
Abstract
Convolutional neural networks (CNNs) and mammalian visual systems share architectural and information processing similarities. We leverage these parallels to develop an in-silico CNN model simulating diseases affecting the visual system. This model aims to replicate neural complexities in an experimentally controlled environment. Therefore, we examine object recognition and internal representations of a CNN under neurodegeneration and neuroplasticity conditions simulated through synaptic weight decay and retraining. This approach can model neurodegeneration from events like tau accumulation, reflecting cognitive decline in diseases such as posterior cortical atrophy, a condition that can accompany Alzheimer’s disease and primarily affects the visual system. After each degeneration iteration, we retrain unaffected synapses to simulate ongoing neuroplasticity. Our results show that with significant synaptic decay and limited retraining, the model’s representational similarity decreases compared to a healthy model. Early CNN layers retain high similarity to the healthy model, while later layers are more prone to degradation. The results of this study reveal a progressive decline in object recognition proficiency, mirroring posterior cortical atrophy progression. In-silico modeling of neurodegenerative diseases can enhance our understanding of disease progression and aid in developing targeted rehabilitation and treatments.
Similar content being viewed by others
Introduction
The intersection of computational neuroscience and deep learning-based machine learning is a rapidly evolving field that seeks to leverage insights from the brain’s structure and functions to enhance the design and performance of deep learning algorithms and models. Notably, convolutional neural networks (CNNs) are the predominant choice for computationally modeling the biological visual cortex1,2,3. CNNs have proven highly effective in solving many computer vision tasks by using local receptive fields that are hierarchically aggregated and whose functions closely resemble the organization of visual processing in the mammalian visual cortex. More recently, the research dynamic has started to shift towards investigating the reverse direction as well, namely to develop a better understanding of the human brain by analyzing deep learning models1,4,5,6. Moreover, deep learning has not only been used to draw parallels with the healthy brain but also to simulate the cognitive deficits that accompany neurodegenerative disorders (Fig. 1), such as Alzheimer’s disease (AD), through the introduction of axonal or neuronal injury to deep learning models7,8,9. Broadly summarized, neuroscience seeks to uncover the principles underlying neural information processing in the brain, while deep learning researchers focus on developing algorithms that can extract useful representations and perform complex tasks inspired by the brain’s computational capabilities. In this work, we incorporate a transdisciplinary approach to neuroscience and deep learning with the aim of developing a disease progression model of the visual system with more biologically plausible deep learning and retraining methods.

Convolutional neural networks can be used to simulate the degeneration of the visual cortex. When initially trained for object recognition tasks, they perform with high accuracy, similar to the performance of humans. Inflicting progressive synaptic damage to a CNN results in decline of visual cognitive abilities, similar to patients with neurodegenerative diseases.
Previous work has attempted to simulate the effects of neurodegenerative disease with the complete removal of synapses and neurons from ‘cognitively healthy’ CNNs trained for object classification tasks8,9. However, this simulation is not necessarily biologically realistic. For example, the accumulation of hyperphosphorylated tau in the brain occurs for years before eventual synaptic death10,11. Tau aggregates can accumulate within presynaptic terminals, interfering with the release of neurotransmitters12. This disruption reduces the efficiency of neurotransmission and compromises the communication between neurons in the brain. Thus, tau pathology gradually and progressively interferes with signaling pathways that are critical for synaptic function, and is a process that can be observed well before complete synaptic injury. For example, tau aggregates can dysregulate synaptic plasticity, a process crucial for learning and memory. This disruption hampers the ability of synapses to adapt and modify their strength, impairing cognitive processes. These pathological effects contribute to the cognitive deficits observed in individuals with AD, such as impaired visual cognition, memory, and executive function13,14,15.
In this work, we address the shortcomings of previous works8,9 by modeling the process of tau accumulation leading to eventual synaptic loss in a CNN. In patients with posterior cortical atrophy (PCA), clinical evidence shows that tau deposition starts in the posterior cortical regions within the visual network and gradually propagates in anterior direction. As a result of this propagation, visuospatial cognition abilities decline16. Therefore, we simulate the effects seen in the visual cortex in patients with PCA by progressively decaying the weights of an increasing number of a CNN model’s synapses in conjunction with a biologically-inspired retraining processes. The progressive synaptic decay is motivated by the synaptic degeneration and impaired synaptic transmission caused by tau aggregates in the diseased brain17,18,19. Importantly, after model synapses are decayed, they are not included in the retraining of the remaining intact model weights. By doing so, we simulate synaptic dysfunction and loss of plasticity due to the accumulation of hyperphosphorylated tau in the visual system in patients with PCA. The pipeline used for model degeneration is illustrated in Fig. 2. Moreover, by the end of the complete cycle of model degeneration, the majority of synapses have been degenerated so severely that they can be essentially considered to be ablated. For quantitative analysis, we measure levels of object recognition accuracy as a proxy for cognitive function as the models are decayed and retrained with different subsets of retraining data. By measuring levels of accuracy and internal representations as a compounding number of synapses are included in the model decay, we are able to simulate a pattern of visual cognitive decline akin to that seen in patients with AD20.
Moreover, we examine how continual weight decay and retraining affect the internal representations that the model learns. In this set-up, internal representations of our injured model offer insights to how the injured brain hierarchically embeds visual information and the dynamics of these embeddings with respect to disease progression. Indeed, it has been shown that internal representations of CNNs are highly similar to those of primate brains2,4. Like the ventral visual pathway, CNNs develop hierarchical representations of visual information, with early layers capturing low-level features and later layers encoding increasingly complex and abstract concepts21. We investigate the internal representations to determine whether the neural similarities to the healthy model are preserved during injury and retraining. Using this approach, it becomes possible to identify the layers in the model at which there is a large divergence in similarity to ‘healthy’ processing. By investigating the model’s latent space, or the model’s learned representations of data, we seek insight into whether the decayed model represents visual information in a similar way to the uninjured model and if it is capable of innate plasticity and cognitive reserve with retraining to maintain performance on the same task with variable encoding.
Our contributions can be summarized as follows:
-
First, we introduce synaptic decay rather than removal to encompass a more realistic progression of neurodegeneration.
-
Second, we probe the differences in retraining dataset size to investigate how robust the model is to injury given larger amounts of ’continual learning’.
-
Third, we investigate specific layer dynamics when subjected to injury and retraining to explore which areas of the model are robust to injury.

Pipeline of synaptic degeneration of the CNN model. Grey lines represent synapses that have been decayed. Only healthy, unaffected weights are subjected to retraining. All decayed weights remain frozen and are subject to subsequent decays in the following iteration of model degeneration.
Results
Decreased object recognition accuracy as a proxy for declining cognitive function
The performance of our model was systematically evaluated over \(N=10\) iterations of synaptic decay and retraining. We repeated the simulations ten times for each iteration to account for biasing effects that may be present due to the order in which the synapses are degenerated. The initial, intact, healthy model (a compressed VGG19 CNN) achieved an accuracy of 93.3% in object classification on the CIFAR10 test dataset, a commonly used computer vision dataset containing ten naturalistic image classes22. After the first 10% of model synapses were decayed, the accuracy of the model decreased by approximately 25%. After degeneration, three different training regimes were used to simulate neuroplasticity, and accuracy improvements were measured. More precisely, retraining of the unaffected synapses was performed using one epoch with 5000, 1000, and 100 images (randomly selected). It was found that overall accuracies improved to \(83.0 \pm 1.87\%\), \(76.6 \pm 6.06\%\), and \(70.8 \pm 21.8\%\) (mean ± SD), respectively (5000, 1000, and 100 images used for retraining). It is intuitive that during neurodegenerative disease progression, cognitive abilities remain more stable if a patient is subjected to more cognitive stimulation23. Similar findings were observed in our work, in that a smaller amount of training data leads to a lower level of accuracy recovery. As a baseline comparison, we also included experiments of the progression of synaptic decay in a static model without any retraining. These results can be found in Supplementary Information, S5 and show that the model accuracy is reduced to chance level after just 20% of synapses are decayed. However, even at high levels of injury (i.e., 60% of synaptic decay), we found that one epoch of retraining with 1000 images led to an increase in performance from \(15.0 \pm 7.01\%\) in its decayed state to \(66.4 \pm 6.12\%\) accuracy after retraining. These trends continue throughout the full cycle of degeneration, although at injury levels of 70% and higher, the model retrained with only 100 images fails to recover an accuracy above chance-level (10% accuracy), as seen in Fig. 3. As the injury progresses, an interesting trend emerges regarding the standard deviations in accuracy levels. As decay levels increase, so do the standard deviations, especially with fewer retraining data. The large standard deviations in accuracy at high injury levels can be attributed to the extreme synaptic sparsity that arises as most of the model is injured. If a salient weight is decayed and frozen, the models may not be able to recover to the original accuracy, even with retraining. These findings imply that the cognitive reserve of the model can lead to some recovery of function after damage, but this is largely dependent on the pathways that have been injured. To verify the consistency of these findings, we completed the same analysis using a compressed ResNet-56 model, which achieved an initial accuracy of 92.5% on object classification of CIFAR10, and found a very similar trajectory of loss of model accuracy as a function of synaptic decay. ResNet-56 was chosen as an additional model for its distinction in architecture from VGG19 and its predictivity of neural data, as measured by BrainScore24. These results can be found in Supplementary Information, S1.

Graph showing the model accuracy ± SD as an increasing number of weights are decayed with subsequent retraining of the remaining ‘healthy’ weights. Each panel displays the accuracy trajectory for a different number of retraining images. Each retraining subset of data is randomly sampled from the initial training data and is not balanced between classes. Intuitively, we found that more retraining images give way to higher levels of accuracy gained after each retraining step. The data points on the graphs are from the same (n=10) networks, showing the average progressive impairment from 0 to 100% synapses affected, in 10% steps. Average accuracies are reported for 10 different cycles of model degeneration.
Layer-wise analysis of representational similarity
In addition to the vision task performance, we investigated whether the injured CNN model can recover internal representations similar to those of the initial healthy state by quantifying the evolution of centered kernel alignment (CKA) states in each layer throughout the degeneration process. CKA represents a correlation value with an upper bound of 1.0, indicating perfect similarity. This is a metric to compare layer-wise representations of architecturally identical models. Figure 4 shows the CKA analysis of the model retrained on the subset of 1000 images at four different levels of injury. It can be seen that even at high levels of model degeneration (i.e., 80% of synaptic decay), the early layers of the model retain high representational similarity to those of the healthy model. In nontrast to that, representations in later layers were found to be more vulnerable to change as injury is applied. Figure 4 shows that at substantial levels of weight decay (40–60%), the deeper layers of the model begin to show larger deviations in similarity compared to the healthy model. Thus, as more simple features are disrupted in early layers, the complex, deeper features are affected even more. This can be explained by the locality in earlier layers compared to more compressed and abstract global information in deeper layers, where representational similarity is essentially a function of model accuracy. When 60% of model weights are decayed and frozen, layers 22–48 only retain CKA similarity values of 0.4 and below (third panel of Fig. 4C). However, even with only a single epoch of retraining, the CKA values of the later layers recover moderate levels (approximately CKA of 0.6) of representational similarity. These patterns of results are consistent for all three retraining regimes, while a larger retraining subset leads to a higher resemblance to the healthy model, and a smaller subset results in less similarity. Notably, even as the internal structure of the model changes, similar representations are recovered each time the model undergoes retraining. Therefore, internal representations seem not to rely as much on model structure but on the training task.

CKA matrices of layer-wise representational similarities between the baseline (healthy) model and the model with the retraining subset of 1000 images for increasing levels of synaptic decay and retraining. Row (A) shows the CKA analysis comparing the healthy model and the decayed model. Row (B) shows the CKA analysis comparing the healthy model and the iteratively retrained model. Row (C) displays the diagonal (1:1 layer comparison) CKA values for each injury level. Blue and orange indicate the decayed model’s layers and post-retraining layers, respectively. CKA results show that the later layers are more susceptible to “loss” of similarity with the healthy model when subjected to decay.
Diseased models embed object category information
We found that there were large deviations in representational similarity, according to CKA analysis, in layer 36, which is a 2D convolutional layer in the fifth convolutional block of the model. Thus, we extracted model activations from the test set images and concatenated them to form feature representations specific to that layer at an injury level of 50% of the weights decayed and using a subset of 1000 images for retraining. Further analysis on additional layers is provided in the Supplementary Information. Figure 5A shows the first two principal component analysis modes of feature representations from the healthy, decayed, and retrained models, respectively, and overlaid with object class labels. We also show higher-level class information in Fig. 5B, where the same representations are labeled as either “animate” or “inanimate” based on their class. Overall, Fig. 5 reveals that within this layer, the healthy model has clustered representations that reflect distinct object classes according to the ground truth labels (Fig. 5A (left)). The healthy model also shows distinct clustering of inanimate and animate objects, but not in a bimodal distribution as seen in the data projection along the axes (Fig. 5B (left)). These results indicate that at this depth in the model architecture, the healthy model is representing and distributing information according to concepts or features that align with class-specific information rather than the higher-level categories of animate (living) or inanimate (non-living) objects. Conversely, once 50% of the model weights have been decayed (i.e., the accuracy of \(19.4\pm 12.3\%\)), feature representations are visibly more convoluted with less distinct grouping. As seen in Fig. 5A, B in the middle panels, representations tend to overlap more, regardless of class and inanimate/animate information. With retraining, the re-emergence of more organized groupings of representations can be observed. At this iteration of injury, retraining leads to a model accuracy of \(73.8 \pm 2.42\%\). Thus, the model regains more discriminatory abilities in object recognition, which is reflected in the clustering of representations that correspond to class labels.
Another layer of interest according to the CKA analysis was layer 26, as it seems to be consistently heavily affected by injury. We completed the same unsupervised clustering analysis as described above. Detailed descriptions of the results of this experiment can be found in the Supplementary Information, S4. Briefly described, this analysis showed that there seems to be a more dense clustering in the retrained class-wise representation of this layer when compared to the deeper layer displayed in Fig. 5. However, it need to be acknowledged that it is difficult to disentangle what makes one layers more susceptible to cognitive breakdown than another. Also, this layer seems to more distinctly encode a bimodal distribution according to animate or inanimate information. This appears reasonable given its intermediate layer position, where clusters are still defined by more low-level features.

Scatter plots of the first two modes of principal component analysis applied to internal feature representations of model activations on test set data from a deep layer in the network (layer 36) for the healthy model, 50% decayed model, and subsequently retrained model. (A) Scatter plots of feature representations overlaid with ground truth class labels. Marginal distributions of individual classes are seen on both sets of axes. (B) Scatter plots of feature representations overlaid with inanimate or animate labels. This characteristic is a higher-level concept that models tend to learn in earlier layers. This analysis utilizes visualization methods inspired by the work described by Glocker et al.25.
Discussion
The main objective of this research was to establish and test an in silico model to simulate the progression of neurodegenerative diseases in the visual cortex, particularly in the context of posterior cortical atrophy, a condition that can accompany Alzheimer’s disease and primarily affects the visual cortex. In this work, we presented a biologically reasonable in silico CNN model, equipped with a retraining procedure to loosely represent continual learning as well as an increasing load of synaptic decay that precedes full atrophy to simulate tau accumulation. By causing rapid deterioration and thinning of visual cortical areas such as V1, V2, V3, and V4, posterior cortical atrophy leads to loss of visual recognition abilities in patients26,27,28. In this work, we take first steps toward simulating the effects of increasing levels of hyperphosphorylated tau accumulation, which according to the current prevailing theory leads to neurofibrillary tangles that disrupt typical brain function. The density of these tangles has been shown to correlate well with features of cognitive decline during AD progression29. Therefore, we incorporated two levels of decay progression to simulate an increasing burden of tau aggregates: (1) increasing the number of synapses that get decayed and frozen for retraining and (2) iteratively increasing the magnitude by which the synapses decay until they have essentially been removed from the network.
The results of our simulations show that even with high levels of synaptic decay and limited retraining data, the CNN model is able to recover substantial levels of object classification accuracy and similarities in internal representations compared to those of the unaffected healthy model. This results in a more gradual decline in object recognition accuracy of the retrained model, much like the continuous cognitive decline seen in patients with Alzheimer’s disease and posterior cortical atrophy30,31,32. This trend is also supported by human data (Fig. 6), which shows that increasing levels of phosphorylated-tau181 (P-tau181) are associated with a gradual loss of performance on the Boston Naming test and the Mini Mental State Exam (MMSE). These cognitive assessments measure patient recall of objects and general cognitive function, and are similar to the vision tasks we use in this research33.

Scatter plots of a fluid biomarker measure levels of P-tau181 versus patient performance for two cognitive assessments, the Boston Naming Test (BNT) and the Mini Mental State Exam (MMSE) with fitted locally estimated scatter-plot smoothing curves. The MMSE is overlaid with the model’s accuracy trajectory from Fig. 3A, showing that the model estimates the cognitive decline as a function of tau accumulation to an abstracted degree. This figure is modified from Memon et al.20.
Additionally, we demonstrated that throughout a complete cycle of model injury, the early layers of the model retained high levels of CKA similarity, while later layers, which capture more class-specific information, were much more susceptible to breakdown. This is a promising result, from a neural data standpoint. Recently, Katsumi et al. have shown that in patients with PCA, elevated uptake of tau begins in the visual cortex and spreads to the dorsal attention network, directly anterior to the visual network. It was also shown that elevated tau levels in the dorsal attention network are associated with higher levels of cognitive impairment on visuospatial tests16. This is in line with our findings from a behavioral standpoint as can be seen in Fig. 5 that shows that as disease progresses, class representations becomes less distinct. Patients with diseases such as posterior cortical atrophy do not lose the ability to process low-level visual information such as edges and curvature, but rather have decreased full object perception abilities34. It has been established that early layers of CNNs essentially act as low level feature (i.e., edge) detectors, while subsequent deeper layers detect more complex image features, and the deepest layers in the network can detect full objects2,3. Thus, the disruption of the deeper layers in this in silico model of disease offers further evidence of biological plausibility in the degradation of visual cognitive abilities.
The progressive synaptic decay employed in this work to simulate neurodegenerative diseases bears some resemblance to regularization techniques, like L1 and L2 regularization. More precisely, these approaches encourage sparsity and smaller weight magnitudes in neural networks during training, which can lead to simpler, more generalizable models35. However, our method differs in key ways. Rather than applying a global regularization term during training, we explicitly decay and freeze weights in a progressive manner after the model has been fully trained to mimic the biological processes of neurodegeneration. This targeted approach allows us to study the specific impacts of synaptic degradation on network performance and examine potential mechanisms of resilience. Indeed, the results of this work also show that, while weight decay led to initial reductions in accuracy, the model exhibited a degree of resilience and adaptability as a result of retraining, even with limited amounts of unbalanced retraining data. These results raise the question if the retraining procedure indeed results in the formation of new pathways as a result of increasing number of decayed and frozen parameters. Alternatively, retraining could simply train the model on how to effectively transmit information through these weight-decayed synapses. Analyzing the indices of the highest magnitude weights within various layers demonstrated that both scenarios appeared to be occurring. More precisely, we found that the indices of maximal value weights changed, suggesting that a new pathway for information flow may have been utilized. However, we also occasionally observed instances where the indices of maximal weights did not change, while the values of the weights themselves did. Thus, it may be posited that the model is learning new weight distributions to accomplish object recognition. It is assumed that the cognitive reserve and plasticity in the brain allow for compensation of these decaying pathways36. The results of this work show that CNNs can behave in similar ways as the mammalian brain by finding new pathways and scaling information to retain object recognition performance.
Further examination of internal representations using an unsupervised approach allowed us to visualize the specific differences in which internal representations of the model are changing with weight decay and retraining. We found that in deeper layers of the network (i.e., layer 36, as seen in Fig. 5), decaying the model weights caused extreme degeneration of distinct class-wise groupings. Subsequently, retraining leads to partial recovery and organization of these groupings. Nonetheless, larger overlaps between different class distributions remained when compared to the healthy model. Additionally, it can be seen that the healthy model does not necessarily group high-level information (inanimate/animate) into meaningful distributions. While slightly unexpected, it is apparent that the healthy model relied on features specific to each class to identify object categories that did not necessarily correlate with higher-level group features. However, after decay and retraining, a bimodal distribution of this high-level information emerges. This indicates that as a result of disease progression, model accuracy or ‘cognition’ becomes more reliant on high-level image information to classify objects. This retention of ‘super-class’ information as specific object recognition diminishes is similar to that of patients with semantic dementia30. In the case of semantic dementia, patients lose object-specific knowledge yet retain object-category knowledge for much longer. These results are indicative of this same phenomenon emerging in our in silico model of disease progression, in that as the model lost plasticity and synaptic strength, it became more reliant on object-category information in order to try to identify specific objects.
Considerations and future work
A notable consideration in this study is the remaining discernible differences in information processing between CNNs and the biological visual system37. For instance, CNNs employ global convolutional filters, whereas the human visual system encompasses filtering units responsible for specific regions within the receptive field38. Additionally, CNNs are trained with backpropagation and contain functions such as batch normalization and softmax activation, which are processes that are different from the learning and computational mechanisms of the mammalian visual system. Despite these differences, it should be noted that CNNs achieve object recognition performance comparable to that of humans for many visual tasks. It also has been shown previously that CNNs trained only for object recognition tasks possess a remarkable capacity to be predictive of neural activation in the primate visual cortex, surpassing the performance of any other computational model to date4,39,40. Although CNNs operate distinctly at the neural level, the hierarchical nature broadly mirrors that of human visual processing.
While our CNN-based model provides insights into how progressive weight decay affects hierarchical visual processing, it has important limitations. Notably, in neurodegenerative diseases like Alzheimer’s disease, tau pathology typically originates in the hippocampus—a structure that processes memory and spatial information through recurrent connections rather than the feed-forward hierarchical processing found in CNNs and the ventral visual stream. However, in this work we specifically aimed to model the progression of posterior cortical atrophy, sometimes referred to as the visual variant of Alzheimer’s disease41. In posterior cortical atrophy, the earliest signs of synaptic/neuronal loss and dysfunction are observed in the posterior regions of the cerebral cortex. As the disease advances, this degenerative process spreads to more anterior cortical areas. The current understanding of Alzheimer’s disease pathology suggests that the accumulation of tau protein in a given brain region precedes the onset of neurodegeneration. Therefore, in the case of posterior cortical atrophy, it is probable that tau pathology also follows a posterior-to-anterior progression, with initial build-up in the posterior cortical regions followed by the gradual involvement of more anterior regions as the disease progresses16. Therefore, our model does not aim to capture the initial hippocampal pathology or the spread of typical amnestic AD dementia, but rather focuses on understanding how progressive spread of degeneration affects hierarchical visual systems. Future work could explore how neurodegeneration affects recurrent networks modeling hippocampal function, potentially providing insights into earlier stages of typical AD disease progression. Although out of scope for this study, layer-specific injury would be a valuable future direction for research in modeling both, PCA and AD. Propagating injury through cascading layers could somewhat topographically capture the diffuse but directional spread of tau deposition and neurodegeneration.
There is a wide avenue for other explorations in future work. For example, it would be important to validate these in silico model results in more detail with neural biomarkers from patients suffering from neurodegenerative diseases. Although challenging to draw direct comparisons, AD patients typically complete cognitive assessments that target measuring visual object recognition and recall abilities such as the MMSE and the Boston Naming test. Classifying objects in pictures, as done by the models in this work, is a skill also tested in these examinations. A joint modality approach, which pairs biomarkers such as neurofibrillary tangles and cortical atrophy with assessment scores, would allow us to more accurately calibrate the in silico disease progression as a function of disease load. In the future, model predictions should be corroborated with longitudinal neural data from visual cortical areas in patients with posterior cortical atrophy. Structural or functional connectivity imaging studies could also provide insight into the stability of different areas of visual processing under different loads of disease accumulation. This may also be done on a personalized basis, so that the model can get fine-tuned to be more indicative of how an individual’s neurodegenerative disease could progress based on a few longitudinal measurements of atrophy and cognition.
Examining variations in network plasticity and recovery opens avenues for identifying optimal and personalized intervention strategies, potentially connecting these insights to rehabilitation techniques employed in individuals with dementia. Our framework provides a flexible computational model to simulate individual patient differences by adjusting both, different retraining abilities and decay strategies. In our current approach, varying the parameter \(\gamma\) from Equation 2 allows us to probe how disease trajectories vary by inducing heavier or lighter dampening of synapses. In this analyses, an increased value of gamma leads to lighter weight decay, as the gamma value is multiplied by the magnitude of the original synaptic weight values. The results of these experiments are intuitive in that a lighter weight decay (\(\gamma =0.75\)) results in less degradation to the accuracy of the network. Conversely, we see the opposite trend with \(\gamma =0.25\), leading to a stronger affect on synapses and, thus, on network accuracy as more synapses are affected. In addition, we included the results of an experiment of the extreme case of setting \(\gamma =0\), effectively simulating immediate and complete synaptic ablation. This experiment results in the most rapid rates of cognitive decline over time (modeled by injury percent). The results of these experiments can be found in the Supplementary Information (S2, S3, and S4), and show that both gradual decay and immediate ablation lead to qualitatively similar patterns of degradation in network performance. While our decay-based approach attempts to model the gradual synaptic changes observed in neurodegenerative diseases more closely and realistically, we note that both decay and ablation methods represent simplified models of complex pathological processes. The framework presented in this work allows for modeling different combinations of injury mechanisms, which may be relevant given the heterogeneous manifestation of pathologies in neurodegenerative diseases. Overall, this research contributes to ongoing efforts to employ deep learning models for studying disease progression, while acknowledging that direct validation against neural data would be needed to assess the biological relevance of different modeling approaches. The in silico model presented in this work aims to simulate the progression of tau pathology in neurodegenerative diseases and offers a promising avenue for precise disease modelling. The development of these computational techniques holds significant potential to revolutionize diagnosis, treatment, and management of neurodegenerative diseases.
Material and methods
Model architecture and data
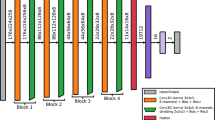
As the first step, we modeled ‘healthy’ cognition using a VGG19-like architecture, a CNN with a high similarity to the human brain as measured by the widely used Brain-Score 24, and trained it using the CIFAR10 dataset22,42. CIFAR10 consists of 60,000 \(32\times 32\) naturalistic, color images with ten object classes (e.g., airplane, bird, cat). The initial train and test split was 50,000/10,000 images, respectively. While VGG19 is named for its 19 weight (convolutional and linear) layers, our analysis encompasses all operational layers including convolutional layers, batch normalization layers, ReLU activations, max pooling layers, and the final classifier, totaling 48 distinct layers. The network contains five blocks with convolutional and batch normalization layers, five pooling layers (four max pooling and one adaptive average pooling), and a final linear classification layer. The final layer is a softmax activation with ten nodes corresponding to the ten classes in the dataset. Previous studies have indicated that standard VGG19 models exhibit a significant degree of overparameterization, as evidenced by their ability to maintain high levels of accuracy even after undergoing optimized pruning43,44. This overparameterization can be attributed to the excess number of neurons and their highly dense weight (synaptic) connections, leading to pathways that learn redundant information. On the contrary, the brain, while also displaying overparameterization to some extent that enables it to learn and make use of new neural pathways, operates under more stringent constraints such as energy consumption and physical space limitations45,46. Thus, to account for potential spurious outcomes resulting from overparameterization rather than the plasticity of the model, we performed experiments with a highly compressed version of VGG19. This is another limitation of previous work directly addressed in this work, as the previous studies performed experiments with unpruned networks.
Compression of the original VGG19 architecture was performed with structured pruning techniques as described in Li et al. and Fang et al.47,48. More precisely, filters and their corresponding weights were simultaneously eliminated based on their L1 norm until the model’s inference speed, measured in floating-point operations per second (FLOPS), was enhanced by a predefined margin. We found that increasing FLOPS by three-fold led to a compressed model with only 8.54% of the number of weights of the original VGG19 model (1.71 million weights compared to the original 20.04 million). Despite the significant reduction in the number of weights, the model achieved an accuracy of 93.3% on the test set, compared to the full VGG19 model achieving 93.7%. Therefore, all experiments described in the following were conducted using this highly optimized and constrained version of VGG19 as the baseline healthy model. Our model was pre-trained on ImageNet and fine-tuned on CIFAR10 for \(E=100\) epochs with a learning rate of \(\alpha = 0.001\), using a batch size of \(B=128\), and a stochastic gradient descent optimizer with momentum \(\eta =0.9\).
Synaptic decay and retraining
Instead of a simple removal of model synapses as done previously8,9, in this work, synapses were decayed in a dynamic process described in the following.
Let \(w_0 \in \mathbb {R}^n\) be the initial vector of all synaptic weights for the pre-trained CNN. For each synaptic decay and retraining iteration \(i \in \{0,\ldots ,N\}\), we define \(w_i^{\text {pre}} \in \mathbb {R}^n\), \(w_i^{\text {dec}} \in \mathbb {R}^n\), and \(w_i^{\text {ret}} \in \mathbb {R}^n\) to be the weights before the decay process, after the decay, and after retraining respectively, with \(w_0=w_0^{\text {pre}}\). Then, the dynamics of the synaptic decay and retraining for each i can be described by the following:
where \(\gamma\) is the decay rate, \(\odot\) is an element-wise multiplication, SGD indicates stochastic gradient descent-based retraining of the model, and \(s_i\in \{0, 1\}^n\) is the binary indicator vector for step i (\(s_0\) is the zero vector). The injury indicator for weight synapse j follows the stochastic process:
where \(X_{i}^{j} \sim {\text {Bern}}(\theta _i)\) with \(\theta _i\) chosen such that \(|s_i|_0 = \xi n\). That is, exactly \(\xi\) fixed percentage of previously healthy synapses are randomly selected for injury at each iteration.
In this study, we performed experiments with \(N=10\), \(\xi =0.1\), and \(\gamma =0.5\). While these values are hyperparameters and may be adjusted to specific modeling scenarios, these values were chosen as this was found to be representative of general injury resolution, as seen in Fig. 6, while remaining computationally feasible. Results for \(\gamma =0.25\) and \(\gamma =0.75\) may be found in the Supplementary Information, S2 and S3. Therefore, in the first iteration of injury, 10% of model synapses were decayed by a factor of 0.5. In the second iteration of injury, an additional 10% of model synapses were decayed by a factor of 0.5, while the initially decayed weights were decayed by an additional factor of 0.5. Therefore, the weights that are decayed early on in the process practically become ablated as the cycle of degeneration completes. This process is repeated until all potential weights have been affected, as visualized in Fig. 2.
After each iteration of injury, the remaining healthy weights in the model were retrained, while the decayed weights remained frozen in their decayed state. This simulates the continual neuroplasticity of the healthy areas of the brain, while the diseased areas have compromised plasticity abilities. Random subsets of 5000, 1000, and 100 images were chosen for retraining without considering class balance from the initial training set. This random selection aimed to mimic a more realistic continual learning method of information processing in the visual cortex.
Centered kernel alignment
Analyzing similarity between and within artificial neural networks remains to be an active area of study. Some of the challenges arise due to the distributed nature of representations across large numbers of neurons. Centered kernel alignment (CKA) is an established method that addresses these challenges and is frequently used to compare internal model representations within and across deep neural networks. CKA identifies correspondences or similarities between models on a layer-wise basis and is invariant to orthogonal transformations and isotropic scaling of model representations of data49. Although similarities between representations of deep neural networks have been measured in various ways, CKA is a powerful tool for correlation discovery in both, architecturally identical and different models50,51,52,53. As described in49 , CKA is denoted:
where \({\mathbf {K}}\) and \({\mathbf {L}}\) are the Gram matrices of two layer representations and HSIC denotes the Hilbert-Schmidt Independence Criteria, which measures the independence between two distributions. In this study, we used CKA to determine if the compressed VGG19 model can reestablish and retain similar internal representations as the model undergoes synaptic decay and retraining. This approach effectively quantifies a model’s capacity for cognitive reserve and its plasticity in learning new pathways for object recognition tasks, under escalating injury. Additionally, this approach allows for a further investigation as to whether the model learns new pathways to perform the same object recognition task or forms new representations while achieving similar accuracy levels.
Unsupervised feature representation analysis
As previously described, CKA quantifies layer-to-layer similarities in the representational feature space. We inspected CKA output patterns for specific model layers and examined layers that are most affected by degeneration. We then performed a more in-depth analysis on the most affected layers’ internal representations to investigate how they encode and distribute visual feature information. For this analysis, we employed an unsupervised technique that allowed us to investigate how information is encoded and distributed in specific layers with synaptic decay and retraining. More precisely, we extracted and concatenated activations for all 10,000 images in the test set from these individual layers in the model with the retraining subset of 1000 images at 50% injury. To analyze the high-dimensional data, we employed principal component analysis (PCA) to capture the primary modes of variance within the layer activations. Subsequently, we plotted the feature representations along the first two PCA modes (75.4% variance explained) for visualization purposes25.
Data availability
The datasets generated and/or analysed during the current study are available in the CIFAR10 repository, at https://www.cs.toronto.edu/ kriz/cifar.html.
Code availability
The underlying code for this study will be made publicly available upon publication.
References
Richards, B. A. et al. A deep learning framework for neuroscience. Nat. Neurosci. 22(11), 1761–1770 (2019).
Kriegeskorte, N. Deep neural networks: A new framework for modeling biological vision and brain information processing. Annu. Rev. Vis. Sci. 1, 417–446 (2015).
Lindsay, G. W. Convolutional neural networks as a model of the visual system: Past, present, and future. J. Cognit. Neurosci. 33(10), 2017–2031 (2021).
Yamins, D. L. K. et al. Performance-optimized hierarchical models predict neural responses in higher visual cortex. Proc. Natl. Acad. Sci. USA 111(23), 8619–8624 (2014).
Yamins, D. L. K. & DiCarlo, J. J. Using goal-driven deep learning models to understand sensory cortex. Nat. Neurosci. 19(3), 356–365 (2016).
Saxe, A., Nelli, S. & Summerfield, C. If deep learning is the answer, what is the question?. Nat. Rev. Neurosci. 22(1), 55–67 (2021).
Tuladhar, A., Moore, J. A., Ismail, Z. & Forkert, N. D. Modeling neurodegeneration in silico with deep learning. Front. Neuroinform. 15, 748370 (2021).
Moore, J. A. et al. Dementia in convolutional neural networks: Using deep learning models to simulate neurodegeneration of the visual system. Neuroinformatics 21, 45–55 (2022).
Moore, J. A. et al. Simulation of neuroplasticity in a CNN-based in-silico model of neurodegeneration of the visual system. Front. Comput. Neurosci. 17, 1274824 (2023).
Moghekar, A., Li, S., Lu, Y., Li, M., Wang, M.-C., Albert, M., O’Brien, R. & For the BIOCARD Research Team. CSF biomarker changes precede symptom onset of mild cognitive impairment. Neurology, 81(20), 1753–1758 (2013).
Moreno, H. et al. Tau pathology-mediated presynaptic dysfunction. Neuroscience 325, 30–38 (2016).
Lim, S., Haque, Md. M., Kim, D., Kim, D. J. & Kim, Y. K. Cell-based models to investigate tau aggregation. Comput. Struct. Biotechnol. J. 12(20), 7–13 (2014).
Cook, C. et al. Tau deposition drives neuropathological, inflammatory and behavioral abnormalities independently of neuronal loss in a novel mouse model. Hum. Mol. Genet. 24(21), 6198–6212 (2015).
Albin John, P. & Reddy, H. Synaptic basis of Alzheimer’s disease: Focus on synaptic amyloid beta, P-tau and mitochondria. Ageing Res. Rev. 65, 101208 (2021).
Pelkmans, W. et al. Tau-related grey matter network breakdown across the Alzheimer’s disease continuum. Alzheimer’s Res. Therapy 13(1), 138 (2021).
Katsumi, Y. et al. Anterior dorsal attention network tau drives visual attention deficits in posterior cortical atrophy. Brain 146, 295–306 (2023).
Alonso, A. D. et al. Hyperphosphorylation of tau associates with changes in its function beyond microtubule stability. Front. Cell. Neurosci. 12, 338 (2018).
Gendron, T. F. & Petrucelli, L. The role of tau in neurodegeneration. Mol. Neurodegener. 4(1), 13 (2009).
Koller, E. J. & Chakrabarty, P. Tau-mediated dysregulation of neuroplasticity and glial plasticity. Front. Mol. Neurosci. 13, 151 (2020).
Memon, A., Moore, J.A., Kang, C., Ismail, Z. & Forkert, N.D. Visual functions are associated with biomarker changes in Alzheimer’s disease. J. Alzheimer’s Dis. (in-press) (2024).
Cohen, U., Chung, S. Y., Lee, D. D. & Sompolinsky, H. Separability and geometry of object manifolds in deep neural networks. Nat. Commun. 11, 746 (2020).
Krizhevsky, A. & Hinton, G. Learning multiple layers of features from tiny images (2009).
Woods, B. et al. Cognitive stimulation to improve cognitive functioning in people with dementia. Cochrane Database Syst. Rev. 15(1), CD005562 (2023).
Schrimpf, M., Kubilius, J., Hong, H., Majaj, N.J., Rajalingham, R., Issa, E.B., Kar, K., Bashivan, P. & Prescott-Roy, J. Brain-score: Which artificial neural network for object recognition is most brain-like?. BioRxiv 407007 (2018).
Glocker, B., Jones, C., Bernhardt, M. & Winzeck, S. Algorithmic encoding of protected characteristics in chest X-ray disease detection models. eBioMedicine 89, 104467 (2023).
McMonagle, P., Deering, F., Berliner, Y. & Kertesz, A. The cognitive profile of posterior cortical atrophy. Neurology 66(3), 331–338 (2006).
Lehmann, M. et al. Cortical thickness and voxel-based morphometry in posterior cortical atrophy and typical Alzheimer’s disease. Neurobiol. Aging 32(8), 1466–1476 (2011).
Borruat, F.-X. Posterior cortical atrophy: Review of the recent literature. Curr. Neurol. Neurosci. Rep. 13(12), 406 (2013).
Perl, D. P. Neuropathology of Alzheimer’s disease. Mount Sinai J. Med. J. Transl. Personal. Med. J. Transl. Personal. Med. 77(1), 32–42 (2010).
Hodges, J. R., Graham, N. & Patterson, K. Charting the progression in semantic dementia: Implications for the organisation of semantic memory. Memory 3(3–4), 463–495 (1995).
Fox, N. C., Scahill, R. I., Crum, W. R. & Rossor, M. N. Correlation between rates of brain atrophy and cognitive decline in AD. Neurology 52(8), 1687–1689 (1999).
Jefferson, A., Barakat, L., Giovannetti, T., Paul, R. & Glosser, G. Object perception impairments predict instrumental activities of daily living dependence in Alzheimer’s disease. J. Clin. Exp. Neuropsychol. 28(6), 884–897 (2006).
Williams, B. W., Mack, W. & Henderson, V. W. Boston naming test in Alzheimer’s disease. Neuropsychologia 27(8), 1073–1079 (1989).
Yong, K. X. X. et al. Diagnosis and management of posterior cortical atrophy. Curr. Treatment Options Neurol. 25(2), 23–43 (2023).
Bengio, Y., Goodfellow, I. & Courville, A. Deep Learning Vol. 1 (MIT Press, 2017).
Esiri, M. M. & Chance, S. A. Cognitive reserve, cortical plasticity and resistance to Alzheimer’s disease. Alzheimer’s Res. Therapy 4(2), 7 (2012).
Lonnqvist, B., Bornet, A., Doerig, A. & Herzog, M. H. A comparative biology approach to DNN modeling of vision: A focus on differences, not similarities. J. Vis. 21(10), 17 (2021).
Wilson, C. L., Babb, T. L., Halgren, E. & Crandall, P. H. Visual receptive fields and response properties of neurons in human temporal lobe and visual pathways. Brain 106(2), 473–502 (1983).
Cadieu, C. F. et al. Deep neural networks rival the representation of primate IT cortex for core visual object recognition. PLoS Comput. Biol. 10(12), e1003963 (2014).
Khaligh-Razavi, S.-M. & Kriegeskorte, N. Deep supervised, but not unsupervised, models may explain it cortical representation. PLoS Comput. Biol. 10(11), e1003915 (2014).
Levine, D. N., Lee, J. M. & Fisher, C. M. The visual variant of Alzheimer’s disease. Neurology 43, 305–305 (1993).
Russakovsky, O. et al. ImageNet large scale visual recognition challenge. Int. J. Comput. Vis. 115(3), 211–252 (2015).
Frankle, J. & Carbin, M. The Lottery ticket hypothesis: Finding sparse, trainable neural networks. In International Conference on Learning Representations (2018).
Ayinde, B. O., Inanc, T. & Zurada, J. M. Redundant feature pruning for accelerated inference in deep neural networks. Neural Netw. 118, 148–158 (2019).
Drachman, D. A. Do we have brain to spare?. Neurology 64(12), 2004–2005 (2005).
Mizusaki, B. E. P. & O’Donnell, C. Neural circuit function redundancy in brain disorders. Curr. Opin. Neurobiol. 70, 74–80 (2021).
Li, H., Kadav, A., Durdanovic, I., Samet, H. & Graf, H.P. Pruning filters for efficient ConvNets. In Conference on Computer Vision and Pattern Recognition (2016).
Fang, G., Ma, X., Song, M., Mi, M.B., & Wang, X. DepGraph: Towards Any Structural Pruning. arXiv (2023).
Kornblith, S., Norouzi, M., Lee, H. & Hinton, G. Similarity of neural network representations revisited. In International Conference on Machine Learning, 3519–3529. (PMLR, 2019).
Kriegeskorte, N., Mur, M. & Bandettini, P. Representational similarity analysis—Connecting the branches of systems neuroscience. Front. Syst. Neurosci. 2, 4 (2008).
Raghu, M., Gilmer, J., Yosinski, J. & Sohl-Dickstein, J. SVCCA: Singular vector canonical correlation analysis for deep learning dynamics and interpretability. In Advances in Neural Information Processing Systems 30 (eds Guyon, I. et al.) 6076–6085 (Curran Associates Inc, 2017).
Morcos, A.S., Raghu, M. & Bengio, S. Insights on representational similarity in neural networks with canonical correlation. In Advances in Neural Information Processing Systems 31 (2018).
Raghu, M., Unterthiner, T., Kornblith, S., Zhang, C. & Dosovitskiy, A. Do vision transformers see like convolutional neural networks?. Adv. Neural. Inf. Process. Syst. 34, 12116–12128 (2021).
Author information
Authors and Affiliations
Contributions
J.A.M. wrote the main code and manuscript. V.V. and E.A.M. provided code contributions. A.M. made Figure 6. J.A.M., C.K., M.W., N.D.F. conceptualized the project. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Moore, J.A., Kang, C., Vigneshwaran, V. et al. Towards realistic simulation of disease progression in the visual cortex with CNNs. Sci Rep 15, 6099 (2025). https://doi.org/10.1038/s41598-025-89738-y
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-89738-y
Keywords
This article is cited by
-
Digital dementia and testing of cognitive intervention for degenerating neural networks
npj Systems Biology and Applications (2025)
