
Abstract
BRD4, a member of the BET family proteins, is extensively studied in cancer and is known for its complex network within tumor cells, interacting with various transcription factors and epigenetic regulatory proteins. However, the impact of BRD4 and its associated genes on hepatocellular carcinoma (HCC) remains unclear. HCC is one of the leading causes of cancer-related deaths globally, often diagnosed at an advanced stage, limiting treatment options. In this study, we curated BRD4-interacting genes and, through analysis of the TCGA-LIHC dataset, developed a four-gene prognostic model for HCC comprising EZH2, KIF20A, G6PD, and KIF2C. This model demonstrated strong predictive power in both training and validation cohorts, with high gene expression levels significantly correlating with poor prognosis. Furthermore, our analysis revealed significant immunological differences between risk groups and identified increased drug sensitivity in high-risk patients to certain medications, while decreased sensitivity to others. Experimental validation further indicated that the combination of BRD4 inhibitor ZBC260 and EZH2 inhibitor CPI-169 synergistically enhanced apoptosis in HCC. Collectively, this study provides a scientific rationale for early HCC diagnosis and personalized therapy, offering new insights into drug resistance in treatment.
Similar content being viewed by others


Introduction
Hepatocellular carcinoma (HCC) is recognized as the fourth leading cause of cancer-related mortality globally, with an increasing incidence rate, particularly prevalent among males1,2. HCC development is linked to viral hepatitis (HBV, HCV), non-alcoholic steatohepatitis (NASH), and habits like heavy drinking, smoking, and diabetes2. Despite continuous advancements in therapeutic approaches, the complexity of HCC results in a five-year survival rate below 20%, and the changing epidemiological characteristics pose challenges for early diagnosis3.
BRD4, a BET family protein, modulates gene expression through interaction with acetylated chromatin via its bromodomain and plays a pivotal role in maintaining genomic integrity, DNA repair, regulating cell death, and telomere length4,5. Previous studies have revealed elevated BRD4 expression in HCC tissues. Inhibition of BRD4 has been demonstrated to suppress the proliferation of HCC cell lines, induce apoptosis, and retard tumor growth in mouse HCC xenograft models6,7. While BRD4 inhibitors have shown efficacy in hematological malignancies, they face the challenge of drug resistance in solid tumor therapy8. Given the intricate network formed by BRD4’s interaction with various transcription factors and epigenetic regulatory proteins within tumor cells, there is a growing interest in the exploration of combination therapies involving BRD4 inhibitors and inhibitors of its interacting proteins9,10. This approach aims to enhance therapeutic efficacy and mitigate the development of resistance by targeting distinct molecular mechanisms.
The objective of this study was to unravel the prognostic significance of BRD4-related genes in HCC and to construct an effective prognostic model. By juxtaposing BRD4-related genes with differentially expressed genes (DEGs) within the TCGA-LIHC database, we have developed a four-gene signature prognostic model, the accuracy of which was validated. This prognostic model facilitates the early detection of high-risk HCC patient populations and anticipates their responsiveness to therapeutic interventions, thereby offering exquisite clinical decision support to augment patient survival and quality of life. Moreover, this investigation has proposed a potentially efficacious novel combinatorial treatment strategy through synergistic drug experimentation, charting a new course for HCC therapeutics.
Materials and methods
Data acquisition
RNA sequencing data and clinical information of 377 TCGA-LIHC HCC patients were obtained from The Cancer Genome Atlas (TCGA) database (https://portal.gdc.cancer.gov). Concurrently, the GSE14520 and E-TABM-36 datasets were accessed from the Gene Expression Omnibus (GEO) database (http://www.ncbi.nlm.nih.gov/geo/) and EMBL-EBI (www.ebi.ac.uk), respectively. Drug response predictions were based on the Genomics of Drug Sensitivity in Cancer (GDSC) database (https://www.cancerrxgene.org/). The BRD4 interaction gene set was sourced from BioGRID (www.thebiogrid.org) (Supplementary Table S1).
Preliminary identification of prognostic genes associated with BRD4
Utilizing the “DESeq2” R package, we processed the TCGA-LIHC dataset, identified differentially expressed genes (DEGs) with |log2FC|≥2, and intersected these with the BRD4 interaction gene set. Volcano plots and Venn diagrams were created with Hiplot (https://hiplot.com.cn/). The protein-protein interaction (PPI) network was established using the STRING database (https://string-db.org/).
Risk score model establishment and confirmation
This study employed the LASSO algorithm, integrated with cross-validation, to optimize the λ parameter for identifying prognosis-related signature genes. With the R packages “survival” and “glmnet,” a risk score model was constructed. Risk Score = Σ(Εi * γi), where Εi denotes the risk coefficient and γi denotes gene expression levels. Survival was analyzed with “survival” and “survminer,” and the model’s discrimination was evaluated by AUC via “pROC.” By using the “rms”, “caret”, and “replot” packages in R, and calibrating the nomogram.
Enrichment and somatic mutation analysis
Gene Set Variation Analysis (GSVA) was applied to assess the variation in gene set expressions across different pathways11. Pathway enrichment scores were determined using the R package “GSVA,” contrasting groups categorized as high risk and low risk. GO and KEGG enrichments were conducted using the Metascape (https://metascape.org/) and KOBAS (http://bioinfo.org/kobas/), respectively, with significance set at P < 0.0512. Somatic mutation spectra were analyzed for the TCGA-LIHC dataset using an online platform (http://www.sxdyc.com/index).
Tumor microenvironment analysis and drug sensitivity prediction
Gene expression and the infiltration of immune cells were analyzed utilizing the TIMER, MCP-COUNTER, EPIC, and CIBERSORT algorithms13. we determined immune and stromal scores using the ESTIMATE algorithm. Furthermore, the Tumor Immune Dysfunction and Exclusion (TIDE) method was applied to assess the potential for immunotherapy response. Drug sensitivity was analyzed with the “oncopredict” package.
Drug treatment and cell culture
Cell lines MHCC-97 H and HepG2 were obtained from the China Center for Type Culture Collection (CCTCC). Under the same conditions at 37 °C with 5% CO2, MHCC-97 H cells were cultured in DMEM medium (Servicebio, China) containing 10% FBS (Every Green, China), while HepG2 cells were cultured in MEM medium (Servicebio, China). Drug treatments included ZBC260 (10 nM/mL; MCE, China) and CPI-169 (5 µg/mL; APExBIO, USA) for single treatments, and their combination (5 nM/mL ZBC260, 2.5 µg/mL CPI-169). Transcriptional factor motif enrichment was analyzed using R packages “rcisTarget”, “reshape2”, “visNetwork”, and “tidyverse”.
Western blot analysis, cell viability and apoptosis detection assays
Western blot was conducted following standard procedures, with specific antibodies used to detect protein expression, and results were visualized using chemiluminescence14. Cell viability was determined using the Cell Counting Kit-8 (CCK-8) (Biosharp, China). Apoptosis was detected in cells using the Annexin V-PE Apoptosis Detection Kit (Beyotime Biotechnology, China).
Statistical analysis
Statistics were performed using R (version 4.1.2) and GraphPad Prism (version 8.0.1). We obtained data from at least three independent experiments and presented them as mean ± standard deviation. Comparisons between groups were performed using two-tailed Student’s t-test, chi-square test, or univariate analysis of variance (ANOVA). Statistical significance was set at P < 0.05 (*P < 0.05, **P < 0.01, ***P < 0.001).
Results
Identification and analysis of BRD4-associated DEGs in HCC
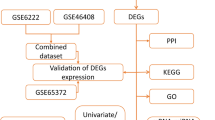
Figure 1 presents a detailed flowchart that illustrates the progression of our research. To develop a prognostic model, we analyzed the expression profiles of genes in HCC compared to non-cancerous liver samples within the TCGA database. Identifying 3,257 DEGs, of which 2,879 were upregulated and 378 downregulated (Fig. 2A). We identified 46 BRD4-interacting DEGs (BI-DEGs) by querying the BIOGRID database for BRD4 interactors and intersecting with our DEGs list (Fig. 2B). Expression patterns were visualized in a heatmap (Fig. 2C), and chromosomal localization was analyzed to determine their genomic distribution (Fig. 2D). PPI network analysis uncovered significant interactions among BI-DEGs, notably those involving CDK1, CCNA2, KIF2C, AURKA, and KIF20A (Fig. 2E).

Flow chart.

Selection and functional analysis of BRD4-associated prognostic genes. (A) DEGs between HCC and non-tumorous tissues in the TCGA-LIHC dataset. (B) Venn diagram of the intersection between differentially expressed genes and the BRD4-related gene set. (C) Expression heatmap of the 46 intersecting genes across samples. (D) Chromosome localization of the intersecting genes. (E) Hub gene PPI network of the intersecting genes. (F) KEGG pathway enrichment analysis and (G) GO functional enrichment analysis of the intersecting genes.
Enrichment analysis implicated BI-DEGs in cancer-related biological processes, encompassing cell cycle regulation, tumorigenesis, immune response, DNA repair, and catalytic activity regulation (Fig. 2F–G). Somatic mutation profiling within the TCGA-LIHC cohort revealed that MKI67 had the highest mutation rate at 3%, surpassing other genes with rates below 1% (Supplementary Fig. S1A). Copy number variation (CNV) analysis also indicated frequent copy number alterations among these genes (Supplementary Fig. S1B).
Establishment and confirmation of a HCC Prognostication Model utilizing BI-DEGs
In our investigation, we initially utilized univariate Cox regression to identify candidate genes related to survival. we employed LASSO survival analysis to develop a model based on a 4-gene signature, including EZH2, KIF20A, G6PD, and KIF2C (Fig. 3A,B). These genes were significantly upregulated in tumors (Fig. 3C), suggesting their potential role in tumor progression. Kaplan-Meier assessments indicated that elevated levels of these genes are associated with an unfavorable prognosis (Fig. 3D).

Development and validation of a HCC prognostic model. (A, B) Selection of four signature genes via LASSO regression. (C) Signature gene expression in tumor versus normal tissues. (D) Kaplan-Meier OS curves for BRD4, EZH2, KIF20A, G6PD, and KIF2C. (E) Kaplan-Meier curves for OS, DFS, PFS, DSS in TCGA-LIHC risk groups. (F) ROC curves for the training cohort. (G–H) Kaplan-Meier and ROC curves for validation cohorts E_TABM_36 and GSE14520. (I) GSVA analysis of risk groups.
For the purpose of confirming our model’s prognostic accuracy, we determined a risk score for each participant and stratified them into groups with either elevated or reduced risk within the TCGA-LIHC dataset, using the median value of the risk score to distinguish between the groups. Risk score = Expression of EZH2 × 0.0002482 + Expression of KIF20A × 0.0833646 + Expression of G6PD × 0.1511231 + Expression of KIF2C × 0.0364607. Our findings demonstrated that individuals classified in the low-risk category exhibited superior overall survival (OS), disease-free survival (DFS), progression-free survival (PFS), and disease-specific survival (DSS), with particularly accurate predictions for OS (Fig. 3E,F). Validation in an independent cohort confirmed the model’s efficacy, with low-risk patients showing superior outcomes (Fig. 3G,H). The GSVA analysis highlighted that the high-risk group was primarily enriched for cell growth-related processes, DNA repair, and signal transduction, while the low-risk group showed substantial correlations with inflammation, cell communication, and immune response (Fig. 3I).
Integrative analysis of immune landscape and clinical outcomes
In our investigation of TCGA-LIHC samples, a notable association was observed between BRD4 and the expression of four prognostic genes along with their relationship to tumor stage (T category) and disease progression (Supplementary Fig. S2A–B). This correlation suggests these genes’ upregulation is linked to increased tumor malignancy. Additionally, we conducted univariate Cox regression analysis for these genes (Fig. 4A). After adjusting for confounding factors, single-variable and multi-variable Cox regression assessments confirmed the significance of the risk score as an independent prognosticator for individuals with LIHC (Supplementary Fig. S2C–D). We then developed a nomogram incorporating alcohol consumption combined with risk scores to forecast 1-, 3-, and 5-year overall survival rates (Fig. 4B), and validated its predictive accuracy through calibration curves (Supplementary Fig. S2E).

4-gene prognostic model analysis in HCC. (A) Univariate Cox regression analysis of signature genes. (B) Nomogram integrating risk scores with clinical factors to predict 1-, 3-, and 5-year OS. (C) Comparison of immune cell infiltration between high- and low-risk groups. (D) Correlation of immune cells with signature genes (purple = positive, orange = negative); (E) Correlation analysis between BRD4 and signature genes. (F) Drug sensitivity to select agents in different risk groups and correlation with risk scores.
The analysis of immune contexture within the TCGA-LIHC dataset revealed marked disparities in the infiltration of quiescent mast cells, M0 macrophages, activated CD4 memory T cells, and follicular helper T cells between patients stratified into elevated and diminished risk categories (Fig. 4C). A negative correlation was observed between prognostic genes and immune cells linked to antitumor immunity (Fig. 4D). The TME and TIDE scores indicated an elevated TIDE value within the high-risk group (Supplementary Fig. S2F–G). Further, BRD4 and the four signature genes had a significant direct association (Fig. 4E). These results imply that these genes may contribute to immune evasion in tumors by suppressing immune responses, potentially enhancing tumor immune escape with an increasing risk score.
Anticancer drug sensitivity
The relationship between the four-gene risk score and cancer drug responsiveness was assessed using the GDSC database, projecting variations in sensitivity between different risk groupings (Supplementary Figure S3). Our analysis indicates that high-risk groups demonstrate heightened sensitivity to drugs such as SB216763, KRAS(G12C) inhibitor-12, and PLX-4720, whereas their sensitivity to Crizotinib, GDC0810, and MIRA-1 is comparatively diminished (Fig. 4F).
Validation experiments
By analyzing immunohistochemical images from the Human Protein Atlas (HPA) database, we found that tumor tissues have overexpressed BRD4, EZH2, KIF20A, G6PD, and KIF2C proteins (Fig. 5A). Western blots corroborated the increased expression of these proteins in tumor (Fig. 5C, Supplementary File S1). Enrichment analysis of transcription factor binding motifs revealed a substantial shared transcription factor network between EZH2 and BRD4 (Fig. 5B, Supplementary Fig. S4). Therefore, we initiated our experiments by targeting this pair of proteins rather than other BRD4-interacting proteins. Subsequently, the impact of the BRD4 inhibitor ZBC260 and the EZH2 inhibitor CPI-169 on cell lines MHCC-97 H and HepG2 proliferation was evaluated by the CCK8 assay. Data demonstrated a marked decrease in optical density (OD) values following drug treatment, suggesting a suppression of cellular growth. Notably, the most pronounced inhibition was observed with the concurrent use of ZBC260 and CPI-169 (Fig. 5D).

Validation of signature gene functions in vitro. (A) Protein expression of signature genes in HCC versus normal tissue (HPA database). (B) Enrichment prediction analysis of transcription factor binding motifs for BRD4, EZH2, KIF20A, G6PD, and KIF2C. (C) Western blot analysis of EZH2, KIF20A, G6PD, and KIF2C proteins in clinical samples. (D) CCK8 assay to evaluate the growth curves of MHCC-97 H and HepG2 cells following drug treatment. (E) Apoptosis in HepG2 cells assessed by flow cytometry after 48 h of drug treatment. (F) Western blot analysis of EZH2, BRD4, and apoptosis-regulating proteins following drug treatment. Drug treatments included 10 nM/mL ZBC260 and 5 µg/mL CPI-169 for single treatments, and their combination at 5 nM/mL ZBC260 and 2.5 µg/mL CPI-169.
Annexin V/PI dual staining flow cytometry was conducted to assess apoptosis following a 48-hour drug treatment. The results showed significantly more apoptotic cells after treatment, with the ZBC260 and CPI-169 combination leading to the highest apoptosis rate (Fig. 5E, Supplementary File S1). This suggests that ZBC260 and CPI-169 have a synergistic role in promoting cell apoptosis. By immunoblotting, treatment reduced BRD4 and EZH2 protein expression, induced an increase in Bax and Bcl-2, and promoted the activity of caspase-3. These findings indicate that BRD4 and EZH2 are pivotal in the apoptotic pathway, the drugs target BRD4 and EZH2, affecting the expression of downstream proteins and activating the apoptotic process (Fig. 5F).
Discussion
In the current landscape of HCC diagnosis, imaging studies and serological biomarkers face challenges in early detection15. The range of therapeutic options includes surgical resection, radiotherapy, chemotherapy, targeted therapies, and immunotherapy. However, a subset of patients exhibits resistance to specific drugs, particularly in cases where drug resistance is a factor, which limits the efficacy of treatment and exacerbates the toxicity profile and economic burden associated with such interventions16. To overcome these challenges, innovative therapeutic strategies are needed urgently. Our research has developed a prognostic model for HCC, designed to identify high-risk patients at an early stage, predict treatment responses to guide personalized therapeutics, and facilitate the development of novel drug strategies to overcome drug resistance, thereby enhancing therapeutic efficacy, improving patient survival rates.
In this study, using the TCGA-LIHC dataset, we identified 3,259 DEGs and narrowed down 46 BI-DEGs, which are hypothesized to play crucial roles in tumor proliferation, drug resistance, and invasiveness. A four-gene prognostic model was developed via LASSO analysis, including EZH2, KIF20A, G6PD, and KIF2C. EZH2, a histone methyltransferase, is central to oncogenesis, EZH2 inhibitors such as Tazemetostat and Valemetostat may hold therapeutic potential for HCC17. KIF20A enhances immunotherapy response by modulating FBXW7-c-Myc18. High expression levels of G6PD and KIF2C correlate with increased tumor proliferation and invasion19,20. There was robust accuracy in our prognostic model across both training and validation cohorts. The independent prognostic value of the risk score, constructed based on patient gene expression levels, was also confirmed by nomogram analysis.
Infiltration analysis revealed significant differences among risk groups. In the group classified as higher risk, resting mast cells and Tregs may dampen the immune system’s ability to fight the tumor and facilitate the tumor’s evasion21,22. Moreover, TIDE score analysis substantiates the high-risk group’s enhanced immune escape capability. These findings validate the predictive accuracy of our model for prognosis and underscore the potential value of targeting specific immune cells to augment antitumor immunity.
In drug susceptibility tests, individuals with elevated risk exhibit heightened response to medications such as SB216763, KRAS(G12C) inhibitor-12, and PLX-4720, whereas their sensitivity to Crizotinib, GDC0810, and MIRA-1 is diminished. The utilization of risk scoring systems can assist in identifying patients who may be sensitive or resistant to specific drugs, thereby providing personalized guidance for clinical cancer treatment.
Analysis of transcription factor binding sites uncovered extensive BRD4-EZH2 shared sites, highlighting their collaborative role in the regulation of gene expression. BRD4, a key epigenetic regulator in multiple cancers, is targeted by inhibitors that block its interaction with acetylated histones to repress oncogenes23. BRD4 regulates gene expression through super-enhancers and plays a key role in the development of drug resistance. BET inhibitors, such as PLX51107, can trigger resistance through activation of the Wnt/β-catenin signaling pathway. In contrast, the multi-CDK inhibitor dinaciclib can overcome this resistance by targeting critical components of the Wnt pathway, including β-catenin and LRP624. Clinical use is hampered by resistance and off-target effects. To surmount these challenges, investigators have proposed combination therapies involving BRD4 and its interacting protein inhibitors. For instance, the concurrent inhibition of CDK7, CBP/p300, and histone deacetylases (HDACs) can elicit synergistic anticancer effects25. Research suggests that dual targeting of BRD4 and EZH2 synergistically enhances tumor cell suppression, potentially overcoming drug resistance and amplifying the anti-tumor response. This is achieved through the modulation of key signaling pathways, including PI3K/AKT and mTOR, across various cancer types, particularly in lung and pancreatic cancers26. This combined inhibition strategy holds substantial promise in overcoming drug resistance, offering a novel perspective for cancer therapeutics.
The combinatorial use of BRD4 inhibitor ZBC260 and EZH2 inhibitor CPI-169 has demonstrated significant synergistic effects. Experimental data indicate that both ZBC260 and CPI-169, when used individually, effectively inhibit the proliferation of HCC cell lines and induce apoptosis. Using Western blot analysis, treatment with these drugs increased the levels of pro-apoptotic proteins Bax and Cleaved-caspase-3, while decreasing the levels of anti-apoptotic proteins Bcl-2, thereby enhancing the regulatory effects of the drugs on apoptotic pathways. Notably, the co-administration of ZBC260 and CPI-169 showed a marked synergistic effect in inducing cellular apoptosis, suggesting that BRD4 and EZH2 play crucial roles in apoptotic signaling pathways. Targeting these proteins with drugs can modulate the expression of downstream proteins and trigger apoptosis, offering a promising strategy for cancer therapy.
In this study, we first explored the combined inhibition of BRD4 and EZH2 in HCC, optimizing drug dosages to reduce potential toxicity, which has been overlooked in previous research. The findings of our study provide new insights into HCC prognosis and clinical treatment strategies, highlighting the advantages of combined BRD4 therapy in reducing drug resistance and enhancing therapeutic effects. However, we acknowledge the need for future research to validate our four-gene prognostic model in a broader patient population and to investigate the interactive mechanisms of BRD4 and EZH2 with other signaling pathways. This study, based on bioinformatics and in vitro experiments, suggests that dual inhibition of BRD4 and EZH2 may be effective for liver cancer treatment. Although primarily reliant on database and in vitro data, we plan to validate these findings further using xenograft and organoid models to more accurately simulate patient responses and explore innovative therapeutic strategies.
Conclusion
In conclusion, our analysis of the TCGA-LIHC dataset identified 46 BRD4-interacting DEGs with potential as early diagnostic and prognostic biomarkers in HCC. Our four-gene signature (EZH2, KIF20A, G6PD, KIF2C) precisely forecasts HCC prognosis and highlights their roles in tumorigenesis. Moreover, our immunoinfiltration analysis elucidated significant differences in immune characteristics between risk groups, and our risk scoring system predicts drug responses in patients, providing a scientific basis for personalized therapeutic strategies. Notably, the synergistic pro-apoptotic effect of BRD4 and EZH2 inhibitors in HCC suggests a new approach to overcoming drug resistance.
Data availability
Public data used in this work can be acquired from the TCGA Research Network portal (https://portal.gdc.cancer.gov/) and Gene Expression Omnibus (GEO, http://www.ncbi.nlm.nih.gov/geo/).
Abbreviations
- HCC:
-
Hepatocellular carcinoma
- BI-DEGs:
-
BRD4-interacting differentially expressed genes
- TCGA:
-
The Cancer Genome Atlas
- GO:
-
Gene ontology
- KEGG:
-
Kyoto encyclopedia of genes and genomes
- LASSO:
-
Least absolute shrinkage and selection operator
- TIDE:
-
Tumor mutation burden
- TME:
-
Tumor microenvironment
- PPI:
-
Protein–protein interaction
- AUC:
-
Area under the curve
- STRING:
-
Search tool for the retrieval of interacting genes
- GSVA:
-
Gene set variation analysis
References
Yang, J. D. et al. A global view of hepatocellular carcinoma: Trends, risk, prevention and management. Nat. Rev. Gastroenterol. Hepatol. 16, 589–604. https://doi.org/10.1038/s41575-019-0186-y (2019).
Han, B. et al. Cancer incidence and mortality in China, 2022. J. Natl. Cancer Cent. 4, 47–53. https://doi.org/10.1016/j.jncc.2024.01.006 (2024).
Rich, N. E. Changing epidemiology of hepatocellular carcinoma within the United States and worldwide. Surg. Oncol. Clin. N Am. 33, 1–12. https://doi.org/10.1016/j.soc.2023.06.004 (2024).
Pecharromán, I. et al. IκB kinase-α coordinates BRD4 and JAK/STAT signaling to subvert DNA damage-based anticancer therapy. Embo J. 42, e114719. https://doi.org/10.15252/embj.2023114719 (2023).
Wang, S. et al. BRD4 inhibitors block telomere elongation. Nucleic Acids Res. 45, 8403–8410. https://doi.org/10.1093/nar/gkx561 (2017).
Wu, X. et al. Inhibition of BRD4 suppresses cell proliferation and induces apoptosis in renal cell carcinoma. Cell. Physiol. Biochem. 41, 1947–1956. https://doi.org/10.1159/000472407 (2017).
Li, G. Q. et al. Suppression of BRD4 inhibits human hepatocellular carcinoma by repressing MYC and enhancing BIM expression. Oncotarget 7, 2462–2474. https://doi.org/10.18632/oncotarget.6275 (2016).
Chen, X. et al. Design, synthesis and mechanism studies of dual EZH2/BRD4 inhibitors for cancer therapy. Bioorg. Med. Chem. 91, 117386. https://doi.org/10.1016/j.bmc.2023.117386 (2023).
Liang, Y., Tian, J. & Wu, T. BRD4 in physiology and pathology: “BET” on its partners. Bioessays 43, e2100180. https://doi.org/10.1002/bies.202100180 (2021).
Lovén, J. et al. Selective inhibition of tumor oncogenes by disruption of super-enhancers. Cell 153, 320–334. https://doi.org/10.1016/j.cell.2013.03.036 (2013).
Hänzelmann, S., Castelo, R. & Guinney, J. GSVA: Gene set variation analysis for microarray and RNA-seq data. BMC Bioinform. 14, 7. https://doi.org/10.1186/1471-2105-14-7 (2013).
Kanehisa, M. & Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 28, 27–30. https://doi.org/10.1093/nar/28.1.27 (2000).
Li, T. et al. TIMER: A web server for comprehensive analysis of tumor-infiltrating Immune cells. Cancer Res. 77, e108–e110. https://doi.org/10.1158/0008-5472.Can-17-0307 (2017).
Mahmood, T. & Yang, P. C. Western blot: Technique, theory, and trouble shooting. N Am. J. Med. Sci. 4, 429–434. https://doi.org/10.4103/1947-2714.100998 (2012).
Llovet, J. M. et al. Hepatocellular carcinoma. Nat. Rev. Dis. Primers. 7, 6. https://doi.org/10.1038/s41572-020-00240-3 (2021).
Wang, Y. et al. Chemotherapy-induced acetylation of ACLY by NAT10 promotes its nuclear accumulation and acetyl-CoA production to drive chemoresistance in hepatocellular carcinoma. Cell. Death Dis. 15, 545. https://doi.org/10.1038/s41419-024-06951-9 (2024).
Wang, B. et al. EZH2 in hepatocellular carcinoma: Progression, immunity, and potential targeting therapies. Exp. Hematol. Oncol. 12, 52. https://doi.org/10.1186/s40164-023-00405-2 (2023).
Chen, S. et al. Inhibition of KIF20A enhances the immunotherapeutic effect of hepatocellular carcinoma by enhancing c-Myc ubiquitination. Cancer Lett. 598, 217105. https://doi.org/10.1016/j.canlet.2024.217105 (2024).
Liu, B. et al. Pan-cancer analysis of G6PD carcinogenesis in human tumors. Carcinogenesis 44, 525–534. https://doi.org/10.1093/carcin/bgad043 (2023).
Wei, S. et al. KIF2C: A novel link between Wnt/β-catenin and mTORC1 signaling in the pathogenesis of hepatocellular carcinoma. Protein Cell. 12, 788–809. https://doi.org/10.1007/s13238-020-00766-y (2021).
Baran, J. et al. Mast cells as a target-A comprehensive review of recent therapeutic approaches. Cells 12 (2023). https://doi.org/10.3390/cells12081187
Kang, J. H. & Zappasodi, R. Modulating Treg stability to improve cancer immunotherapy. Trends Cancer 9, 911–927. https://doi.org/10.1016/j.trecan.2023.07.015 (2023).
Tian, T., Guo, T., Zhen, W., Zou, J. & Li, F. BET degrader inhibits tumor progression and stem-like cell growth via Wnt/β-catenin signaling repression in glioma cells. Cell. Death Dis. 11, 900. https://doi.org/10.1038/s41419-020-03117-1 (2020).
Marr, A. R. et al. The multi-CDK inhibitor dinaciclib reverses bromo- and extra-terminal domain (BET) inhibitor resistance in acute myeloid leukemia via inhibition of Wnt/β-catenin signaling. Exp. Hematol. Oncol. 13, 27. https://doi.org/10.1186/s40164-024-00483-w (2024).
Qian, H. et al. Super-enhancers and the super-enhancer reader BRD4: Tumorigenic factors and therapeutic targets. Cell. Death Discov. 9, 470. https://doi.org/10.1038/s41420-023-01775-6 (2023).
Guo, Z. et al. Design and synthesis of dual EZH2/BRD4 inhibitors to target solid tumors. J. Med. Chem. 65, 6573–6592. https://doi.org/10.1021/acs.jmedchem.1c01876 (2022).
Acknowledgements
This study was supported by the National Natural Science Foundation of China (grant number 82103211), the Henan Province Medical Science and Technology Research Plan (grant number LHGJ20200357), and the Henan Provincial Charity Federation Hepatobiliary Funding (grant number GDXZ2023015).
Author information
Authors and Affiliations
Contributions
H. Z. Writing - Review and Editing, Resources, Funding acquisition.J. Y. Methodology, Software.F. Z. Software, Formal analysis.Q. L. Validation, Project administration.J. P. Project administration, Formal analysis.P. W. Supervision, Project administration.D. L. Visualization.R. L. Investigation.T. W. Data Curation.S. W. Conceptualization, Writing - Original Draft, Supervision.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Zhang, H., Yu, J., Zhang, F. et al. BRD4 interacting genes as prognostic biomarkers in hepatocellular carcinoma for optimized treatment strategies. Sci Rep 15, 5617 (2025). https://doi.org/10.1038/s41598-025-89614-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-025-89614-9

{kind=link}