum dominated the non-diarrheal group, while the Proteobacteria phylum had a higher proportion in the diarrheal group. In diarrheal samples, frequent flushing and watery stools could lead to the loss of intestinal contents, potentially altering the intestinal environment.
Previous studies have shown that the Bifidobacterium genus is considered a beneficial bacterium with protective effects33,34. It is noteworthy that, although our study did not find a significant statistical difference in the relative abundance of Bifidobacterium between the RV group and the control group, its relative abundance decreased in RV enteritis children (23.51% in the control group vs. 17.87% in the RV group), indicating that Bifidobacterium is still affected by RV infection, although not significantly. Probiotic preparations made from Bacillus have been reported in the treatment of gut dysbiosis35. In this study, the relative abundance of the Clostridium-innocuum-group and Clostridium genus increased significantly in healthy children (Fig. 5), suggesting they could be candidate gut microbiota for RV enteritis treatment, providing guidance for subsequent clinical applications.
The main clinical symptom of rotavirus infection is watery diarrhea, which can easily lead to dehydration and changes in inflammatory indicators36,37,38. Dehydration and inflammatory responses alter the gut microecological environment, frequently resulting in changes to the structure and composition of the microbiota39,40. Spearman correlation analysis showed significant correlations between some differential genera and clinical indicators, such as dehydration levels and CRP (Fig. 6). These findings help further understand the gut microbiota characteristics of children with RV enteritis. The correlation between differential genera and clinical indicators provides important evidence for exploring the mechanism of action between gut microbiota and clinical symptoms in children with RV enteritis.
This study has certain limitations. Firstly, the sample size is relatively small, and there is a significant difference in the number of samples collected before and after treatment. Increasing the sample size and ensuring an equal number of samples before and after treatment could yield more valuable experimental data. Additionally, we only observed the dynamic changes in the gut microbiota of children during hospitalization and did not track their gut microbiota status after discharge. Consequently, we are unable to determine the exact duration required for the gut microbiota to restore to a healthy state post-treatment. Future research should further investigate this issue to fully understand the long-term recovery process of the gut microbiota.
Conclusion
Rotavirus infection disrupts the balance of gut microbiota, leading to dysbiosis. Certain differential microbiota genera are correlated with clinical indicators. Furthermore, the gut microbiota cannot restore to a healthy state in the short term after RV infection, and Further studies are needed to explore the time required for its recovery.
Methods
Trial participants
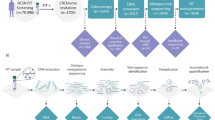
From October 2020 to June 2022, stool samples were collected from 51 children with diarrhea (RV group/pre-treatment group), 29 children post-treatment (post-treatment group), and 38 healthy controls (control group) at the Affiliated Women’s and Children’s Hospital of Ningbo University. The inclusion criteria for the RV group were under 3 years of age, with diarrhea and positive for rotavirus in stool samples by colloidal gold immunochromatography (ENCODE, Zhuhai, China). The post-treatment group consisted of a subset of children from the RV group who received medical treatment, including intravenous nutritional therapy, an appropriate diet, mucosal protectants, and probiotics, with dosages adjusted according to the child’s weight and disease severity. The control group included healthy children under 3 years of age who were asymptomatic for diarrhea, rotavirus-negative by colloidal gold immunochromatography (ENCODE, Zhuhai, China), and matched to the RV group in terms of the study period. Children with known organic diseases, those who had undergone gastrointestinal surgery, had active bacterial infections (e.g., Escherichia, Salmonella, Shigella), stool fungi or parasites, or those who had used immunosuppressants, antibiotics, or probiotics within the 4 weeks prior to sampling, were excluded. All samples tested negative for bacteria (including Escherichia, Salmonella, Shigella, etc.) and other prevalent enteric viruses (including norovirus, adenovirus, astrovirus, etc.). This study was conducted in accordance with the Declaration of Helsinki and was formally approved by the Ethics Committee of the Affiliated Women’s and Children’s Hospital of Ningbo University, Zhejiang, China. Informed consent was obtained from the parents and/or legal guardians for participation in the study.
Sample collection, DNA extraction, and 16 S rRNA gene sequencing
In a clean environment, 2 g of fresh fecal samples were collected using sterile fecal sampling kits and stored at -80 °C for subsequent analysis. For children diagnosed with RV enteritis, fresh fecal samples were collected at the time of diagnosis (pre-treatment) and at discharge (post-treatment). Genomic DNA from the fecal samples was extracted using the TIANGEN Stool DNA Kit (TIANGEN, Beijing, China), following the manufacturer’s instructions, and stored at − 80 °C.
The V3V4 region of the bacterial 16 S rRNA gene was amplified using the forward primer 338 F (5’-ACTCCTACGGGAGGCAGCA-3’) and the reverse primer 806R (5’-GGACTACHVGGGTWTCTAAT-3’). The PCR products were analyzed using 2% agarose gel electrophoresis and purified with the AxyPrep DNA Gel Extraction Kit (Axygen, USA) and Vazyme VAHTS DNA Clean Beads (Vazyme, Nanjing, China). Fluorescent quantification was performed using the Quant-iT PicoGreen dsDNA Assay Kit (Invitrogen, USA) and Microplate Reader (Promega, USA). Sequencing libraries were prepared using the TruSeq Nano DNA LT Library Prep Kit (Illumina, USA), which was then subjected to high-throughput sequencing on the Illumina MiSeq platform (Illumina, USA).
Bioinformatic analysis
The raw data obtained from the Illumina platform were processed to remove primer sequences, trimmed, merged, and filtered, resulting in effective Amplicon Sequence Variants (ASVs). QIIME2 was used to annotate each ASV representative sequence, and the RDP classifier was employed for taxonomic analysis to assess the richness and diversity of related species. Pan/core and rarefaction curves were generated using R vegan package to reflect sequencing depth. Based on the taxonomic analysis results, the compositional differences in community structures at the phylum and genus levels were analyzed.
Alpha diversity analysis was conducted using Mothur software (version v.1.30.1), with Chao1, Ace, Simpson, and Shannon indices calculated to characterize community diversity. Beta diversity analysis was employed to compare microbial community differences between groups, with Principal Coordinates Analysis (PCoA) and Non-Metric Multidimensional Scaling (NMDS) used to visualize structural differences. Linear Discriminant Analysis Effect Size (LEfSe) was performed using default parameters to identify significantly differentially abundant taxa between the two groups. Clustering correlation heatmap with signs was performed using the OmicStudio tools at https://www.omicstudio.cn.
Statistical analyses
All statistical analyses were carried out using SPSS Version 21.0 (SPSS, Inc., Chicago, USA) and GraphPad Prism 8.0 (GraphPad, Inc., California, USA). Normally distributed data were presented as mean ± standard deviation (SD). Comparisons between groups were conducted using independent t-tests or chi-square tests. Beta diversity differences between groups were assessed using PCoA. Spearman’s rank correlation was employed to evaluate associations between the microbiome and clinical indicators. The above correlation statistical analysis, with P < 0.05, indicated that the differences were significant.
Data availability
The sequence data supporting the results of this study are available in the NCBI SRA repository (BioProject ID: PRJNA1185898) at the following link: https://dataview.ncbi.nlm.nih.gov/object/PRJNA1185898?reviewer=e0qio2556noma8erhdp6vuvhvu. The datasets generated and/or analyzed during the current study are available from the corresponding author on reasonable request.
References
Adak, A. & Khan, M. R. An insight into gut microbiota and its functionalities. Cell. Mol. Life Sci. 76, 473–493. https://doi.org/10.1007/s00018-018-2943-4 (2019).
Shoemaker, W. R. A macroecological perspective on genetic diversity in the human gut microbiome. PLoS One. 18, e0288926. https://doi.org/10.1371/journal.pone.0288926 (2023).
Zhao, S. et al. Adaptive evolution within gut microbiomes of healthy people. Cell Host Microbe 25, 656–667 e658 (2019). https://doi.org/10.1016/j.chom.2019.03.007
Sender, R., Fuchs, S. & Milo, R. Revised estimates for the number of human and bacteria cells in the body. PLoS Biol. 14, e1002533. https://doi.org/10.1371/journal.pbio.1002533 (2016).
Guan, L. & Liu, R. The role of diet and gut microbiota interactions in metabolic homeostasis. Adv. Biol. (Weinh). 7, e2300100. https://doi.org/10.1002/adbi.202300100 (2023).
Rowan-Nash, A. D., Korry, B. J., Mylonakis, E. & Belenky, P. Cross-domain and viral interactions in the microbiome. Microbiol. Mol. Biol. Rev. 83, e00044–e00018. https://doi.org/10.1128/MMBR.00044-18 (2019).
Ansaldo, E., Farley, T. K. & Belkaid, Y. Control of immunity by the Microbiota. Annu. Rev. Immunol. 39, 449–479. https://doi.org/10.1146/annurev-immunol-093019-112348 (2021).
Zhou, B. et al. Intestinal flora and disease mutually shape the regional immune system in the intestinal tract. Front. Immunol. 11, 575. https://doi.org/10.3389/fimmu.2020.00575 (2020).
Wu, X., Wei, Q., Wang, X., Shang, Y. & Zhang, H. Evolutionary and dietary relationships of wild mammals based on the gut microbiome. Gene 808, 145999. https://doi.org/10.1016/j.gene.2021.145999 (2022).
Fan, Y. & Pedersen, O. Gut microbiota in human metabolic health and disease. Nat. Rev. Microbiol. 19, 55–71. https://doi.org/10.1038/s41579-020-0433-9 (2021).
Khalil, M. et al. Unraveling the role of the human gut Microbiome in Health and diseases. Microorganisms 12 https://doi.org/10.3390/microorganisms12112333 (2024).
Banyai, K., Estes, M. K., Martella, V. & Parashar, U. D. Viral gastroenteritis. Lancet 392, 175–186. https://doi.org/10.1016/S0140-6736(18)31128-0 (2018).
Florez, I. D., Nino-Serna, L. F. & Beltran-Arroyave, C. P. Acute infectious diarrhea and gastroenteritis in children. Curr. Infect. Dis. Rep. 22, 4. https://doi.org/10.1007/s11908-020-0713-6 (2020).
Yang, Y., An, R., Lyu, C. & Wang, D. Interactions between human norovirus and intestinal microbiota/microbes: A scoping review. Food Microbiol. 119, 104456. https://doi.org/10.1016/j.fm.2023.104456 (2024).
Zhao, T. et al. Influence of gut microbiota on mucosal IgA antibody response to the Polio vaccine. NPJ Vaccines. 5, 47. https://doi.org/10.1038/s41541-020-0194-5 (2020).
Zhang, Y. et al. Signals from intestinal microbiota mediate the crosstalk between the lung-gut axis in an influenza infection mouse model. Front. Immunol. 15, 1435180. https://doi.org/10.3389/fimmu.2024.1435180 (2024).
Robinson, C. M. Enteric viruses exploit the microbiota to promote infection. Curr. Opin. Virol. 37, 58–62. https://doi.org/10.1016/j.coviro.2019.06.002 (2019).
Nagai, M. et al. High body temperature increases gut microbiota-dependent host resistance to influenza a virus and SARS-CoV-2 infection. Nat. Commun. 14, 3863. https://doi.org/10.1038/s41467-023-39569-0 (2023).
Shi, Z. et al. Segmented filamentous Bacteria prevent and cure Rotavirus infection. Cell 179, 644–658. https://doi.org/10.1016/j.cell.2019.09.028 (2019). e613.
Engevik, M. A. et al. Rotavirus infection induces glycan availability to promote ileum-specific changes in the microbiome aiding rotavirus virulence. Gut Microbes. 11, 1324–1347. https://doi.org/10.1080/19490976.2020.1754714 (2020).
Abbasi, A. et al. A critical review on Akkermansia muciniphila: Functional mechanisms, technological challenges, and safety issues. Probiotics Antimicrob. Proteins. 16, 1376–1398. https://doi.org/10.1007/s12602-023-10118-x (2024).
Yamaguchi, M. & Yamamoto, K. Mucin glycans and their degradation by gut microbiota. Glycoconj. J. 40, 493–512. https://doi.org/10.1007/s10719-023-10124-9 (2023).
Kim, A. H., Hogarty, M. P., Harris, V. C. & Baldridge, M. T. The complex interactions between Rotavirus and the gut microbiota. Front. Cell. Infect. Microbiol. 10, 586751. https://doi.org/10.3389/fcimb.2020.586751 (2020).
Zhao, W. et al. Analysis of the intestinal microbial community altered during rotavirus infection in suckling mice. Virol. J. 18, 254. https://doi.org/10.1186/s12985-021-01727-5 (2021).
Barko, P. C., McMichael, M. A., Swanson, K. S. & Williams, D. A. The gastrointestinal microbiome: A review. J. Vet. Intern. Med. 32, 9–25. https://doi.org/10.1111/jvim.14875 (2018).
Bao, S. et al. Viral metagenomics of the gut virome of diarrheal children with Rotavirus A infection. Gut Microbes. 15, 2234653. https://doi.org/10.1080/19490976.2023.2234653 (2023).
Sohail, M. U. et al. Microbiome profiling of rotavirus infected children suffering from acute gastroenteritis. Gut Pathog. 13, 21. https://doi.org/10.1186/s13099-021-00411-x (2021).
Manouana, G. et al. Gut microbiota in vaccine naive Gabonese children with rotavirus A gastroenteritis. Heliyon 10, e28727. https://doi.org/10.1016/j.heliyon.2024.e28727 (2024).
Chung The, H. & Le, S. H. Dynamic of the human gut microbiome under infectious diarrhea. Curr. Opin. Microbiol. 66, 79–85. https://doi.org/10.1016/j.mib.2022.01.006 (2022).
Kumar, A. et al. Impact of nutrition and rotavirus infection on the infant gut microbiota in a humanized pig model. BMC Gastroenterol. 18, 93. https://doi.org/10.1186/s12876-018-0810-2 (2018).
Li, L. et al. Lactic acid and short chain fatty levels of infants following rotavirus infection revealed by illumina miseq high-throughput sequencing and HPLC method. Jundishapur J. Microbiol. 12, e68389. https://doi.org/10.5812/jjm.68389 (2019).
Chen, S. Y. et al. Intestinal microbiome in children with severe and complicated acute viral gastroenteritis. Sci. Rep. 7, 46130. https://doi.org/10.1038/srep46130 (2017).
Xiao, Y., Zhao, J., Zhang, H., Zhai, Q. & Chen, W. Mining Lactobacillus and bifidobacterium for organisms with long-term gut colonization potential. Clin. Nutr. 39, 1315–1323. https://doi.org/10.1016/j.clnu.2019.05.014 (2020).
Laursen, M. F. et al. Bifidobacterium species associated with breastfeeding produce aromatic lactic acids in the infant gut. Nat. Microbiol. 6, 1367–1382. https://doi.org/10.1038/s41564-021-00970-4 (2021).
Elshaghabee, F. M. F., Rokana, N., Gulhane, R. D., Sharma, C. & Panwar, H. Bacillus as potential probiotics: Status, concerns, and future perspectives. Front. Microbiol. 8, 1490. https://doi.org/10.3389/fmicb.2017.01490 (2017).
Crawford, S. E. et al. Rotavirus infection. Nat. Rev. Dis. Primers. 3, 17083. https://doi.org/10.1038/nrdp.2017.83 (2017).
Dian, Z. et al. Rotavirus-related systemic diseases: Clinical manifestation, evidence and pathogenesis. Crit. Rev. Microbiol. 47, 580–595. https://doi.org/10.1080/1040841X.2021.1907738 (2021).
Hellysaz, A. & Hagbom, M. Rotavirus sickness symptoms: Manifestations of defensive responses from the brain. Viruses 16, 1086. https://doi.org/10.3390/v16071086 (2024).
Harper, A. et al. Viral infections, the Microbiome, and Probiotics. Front. Cell. Infect. Microbiol. 10, 596166. https://doi.org/10.3389/fcimb.2020.596166 (2020).
Kim, C. S. & Shin, D. M. Gut microbiota and cognitive development in infant mice: Quantity and source of potable water. PLoS One. 18, e0286951. https://doi.org/10.1371/journal.pone.0286951 (2023).
Funding
This work was supported by the Ningbo Clinical Research Center for Children’s Health and Diseases (no. 2019A21002), the Zhejiang Province medicine and health project (no. 2024KY1572), the Ningbo key discipline Pediatrics (no. 2022-B17), the Innovation Project of Distinguished Medical Team in Ningbo (no. 2022020405). Ningbo Medical and Health Brand Discipline (PPXK2024-06).
Author information
Authors and Affiliations
Contributions
Y.Q: Investigation, Methodology, Formal analysis, Data curation, Writing – original draft. C.Y: Investigation, Methodology, Writing – original draft. G.E: Visualization, Methodology, Data curation, software, Writing – review & editing. L.Y: Investigation, supervision, Data curation, Formal analysis, Writing – review & editing. W.J: Conceptualization, Methodology, Data curation, software, Project administration, Writing – review & editing. Q.H: Conceptualization, Project administration, Funding acquisition, Formal analysis, Writing – review & editing.All authors reviewed the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethical approval
This study was approved by the Ethics Committee of the Affiliated Women and Children’s Hospital of Ningbo University (protocols code No. EC2023-006).
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Yan, Q., Chen, Y., Gao, EB. et al. The characteristics of intestinal microflora in infants with rotavirus enteritis, changes in microflora before and after treatment and their clinical values. Sci Rep 15, 4312 (2025). https://doi.org/10.1038/s41598-025-88312-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-025-88312-w
