
Abstract
Chronic Kidney Disease (CKD) is associated with heightened risk of thrombosis. Prescription of anticoagulants is key to manage it; however, CKD patients have shown an increased risk of bleeding under anticoagulation therapy compared to non-CKD patients. We hypothesized that the sex could modify the metabolism of indoxyl sulfate (IS), a uremic toxin and Apixaban. Our intoxication model shows that higher doses of IS and apixaban accumulate in the plasma of female mice because of expression differences in efflux transporters and cytochromes in the liver, ileum and kidneys, when compared to males. Furthermore, we found that accumulation of apixaban in females contributes to increased bleeding. Transcriptional analysis of liver samples revealed elevated Sult1a1 but reduced Abcg2 and Cyp3a11 in female mice, while in the kidneys the expression rates of Oat1 and Oat3 were respectively lower and higher than those observed in males, potentially affecting drug clearance. Whole proteomics liver analysis confirmed the previous transcriptional results at the protein level and revealed that sex had a major influence in regulating both coagulation and drug metabolism pathways. Thus, our findings underline the need for inclusive clinical and preclinical trials to accurately reflect sex-specific metabolic variations, and to consider CKD-specific changes to optimize dosing, minimize side effects, and improve patient outcomes.
Similar content being viewed by others


Introduction
Chronic kidney disease (CKD) can be defined as structural or functional abnormalities of the kidneys, regardless of presenting a decreased glomerular filtration rate (GFR), or as a GFR < 60 mL/min/1.73m2 for ≥ 3 months irrespective of visible kidney damage1. CKD is a widespread condition with a prevalence between 9.1 and 13.4%2. Sex has been shown to affect both prevalence and disease progression: while women are more prone to develop CKD, men are more likely to reach end-stage kidney failure3 and face a higher risk of death compared to women with CKD4. CKD is associated with several comorbidities such as hypertension, diabetes and most importantly cardiovascular disease (CVD)5 - the main cause of death in developed countries6. Therefore, CKD should be viewed as a whole-body affliction and considered from a global perspective. Traditional cardiovascular risk factors are insufficient to explain the elevated rates of CVD, rising uremic toxins as a potential link between CKD and CVD outcomes. Uremic toxins are compounds excreted by the kidneys under physiological conditions. Nevertheless, they accumulate in the blood and tissues of patients with CKD due to inadequate renal clearance7.
Hemostasis is dysregulated in patients with CKD. Indeed, an increased risk of thrombosis in the arterial and venous bed coexists with an enlarged bleeding risk, further exacerbated in more advanced stages of CKD8,9. All the phases of the hemostasis cascade: primary hemostasis, coagulation and fibrinolysis, are impaired during CKD10. Although the prime cause of this imbalance is yet to be discerned, it is plausible that uremic toxins play a significant role in its development. Two major cardiovascular disorders, atrial fibrillation (AF) and venous thromboembolism (VTE), are associated with thrombotic events and require preventive and/or curative anticoagulation. Both conditions exhibit higher frequencies in CKD patients11,12. Hence, correct coagulation management is vital to avoid stroke on AF or pulmonary embolism on VTE and haemorrhage in both conditions. Different anticoagulants have been used in these patients over the years. Warfarin, a vitamin K antagonist (VKA), is one of the most widely prescribed despite showing enhanced bleeding risk at later CKD stages, calciphylaxis and high discontinuation rates13,14. Lately, apixaban, an oral direct factor Xa inhibitor, has gained increasing popularity as the preferred anticoagulation treatment for CKD patients, in both early and later stages15. This preference stems from two primary reasons: kidney clearance amounts only up to 27%16 while having similar efficacy and a superior safety profile than warfarin17. Furthermore, among CKD patients, apixaban shows a reduction in bleeding risk when compared to VKA treatment. Nonetheless, bleeding risk is increased with the use of apixaban in patients with CKD in comparison with non-CKD patients18. Concerning its pharmacokinetics, apixaban is mainly metabolized by cytochrome 450 (CYP) 3A4 (CYP3A11 in mice) and to a lesser extent by CYP1A2 and excreted by efflux transporters such as P-glycoprotein (P-gp) and breast cancer resistance protein (BCRP) (Fig. 1S)16.

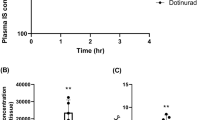
Indoxyl Sulfate levels in mice plasma quantified by HPLC (A), Apixaban levels in mice plasma quantified by LC-MS (B). WT Apixaban-treated females [control (KCl) and IS pooled] in orange, WT Apixaban-treated males WT [control (KCl) and IS pooled] in lilac. Grouped comparisons were made to assess sex related differences in Apixaban plasma levels regardless of treatment. Bleeding time (C). Pairwise comparisons were performed using Mann-Whitney U test, shown in black treatment differences within groups and in green sex related differences. Data are shown as median with interquartile range *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001, ****p ≤ 0.0001 (n = 6–20).
CKD is associated with numerous alterations in drug pharmacodynamics and pharmacokinetics19. Among various mechanisms, activation of Aryl hydrocarbon Receptor (AhR) by uremic toxins is proposed as an essential process to understand how CKD impairs drug metabolism20. AhR is a ligand-inducible transcription factor that regulates both nutrient metabolism and xeno- and endobiotic detoxification by inducing its target genes, among them CYP1A1, CYP1A2 and CYP1B1 (Fig. 1S)21.
Amidst the AhR agonists’ we find Indoxyl sulfate (IS), a protein-bound, tryptophan-derived uremic toxin22. Tryptophan is an essential amino acid, commonly found in human diet, present in poultry, nuts, eggs and other daily consumed products. Tryptophanase-expressing bacteria metabolize tryptophan into indole, an intermediate metabolite which is absorbed in the gut and further processed in the liver by CYP2E1 transforming indole into indoxyl23. Indoxyl is then sulfonated by the Sulfotransferase 1A1 (SULT1A1) to produce IS24. Indole is a highly toxic compound25 so the production of IS could be a mechanism to reduce its toxicity. In healthy individuals, IS is taken up by organic anion transporters (OAT) 1 and 3 (OAT1 and OAT3) in the proximal tubular cells and excreted through the urine (Fig. 1S)26. However, as CKD progresses, the impeded kidney function leads to tissue and plasma accumulation of IS, provoking deleterious effects27.
In excess, IS is considered a pro-inflammatory, pro-oxidative and pro-coagulant molecule, which is also related to endothelial dysfunction28. Plasma levels of IS positively correlate with circulating tissue factor levels, whose concentration and activity increase as CKD advances. Furthermore, indolic uremic solutes increase tissue factor production in endothelial and peripheral blood mononuclear cells via AhR activation, stimulating the activation of the coagulation cascade22. When IS accumulates in the liver, it can activate the expression of efflux transporter: P-gp, in hepatocytes also through AhR activation29. P-gp is responsible for the efflux of neutral and cationic drugs many of which are substrates of CYP3A4 like apixaban30. An alteration in its expression and activity could therefore modify drug metabolism and pharmacokinetics29.
Our goal is to test the hypothesis that indoxyl sulfate accumulation can modulate the expression of key metabolic enzymes and efflux transporters, thereby influencing apixaban metabolism and bleeding risk. Additionally, we aim to explore potential sex-related differences in gene expression, both at baseline and in response to treatment with apixaban, indoxyl sulfate, or their combination.
Materials and methods
Mice trial
C57BL/6J wild-type (WT) mice and Ahr−/− mice (B6.129-Ahrtm1Bra/J)31 were purchased from Jackson Laboratories and maintained as a breeding colony in the animal care facility at the Faculty of Pharmacy of Aix-Marseille Université. Genotypes were confirmed by polymerase chain reaction analysis of DNA from ear clippings obtained at 4 weeks of age (KAPA Mouse Genotyping Kit, KapaBiosystems). Mice were fed ad libitum with a regular diet (A04 standard, SAFE) during the whole trial. Both male and female mice were used in this study and comparisons were made between age-matched groups.
Six groups of mice per sex were created: Ahr −/−KCl, Ahr −/− IS, WT KCl, WT IS, WT KCl + Apixaban and WT IS + Apixaban.
The animal experiments were performed in compliance with the Directive 2010/63/EU of the European Parliament and were approved by the local Ethics Committee (“Comité d’Ethique en Expérimentation Animale de Marseille”, C2EA-14; ethics approval number: APAFIS#29595-2021020514483381 v4; approval date:04/16/2021).
All the animal experiments follow ARRIVE guidelines 2.032. All methods were performed in accordance with the relevant guidelines and regulations.
Indoxyl sulfate (IS) administration
At 10 weeks of age, mice drinking water was substituted by a 5% sucrose water solution with either 0.1% IS (Sigma-Aldrich, France) or KCl (Sigma-Aldrich, France) at the equivalent concentration as control treatment since IS is commercialised as a potassium salt. This solution was administered for 1-week prior to sacrifice.
Apixaban administration
To study the impact of IS on Apixaban metabolism, mice that were following the aforementioned procedure received an Apixaban (Eliquis®, Bristol-Meyers Squibb/Pzizer EEIG) solution (0.16 mg/mL). A 5 mg pill was solubilised in 0.6 mL of DMSO (SIGMA® Life Science) and diluted to the 50th in filtered tap water. Gavage was performed twice a day (morning-afternoon) with a volume of 8 µL per g of weight, starting 48 h before sacrifice. The last dose was given 4 h prior the sacrifice.
Tail bleeding time, blood and tissue collection
At 11 weeks, mice were anesthetized (ketamine 125 mg/kg, xylazine 12.5 mg/kg, atropine 0.25 mg/kg; intraperitoneally (i.p.) and placed on a hot plate maintained at 37 °C. A fragment 5 mm away from the tip of the tail was excised with a razor blade. The tail was immediately immersed in a saline solution at 37 °C. The time to establish cessation of bleeding (no rebleeding within 30 s) was visually determined. If necessary, bleeding was stopped at 5 min, mice that bled beyond this endpoint were counted as 5 min. Immediately after, blood samples (500 µL) were obtained from blood drawn via cardiac puncture. 60 µL of citrate (0,109 mM, BD Vacutainer®, Becton, Dickinson and Company) were added to the blood extracts to avoid coagulation. Mice were sacrificed by cervical dislocation under general anesthesia, before liver, ileum, and kidney samples were extracted. Organs were bathed in PBS to reduce blood contamination and frozen either in an RNAprotect Tissue Reagent (Qiagen) and kept at -20 °C for RNA analysis or Snap frozen in liquid Nitrogen for protein conservation, then kept at -80 °C.
Obtention of platelet-free plasma (PFP)
Whole blood was centrifuged at 2500 G for 15 min and then at 10,000 G for 10 min to obtain platelet-free plasma. Aliquots of PFP were stored at -80 °C.
Indoxyl sulfate plasma concentrations
IS was measured in PFP plasma by high-performance liquid chromatography (HPLC) using a reversed-phase column, an ion-pairing mobile phase, and an isocratic flow, as previously described33. The limit of detection for indoxyl sulfate using this method is 0.72 µM.
Apixaban plasma concentrations
Analyses evaluating apixaban levels were conducted at the Laboratoire de Pharmacocinétique et Toxicologie, CHU de Timone, Assistance Publique–Hôpitaux de Marseille (APHM). PFP diluted 1:2 in a pool of normal human plasma and quantified by LC-MS34. The limit of detection for apixaban is 1 ng/mL.
Organ dissection, RNA extraction and RT-qPCR analysis of mRNA expression
Mice kidneys, liver and ileum that were kept in the RNAprotect Tissue Reagent (Qiagen), were thawed on ice, immersed in QIAzol Lysis Reagent (Qiagen) and ground with TissueRuptor system (Qiagen). RNA was purified by chloroform extraction and isopropanol precipitation. Reverse transcription (RT) was performed on 500ng of total RNA using the PrimeScript™ RT Reagent Kit (Takara®), followed by quantitative polymerase chain reaction (qPCR) on 25 ng of cDNA using the Taqman® Universal Master Mix II, no UNG (Applied biosystems™ by Thermo Fisher Scientific) reaction mixture. Taqman® gene expression assays (Applied biosystems™ by Thermo Fisher Scientific) were used as probes to quantify the mRNA expression of the following genes: Abcb1a, Abcb1b, Abcg2, Cyp1a1, Cyp1a2, Cyp3a11, Cyp1b1, Cyp2e1, Sult1a1, Scl22a6 and Scl22a8 (Table 1SM, Supplementary materials).
All PCR reactions were performed using the Applied Biosystems™ StepOnePlus™ Real-Time PCR System (Thermo Fisher Scientific). The data was acquired by StepOne™software v2.3 (©2012 Life Technologies Corporation) and Relative Quantification (RQ) Application Module on Thermo Fisher Connect Platform (Thermo Fisher Scientific).
mRNA results are presented as normalized mRNA levels that reflect the variation in a target gene across samples. The 2−ΔΔCT normalization method as presented by Livak et al.35 is a derivation of the equation that describes the exponential amplification of PCR where the amount of target gene is normalized to an endogenous reference and expressed relative to a calibrator. This method assumes that the efficiency of the PCR is close to 1 and the PCR efficiency of the target gene is similar to the internal control gene. A modified version of this method described by Smadja et al.36 was used in this study (equation shown below).
Thus, target gene mRNA expression levels were normalized based on the Gusb expression (endogenous control) of each sample: N target = 2−ΔCtsample where ΔCt represents the value of the threshold cycle that leads to the amplification of the target gene minus the value of the threshold cycle that leads to the amplification of the endogenous control (Gusb). Unlike the traditional 2−ΔΔCTapproach, where normalized mRNA expression is relative to a calibrator sample, here the expression is relative to the normalized smallest measurable amount of target gene mRNA in the dataset (Ct value = 35), designated as N target smallest.
Protein extraction and whole proteome analysis of liver samples
Approximately 10 mg of tissue was combined with 150 µL 1× SDT buffer. Samples were disrupted by glass beads in the sonicator Bioruptor® Pico (Diagenode). 50 µg of protein were loaded onto the FASP (Filter assisted sample preparation) membrane, 30 kDa cut-off filters were used. Protein reduction (DTT) and alkylation (iodoacetamide) were performed, and samples were incubated with trypsin for 18 h at 37 °C. The peptide mixture was cleaned-up using ethylacetate extraction to remove potential residual SDS prior to LC-MS analyses. LC-MS/MS analyses were done using nanoElute (Bruker) online connected to timsTOF Pro system (Bruker): 90 min long linear LC gradient. MS and MS/MS spectra were recorded in a time of flight (TOF) analyzer using data-independent acquisition (DIA) mode utilizing trapped ion mobility (TIMS) in the precursor m/z range of 400–1000. All peptide mixtures were analyzed separately.
DIA LC-MS data processing was done using DIA-NN application (version 1.8): https://github.com/vdemichev/DiaNN). Library free search mode was used using the following protein databases:
-
cRAP contaminant database (version 170518).
-
based on http://www.thegpm.org/crap/.
-
111 protein sequences in total.
-
-
UniProtKB_Mouse (version from 16.6.2021).
-
taxonomy: Mus musculus.
-
taxon ID: 10,090.
-
download link: ftp://ftp.uniprot.org/pub/databases/uniprot/current_release/knowledgebase/reference_proteomes/Eukaryota/UP000000589/UP000000589_10090.fasta.gz.
-
22,001 protein sequences in total.
-
Match between runs (MBR) used across the whole dataset. Database search results were set to follow these false discovery rate (FDR) thresholds, precursor level: 1% FDR and protein group level: 1% FDR. The default protein inference algorithm implemented in DIA-NN was used to construct the protein groups list utilizing proteotypic (i.e. peptides unique for the given protein within the whole protein database) and non-proteotypic peptides.
LC-MS data evaluation: More than 6000 proteins were identified across all 60 samples. Proteins with no proteotypic peptides, no UniProtKB ID to gene symbol match (Omnipath37), or with more than 90% of missing values across samples were discarded. Samples: M KO IS 6 and the most prominent F WT IS 4 and F WT IS 6, were considered outliers and excluded from the posterior analysis. These samples exhibited abnormal separation in the PCA plot, clustering distinctly away from their main respective groups. Variance stabilizing normalization (Vsn)38 was used to normalize the filtered dataset, followed by row-mean imputation of missing values. Moderated t-test using LIMMA package in R39 was used to perform differential expression analysis, P values adjustment on multiple hypothesis testing was done using Benjamini & Hochberg method. Comparisons are mentioned as subtractions of two compared sample types as it was done on log2-transformed data. The comparisons performed were the following: M WT KCl – F WT KCl, M WT IS - F WT IS, M KO KCl - F KO KCl, and M KO IS - F KO IS. Volcano plots for the selected comparisons were created to determine differentially expressed proteins (FDR: 0.5 and FC: ±1.5). Pathway enrichment of differentially expressed proteins with cutoff values of Uncorrected P value < 0.05 and logFC > 0.5 or logFC < – 0.5, was performed using univariate linear models (ulm) in DecoupR40 and the MsigDB hallmark database for Mus Musculus41.
Statistical analysis
Data was presented as scatter plots (median +/- interquartile range). Statistical analyses were performed using GraphPad Prism version 9.1.2. (GraphPad Software Inc, San Diego, CA, USA). Statistical tests for non-parametric data were chosen since the number of mice per group was insufficient to assume a Gaussian distribution. Mann–Whitney U test was used for comparisons between two groups, P values ≤ 0.05 were considered significant. For comparisons between more than two groups, Kruskal–Wallis test followed by a Dunn’s test for comparisons between more than two groups was used. Spearman correlation coefficient (r) was used to measure linear correlation between two variables. All methods were performed in accordance with the relevant guidelines and regulations.
Results
IS and apixaban accumulate in WT female mice
IS concentration was increased in all the groups under IS treatment, with the accumulation of IS being higher in females compared to males (Fig. 1A). Females also showed a higher overall accumulation of apixaban in plasma when compared to males despite the dose being adjusted to weight and regardless of being under control or IS treatment (Fig. 1B) (non-pooled data is found in Fig. 2S). These results were also consistent with altered coagulation processes in females, as apixaban-treated females have higher bleeding times than males under the same treatment. If apixaban is administrated together with IS, bleeding time returns to basal levels (Fig. 1C). In males, bleeding time remains stable regardless of treatment.

Sex-dependent differences in mRNA Expression of efflux transporters and metabolic enzymes of apixaban in mice ileum (A), liver (B) and kidney (C) of apixaban-treatment mice. Pairwise comparisons of pooled—Males vs. Females—were performed using Mann-Whitney U test. Data are shown as median with interquartile range *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001, ****p ≤ 0.0001 (n = 12–20).
Apixaban accumulation in female mice is related to sex-dependent differences in mRNA expression of efflux transporters and metabolic enzymes
To further investigate the possible cause of apixaban accumulation in female plasma, we analyzed the expression of efflux transporters Abcb1a, Abcb1b and Abcg2 that mediate the excretion of apixaban as well as that of Cyp1a2 and Cyp3a11, which are the main enzymes that metabolize it (especially the latter). These results presented in the Fig. 3S, which display ungrouped data, demonstrate that most of the observed changes in gene expression were primarily driven by sex rather than by apixaban and/or IS treatment, both of which had relatively weak effects. We highlight sex as the most important source of variability. Hence, to properly analyse the data. We grouped mice by sex to analyze its effect on the expression of genes involved in drug metabolism and apixaban accumulation (Fig. 2).

Scatterplot of IS and Apixaban levels in males (A) and females (C). Spearman correlation coefficient was applied to assess linear correlation between Apixaban and IS levels. Cyp3a11 mRNA Expression levels in male (B) and female (D) liver. Pairwise comparisons were performed using Mann-Whitney U test. Data are shown as median with interquartile range *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001, ****p ≤ 0.0001 (n = 6–12).
The first barrier that apixaban faces before entering the bloodstream is the intestinal epithelium of the ileum: Higher expression of Abcb1a and Abcg2 was observed in males compared to females (Fig. 2A). In addition, we observed an increase in Abcb1a in both males and females treated with apixaban when compared to their respective KCl controls (Fig. 3S).
Due to apixaban being mainly metabolized in the liver, higher Cyp3a11 mRNA expression in males (Fig. 2B) could help reduce circulating apixaban levels. Furthermore, higher Abcg2 levels in males would promote apixaban excretion into the bile. Abcb1a showed higher expression rates in females, but due to overall lower expression in the liver, this is expected to minimally affect apixaban clearance.
Only 27% of apixaban is metabolized by the kidneys16, yet this elimination pathway should not be overlooked. As with the precedent organs, Abcg2 expression in the kidneys was higher in males (Fig. 2C). Concerning, Abcb1a and Abcb1b, despite being higher expressed in females, their contribution to total kidney clearance is minor (lower expression rates).
These results suggest that higher accumulation of apixaban in females could be due to a sex-dependent expression of genes involved in apixaban metabolism: Cyp3a11 and excretion: Abcg2, both less expressed in females.
Apixaban and IS accumulation are correlated in male mice but not in females
Despite lower accumulation in plasma, apixaban and IS showed a positive correlation in males (Fig. 3A). Furthermore, IS seemed to have a modulatory effect on liver Cyp3a11 expression in the presence of apixaban (Fig. 3B). This alteration in Cyp3a11 expression could explain the positive correlation observed between these two molecules in male mice. Nonetheless, in females, no association was found between IS an apixaban concentrations (Fig. 3C), and Cyp3a11 remained unchanged regardless of the treatment (Fig. 3D).
Organic anion transporters (OATs) in the kidneys have major mRNA expression differences regarding sex
Organic anionic transporters Scl22a6 (OAT1) and Scl22a8 (OAT3) are the main efflux transporters that regulate the passage of IS from the circulating blood to the proximal renal tubule towards excretion. Previous research by Jansen et al.42 showed an up-regulation of OAT1 induced by IS in an adenine-induced CKD rat model and in vitro via the AhR and EGFR axis. In our study, neither genotype nor apixaban nor IS treatment modifies the sex dimorphic expression of these transporters (Fig. 4S). Consequently, samples were pooled by sex to facilitate a clearer interpretation of the data. The results revealed that Scl22a6 mRNA expression was more abundant in males, while Scl22a8 expression was notably lower compared to levels observed in females (Fig. 4), which could explain IS accumulation in female plasma.

mRNA Expression of Scl22a6 and Scl22a8 in mice kidney. Mice were pooled regarding treatment. Pairwise comparisons—Males vs. Females—were performed using Mann-Whitney U test. Data shown are as median with interquartile range *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001, ****p ≤ 0.0001 (n = 13–20).
Sexual dimorphism in mRNA expression of efflux transporters and genes related to IS production influences IS accumulation
To try to understand the differences in the accumulation of IS between male and female mice, we studied the expression of the genes implicated in its excretion and production. As in previous analyses, samples were grouped by sex to highlight sex-related differences. Detailed results comparing Control (KCl) vs. IS-treated mice, as well as sex-dependent differences in non-pooled samples that justify the applied grouping, are presented in Figs. 3S and 5S, and 6S.

Sex-dependent differences in mRNA Expression of efflux transporters and metabolic enzymes in mice ileum (A), liver (B) and kidney (C) involved in IS metabolism. Samples with IS or control treatment were pooled by sex. Pairwise comparisons—Males vs. Females—were performed using Mann-Whitney U test (green). Data shown as median with interquartile range *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001, ****p ≤ 0.0001 (n = 11–17).

Differences in mRNA Expression of efflux transporters (A) and metabolic enzymes (B) in liver of Wild-type Vs. AhR−/− mice. For Cyp1a1, Cyp1a2 and Cyp2e1 males and females were pooled. Pairwise comparisons were performed using Mann-Whitney U test (blue). For Abcg2 and Sult1a1, Kruskal Wallis statistical test followed by Dunn’s test was used for sex related comparisons (green). Pairwise comparisons were performed using Mann-Whitney U test (black). Data shown as median with interquartile range *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001 (n = 5—17).
No sex-related differences in mRNA expression were found at the ileum level (Fig. 5A). In the liver, the site of transformation of indole to indoxyl sulfate, we observed higher Sult1a1 in females (Fig. 5B), which could facilitate endogenous IS production. Concerning efflux transporters in the kidney, changes in Abcb1a and Abcb1b were less relevant, due to Abcg2showing higher expression rates overall (Fig. 5C). Abcg2 mRNA remained either higher in males (liver), where higher quantities of IS could be excreted via this efflux transporter to the bile, or unchanged (kidney) compared to females (Fig. 5B and C).
IS effect in mRNA expression of genes modulated by activation AhR in the liver
To further understand, the influence of IS in liver metabolism and detoxification processes, we analyzed several cytochromes and efflux transporters associated with IS production, as well as genes whose expression is modulated by AhR. In this case, males and females were pooled when no sex-related differences were observed to provide a better insight into treatment effects, non-pooled data can be found in Fig. 6S.
IS-treated AhR−/− mice were the only experimental group with similar IS accumulation levels in females and males (Females: 68.2 (3.3; 84.1)* vs. Males: 49.1 (5.5; 140.9)). Abcg2 levels in AhR−/− male mice, although still higher than female levels, were highly diminished, which could influence IS accumulation in AhR−/− males (Fig. 6A). Foreseeably, Cyp1a1 and Cyp1a2 were increased in WT IS-treated mice and greatly diminished in the AhR−/− mice regardless of treatment validating the correct functionality of the model (Fig. 6B). Concerning genes related to IS production, Cyp2e1 showed lower mRNA expression in the AhR−/− mice and Sult1a1 was increased under IS treatment in all females (Fig. 6B) which could lead to induced IS production, an effect that we don’t retrieve in males treated under the same conditions.
Whole proteome analysis revealed that the main agent driving sample separation is sex
It has been extensively described in the literature that the liver shows high sexual dimorphism. Not surprisingly, from a total of 60 liver samples analyzed, constituted by 4 experimental groups per sex (WT-KCl, WT-IS, AhR−/− KCl, AhR−/− IS), sample separation was mainly driven by mice sex, as showed on the principal component analysis (Fig. 7A). Figure 7A, demonstrates that the primary source of variance in this dataset is sex-dependent, while genotype and treatment have minimal influence on the clustering patterns defined by PC1 and PC3, that represent a 23% of the explained variance.

Protein expression changes in the mouse liver. PCA of global expression changes in mice liver (A). Sex is the main clustering variable. Volcano plots of differentially expressed proteins (blue) (B–E), Table with the number of differentially expressed proteins across experimental groups (F), Heatmap containing the pathway enrichment analysis results (G) (Downregulated pathways in blue and upregulated in red).
Differentially expressed proteins amongst sexes lead to remarkable differences in coagulation and xenobiotic metabolism pathways
The Volcano plots in Fig. 7B–E showcase the differential expression of proteins between male and female groups within the same treatment and genotype. Blue dots represent differentially expressed proteins, while red dots represent proteins whose expression does not significantly differ. Additionally, a table that summarizes the exact number of upregulated, downregulated, and total differentially abundant proteins across male and female mice comparisons is presented in Fig. 7F. The total number of differentially expressed proteins highlights the significant proteomic divergence between sexes. It is important to note that the highest number of differentially abundant proteins (298) is observed between M AhR−/− IS and F AhR−/− IS, suggesting that the combination the AhR−/− genotype and IS treatment intensifies sex-based differences, potentially through IS activation of AhR-independent pathways that might differ in males and females.
Most interestingly, pathway enrichment analysis reveals that at the liver level, pathways such as coagulation and bile acid metabolism were upregulated (red) while xenobiotic metabolism and fatty acid metabolism were downregulated (blue) in males compared to females (Fig. 7G). Corroborating the need to study both sexes in preclinical models involving any of the pathways. Proteins contributing to each pathway can be found in Table 1S.
Most of the sex-dependent differences previously identified at the mRNA level, have been confirmed by whole proteome analysis. Notably, significant differences in the expression of Abcg2, upregulated in males, and Sult1a1, upregulated in females, remained consistent amongst all the comparisons (Fig. 8).

Protein expression of efflux transporters and metabolism enzymes related with IS or Apixaban metabolism. Heatmap (A) Downregulated proteins appear colored in blue and upregulated in red. Table containing logFC, t, P.value and FDR (B) for the proteins of interest amongst the different group comparison.
Study limitations
Our study has several limitations that should be considered when interpreting the results. The administration of IS in drinking water bypasses natural endogenous indole production from tryptophan by the gut microbiota and its subsequent liver processing to form IS. Hence, our IS-intoxication model is an artificial construct which may not fully capture the complexity of IS production and metabolism in vivo.
Furthermore, individual water consumption was not measured. This decision was made to prioritize animal well-being, as the most reliable methods for accurately measuring individual water intake—such as daily oral gavage or housing mice in isolation—pose significant stress to the animals and may influence experimental outcomes. Additionally, commonly used drinking devices are prone to leakage, making precise monitoring of water intake challenging. Nonetheless, the scientific literature suggest that water intake is a strain-dependent trait but not sexually dimorphic43. Therefore, not normalizing observed IS accumulation by individual water intake is unlikely to significantly affect the results regarding IS accumulation.
It is important to recognize that CKD is a systemic condition that affects the entire body, not only the kidneys. CKD patients accumulate a wide variety of uremic toxins beyond IS, such as kynurenine and kynurenic acid, which are also AhR ligands and can influence drug metabolism. Hence, the main objective of the paper which is studying IS effect alone in an in vivo context can be consider a drawback as well, as this model focuses on a single uremic toxin, in a non-uremic context. Despite these constraints, this intoxication model provides valuable insights into IS contribution to drug metabolism deregulation, paving the way for more intricate studies.
Our apixaban treated models, is an overdose model that allow us to assess metabolic changes in healthy mice. For reference, an average mice of 25 g was receiving an apixaban dose of 32 µg per gavage, which equals to giving a 60 Kg human being a dose of 76.8 mg per serving (between 15 and 30 times the real dose used in patients)44. Nonetheless, those are the doses widely used for pre-clinical models45,46.
Discussion
Pre-clinical trials tend to be executed in males only alleging that high variability due to the estrous cyclicity (which has been showed to be not relevant for most of the cases47) would mean to increase sample size and costs, leading to lack of female data. This study has stated the importance of sex segregation to get a closer look at the real drug metabolism panorama. Furthermore, we have identified a significant impact of sex in the expression of metabolism enzymes and efflux transporters implicated in the metabolism of IS and apixaban in the ileum, liver and kidneys of both treated and control mice, as basal expression levels already differ in a sex-dependent manner.
Our results in a pre-clinical model highlight the need to document the impact of sex in clinical trials with well-balanced sex ratios more precisely, particularly in the context of cardio-kidney metabolic syndrome and CKD where women are still underrepresented48,49. For instance, in the DAPA-CKD trial, females comprised only 33% of the cohort50. A recent review study concerning sodium glucose cotransporter-2 inhibitors, a class of drug that reduces the risk of cardiovascular events and/or chronic kidney disease progression in CKD patients51, have showed a potential bias linked to sex in efficacy and adverse side effects. When more women were enrolled in the trials the reduction in cardiovascular mortality was smaller and drug-related complications were detected in a higher proportion in women. In the ARISTOTLE trial, females represented only 35% of the clinical trial population, limiting the power to observe specific outcomes related to the drug52 and in another prospective study it was observed that during heart failure treatment, the optimal dose of RAS inhibitors to prevent the latter outcome is not the full dose but the half dose in women compared to men53.
Uremic toxin buildup, including IS, during CKD could exert an influence not only in the elimination of drugs excreted by the kidneys but also on the metabolism of drugs subjected to non-renal clearance, mainly the liver and the gut54. How modifications in these complex processes translate to patient pathology is poorly understood due to the large number of enzymes, transporters and metabolites implicated. Nonetheless, their understanding is essential as CKD patients are often prescribed several medications simultaneously. This poly-pharmacology results in frequently occurring drug-related adverse events, that could, in many cases be avoided if physicians were aware of the changes in drug disposition55. Therefore, future clinical trials should also consider how pre-existing conditions, in this case CKD patients: uremic toxin accumulation, could alter the metabolism of a given drug and whether the dose should be sex-adjusted to minimize side effects and treatment rejection.
A correct management of hemostasis is essential to avoid bleeding and thrombosis in patients with CKD. Excess IS due to poor renal clearance can tip the scale towards a pro-coagulant state and over-activate AhR target genes which could interfere with xeno- and endobiotic metabolism. Understanding how IS interacts with commonly used anticoagulants, such as apixaban, and how it might affect its metabolism is required to better deal with hemostasis disbalance in patients with CKD.
To date, no human studies have revealed a link between IS accumulation and gender. In this study, IS accumulation in females reached a median of 63.34 µM (minimum 32.63 µM, maximum 133.1 µM), while in males it was significantly lower, with a median of 17.48 µM (minimum 8.1 µM, maximum 62.8 µM). Previous research by our team has shown that CKD patients typically exhibit serum IS levels with a median of 43.7 µM (minimum 0.2 µM, maximum 256.2 µM) in CKD stages 3–556. Given the normal renal function of the mice, low to negligible IS accumulation was anticipated. Hence, the observed sex differences—particularly the higher accumulation in females, comparable to levels seen in mild CKD—were surprising. These findings are quite relevant, particularly in the case of females, as IS levels exceed the threshold necessary to activate AhR and its target genes56.
Concerning apixaban, it is unclear whether its levels differ with gender in humans or not, and the implications it might have16,57. According to the review work of Byon et al.16, despite apixaban accumulation in female subjects being up to 18% higher, no clinically relevant effects were observed. This could be due to the adherence not being closely monitored in some of the studies included and to a poorer collection of bleeding events. In our study, female mice showed a higher accumulation of IS (except from the AhR −/− mice treated with IS) and apixaban in plasma. Uneven efflux transporters expression, especially in the liver, could be behind this phenomenon. U.S Food and Drug Administration (FDA) recommends avoiding strong inhibitors and inducers of CYP3A and P-gp during treatment with apixaban (ELIQUIS (apixaban) tablets for oral use Initial U.S. Approval: 2012), nonetheless no dose adjustment is considered for patients at a higher apixaban accumulation/metabolism risk. Yet, several studies show that inhibition or genetic variation in the drug transporter gene ABCG2 affects the pharmacokinetics of apixaban54,59, which in some cases leads to increased bleeding risk60.
The gut, a major drug absorption site, can be affected by sex-dependent differences in efflux transporter expression and metabolism disturbances caused by exogenous molecules like apixaban or IS, as seen in our study: Higher expression of Abcb1a, and Abcg2 was observed in males compared to females under apixaban treatment. could potentially limit apixaban absorption. This might be the first step to explaining the lower levels of apixaban observed in males. Thus, when treating patients on multiple medications, addressing these factors is crucial to prevent treatment failure.
In the liver, females showed higher Abcb1a levels than males, however, this difference could not compensate for the fact that Abcg2 showed greater expression rates in all males, which is over 10 times greater than Abcb1a, and which would contribute to the excretion of apixaban and IS in the bile quicker. Furthermore, apixaban-treated males showed higher Cyp3a11 than females, which implies that apixaban metabolism rates could be enhanced. Moreover, apixaban induced Cyp3a11 expression in male liver, which would lead to even higher metabolism rates that could explain the lower accumulation but could also lead to treatment ineffectiveness. Nonetheless, both effects disappear when apixaban is given in combination with IS, showing how these two compounds interact with each other metabolism.
It’s widely known that AhR activation induces Cyp1a1 and Cyp1a2 transcription21. Hence, an increased in their expression in both male and female IS-treated mice comes as no surprise. CYP1A2 accounts for approximately 13% of the P450 cytochrome enzymes in the human liver61, and plays a key role in the metabolism of several substances, including caffeine, nonsteroidal anti-inflammatory drugs (e.g., naproxen), antidepressants, and antipsychotics62. Its relevance in drug metabolism studies is particularly significant in the context of CKD, where metabolic processes are often altered. For instance, an in vivo study in 5/6 nephrectomised rats indicated that CYP1A2 protein levels in the liver and the kidney increased by 55% and 168% respectively, when compared to non-CKD controls63.
Additionally, the study of CYP1A2 remains important even beyond the context of CKD, as there are clinically relevant CYP1A2 polymorphisms known to alter the cytochrome activity in adults, such as CYP1A2*1F (associated with reduced enzyme activity) (Laika et al., 2009) and CYP1A2*1D (associated with increased activity) (Uslu et al., 2010) that have been shown to influence the metabolism of drugs processed by CYP1A2. Alleles with reduced enzyme activity are mimicked by our AhR−/− mice, that present a significant downregulation of both Cyp1a1 and Cyp1a2.
Furthermore, in the liver of AhR−/− mice, IS decreases Abcg2 expression (main efflux transporter) in both males and females. We hypothesize that when a drug inhibits AhR at the hepatic level, if IS concentration is high enough, a lower clearing rate could occur through the bile duct of such drug or others that are cleared through the affected transporters.
The whole liver proteomic analysis provided valuable insights into sex-dimorphism. In this context, PCA revealed that samples clustered distinctly by sex, rather than by genotype or treatment. It is noteworthy to mention that PC1 and PC3 together accounted only for 23% of the explained variance. While this proportion may seem modest, it reflects the high-dimensional nature of the dataset, where variance is distributed across many dimensions—a common feature in proteomics. Multiple biological factors, such as genotype, treatment, and environmental influences, along with technical noise, also contribute to this distribution. Nonetheless, this level of explained variance is sufficient to illustrate biologically meaningful separations, such as the sex-driven clustering.
In line with the PCA findings, specific proteins of interest, such as Abcg2 (upregulated in males) and Sult1a1 (upregulated in females), displayed patterns consistent with the observations at the mRNA level. These results reinforce the idea that sexual dimorphism has a major impact in drug metabolism and subsequently in the accumulation of compounds in the bloodstream, including apixaban and IS. Beyond individual proteins, pathway enrichment analysis revealed that key liver functions exhibited sex-dependent regulation: Coagulation and bile acid metabolism pathways were upregulated in males, whereas xenobiotic metabolism and fatty acid metabolism pathways were downregulated in males compared to females. Overall, these findings demonstrate that sex remains the most significant driver of liver proteome differences, irrespective of treatment or genotype, which highlights the importance of studying both sexes in pre-clinical models to ensure robust results.
On a different note, proximal tubule cells in the kidney have been previously described by Jansen et al.42 to be implicated in remote metabolite sensing, particularly in response to IS in vivo (healthy volunteers and adenine-induced CKD rats). Organic anion transporters OAT1 and OAT3, that import IS and apixaban from the bloodstream onto the kidney, show sex-dependent differences, a fact widely described in the literature both at the mRNA and the protein level, that seems to be due to testosterone production64,65. If Scl22a6(OAT1) and Scl22a8(OAT3) were known to have dissimilar transportation rates for apixaban and/or IS, they could play a very significant role in the plasma accumulation of the substances in females.
In humans, OAT1 and OAT3 are the second and first transporters of the Scl22a family with the highest mRNA expression, and this would be the equivalent to our female mice situation. Jansen et al. demonstrated that OAT1 activity and expression was modulated via AhR and EGFR signalling. Interestingly, in their adenine-induced CKD male rat model, gavage with IS over seven weeks was able to increase renal IS clearance (4.4 ± 0.7 mL/min compared to 0.1 ± 0.02 mL/min in controls) and upregulate OAT1 mRNA expression. In contrast, in our study, IS was administered in drinking water for one week, the shorter period together with the use of non-CKD mice might explain why a similar rise in OAT1 expression was not observed. Thus, in our case, sex differences prevail over the effects of treatment and genotype also in the kidneys. Furthermore, other studies show that, hOAT1 mRNA was significantly lower in the kidney of patients with renal disease compared to healthy controls66, which could be one of the factors that could explain increasing IS accumulation as CKD progresses.
In addition, several studies demonstrate that Oat1 knockout mice show an increase in IS plasma levels, that has not been observed in Oat3 knockout mice and that IS accumulation has been seen to inhibit OAT1 in vitro67. The kinetic parameters for rat OATs have been previously described: IS had a 10-fold greater Km value for rOat3 than rOat1 (Km = 174 µmol/L and Km = 18 µmol/L respectively). Moreover, kinetic analysis of the IS uptake by human kidney slices revealed two saturable components with Km1 =24 µmol/L and Km2 = 196 µmol/L similar to those of rOat1 and rOat368. Therefore, higher affinity of IS for OAT1 could be playing a major role in its excretion.
Mice bleeding time is higher in females treated with KCl + Apixaban than in males which could be due to the plasma accumulation of apixaban. Females treated with IS + Apixaban showed lower bleeding time than those treated with IS or apixaban alone. An IS-rich milieu, could lead to endothelial damage and increased tissue factor and activation of coagulation22, which could increase platelet activation69 making them more reactive at the time of an injury, and therefore, lowering the bleeding time. These results suggest that during a procoagulant state, like CKD, the hemorrhagic effect of the accumulation of apixaban could be mitigated by the activation of coagulation. However, translating these findings into a CKD context is complex, as there are multiple contributing factors involved and the buildup of various uremic toxins, not just IS, which could further alter haemostasis and drug metabolism. Therefore, monitoring of the thrombotic and bleeding risk during CKD is a real challenge and better identification of specific risk patients could reduce the chances of under- or over-treatment.
IS and apixaban are both known to bind to human serum albumin (HSA) in plasma, the most abundant plasma protein. HSA exhibits a remarkable capacity to bind and transport a wide range of endogenous molecules and drugs regulating their bioavailability and therefore, their therapeutic effect and safety in the latter. The two most important binding sites on HSA are Sudlow sites I (located in subdomain IIA) and II ( located in subdomain IIIA)70. Research shows that IS primarily binds to subdomain IIIA (ratio 1:1) with high affinity, while it can also bind to subdomain IIA (ratio 2:1), albeit with much lower affinity71. In contrast, Apixaban has a high affinity for Sudlow’s site I72. Hence, minimal competition is expected between IS and apixaban under normal conditions, as they do not directly compete for the same binding site72. Nonetheless, one can speculate that the presence of high concentrations of IS, as observed in uremic conditions, could promote IS binding at site I, competing then with apixaban for HSA binding, increasing the free fraction, and therefore significantly altering its bioavailability. Higher apixaban free doses, could lead to higher haemorrhagic risk, which could be related with why CKD patients present higher haemorrhagic risk under anticoagulant treatment than non-CKD individuals18. Furthermore, in the context of CKD, the radical different plasma metabolome may compete for the available binding sites. This competition could explain why IS exhibits much lower affinity in plasma. Additionally, molecules such as fatty acids, typically transported by HSA, can further interfere, impeding IS binding to HSA73. Regarding the effects of sex on the affinity of the various binding sites of HAS we did not find any relevant literature.
Further studies need to be carried out to assess the efficiency of the different efflux transporters and metabolism enzymes to better determine the cause of the apixaban and IS accumulation in female mice. Specifically, since our study is focused on short term effects and non-CKD mice, it would be interesting to see if this accumulation has any deleterious long-term effects. Moreover, recognizing the impact of sex differences in drug management is crucial for improving the care provided to women. It is essential to be correctly represented in clinical trials, so that possible underlying biological divergences related to sex can be regarded to better understand renal pathophysiology, disease progression and drug metabolism in both men and women.
Data availability
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE [67] partner repository with the dataset identifier PXD053404.
References
Kovesdy, C. P. Epidemiology of chronic kidney disease: an update 2022. Kidney Int. Suppl. 12(1), 7–11. https://doi.org/10.1016/j.kisu.2021.11.003 (2022).
Sundström, J. et al. Prevalence, outcomes, and cost of chronic kidney disease in a contemporary population of 2·4 million patients from 11 countries: the CaReMe CKD study. Lancet Reg. Health Eur. 20, 89. https://doi.org/10.1016/j.lanepe.2022.100438 (2022).
Ricardo, A. C. et al. Sex-related disparities in CKD progression. J. Am. Soc. Nephrol. 30(1), 137–146. https://doi.org/10.1681/ASN.2018030296 (2019).
Balafa, O. et al. Sex disparities in mortality and cardiovascular outcomes in chronic kidney disease. Clin. Kidney J. 17(3), sfae044. https://doi.org/10.1093/ckj/sfae044 (2024).
MacRae, C., Mercer, S. W., Guthrie, B. & Henderson, D. Comorbidity in chronic kidney disease: a large cross-sectional study of prevalence in Scottish primary care. Br. J. Gen. Pract. 71(704), e243–e249. https://doi.org/10.3399/bjgp20X714125 (2021).
Jankowski, J., Floege, J., Fliser, D., Böhm, M. & Marx, N. Cardiovascular disease in chronic kidney disease. Circulation 143(11), 1157–1172. https://doi.org/10.1161/CIRCULATIONAHA.120.050686 (2021).
Sallée, M. et al. The aryl hydrocarbon receptor-activating effect of uremic toxins from tryptophan metabolism: a new concept to understand cardiovascular complications of chronic kidney disease. Toxins 6(3), 934–949. https://doi.org/10.3390/toxins6030934 (2014).
Nunns, G. R. et al. The hypercoagulability paradox of chronic kidney disease: the role of fibrinogen. Am. J. Surg. 214(6), 1215–1218. https://doi.org/10.1016/j.amjsurg.2017.08.039 (2017).
Lutz, J., Menke, J., Sollinger, D., Schinzel, H. & Thürmel, K. Haemostasis in chronic kidney disease. Nephrol. Dial. Transplant. 29(1), 29–40. https://doi.org/10.1093/ndt/gft209 (2014).
Addi, T., Dou, L. & Burtey, S. Tryptophan-derived uremic toxins and thrombosis in chronic kidney disease. Toxins 10(10), 10. https://doi.org/10.3390/toxins10100412 (2018).
Gadde, S. et al. Atrial fibrillation in chronic kidney disease: an overview. Cureus 14(8), e27753. https://doi.org/10.7759/cureus.27753 (2023).
Folsom, A. R. et al. Chronic kidney disease and venous thromboembolism: a prospective study. Nephrol. Dial. Transplant. Off. Publ. Eur. Dial. Transpl. Assoc. 25(10), 3296–3301. https://doi.org/10.1093/ndt/gfq179 (2010).
Yu, W. Y. H. et al. Warfarin-associated nonuremic calciphylaxis. JAMA Dermatol. 153(3), 309–314. https://doi.org/10.1001/jamadermatol.2016.4821 (2017).
Hammett, C. et al. Oral anticoagulant use in patients with atrial fibrillation and chronic kidney disease: a review of the evidence with recommendations for Australian Clinical Practice. Heart Lung Circ. 31(12), 1604–1611. https://doi.org/10.1016/j.hlc.2022.09.003 (2022).
Wetmore, J. B. et al. Apixaban dosing patterns Versus Warfarin in patients with Nonvalvular Atrial Fibrillation receiving Dialysis: a retrospective cohort study. Am. J. Kidney Dis. https://doi.org/10.1053/j.ajkd.2022.03.007 (2022).
Byon, W., Garonzik, S., Boyd, R. A. & Frost, C. E. Apixaban: a clinical pharmacokinetic and pharmacodynamic review. Clin. Pharmacokinet. 58(10), 1265–1279. https://doi.org/10.1007/s40262-019-00775-z (2019).
Fatima, H. et al. Safety and efficacy of apixaban vs warfarin in patients with stage 4 and 5 chronic kidney disease: a systematic review. Cureus J. Med. Sci. 14, 10. https://doi.org/10.7759/cureus.30230 (2022).
Parker, K. et al. A systematic review of the efficacy and safety of anticoagulants in advanced chronic kidney disease. J. Nephrol. 35(8), 2015–2033. https://doi.org/10.1007/s40620-022-01413-x (2022).
Vondracek, S. F., Teitelbaum, I. & Kiser, T. H. Principles of kidney pharmacotherapy for the nephrologist: core curriculum 2021. Am. J. Kidney Dis. 78(3), 442–458. https://doi.org/10.1053/j.ajkd.2021.02.342 (2021).
Santana Machado, T., Cerini, C. & Burtey, S. Emerging roles of aryl hydrocarbon receptors in the altered clearance of drugs during chronic kidney disease. Toxins 11(4), E209. https://doi.org/10.3390/toxins11040209 (2019).
Fujii-Kuriyama, Y. & Mimura, J. Molecular mechanisms of AhR functions in the regulation of cytochrome P450 genes. Biochem. Biophys. Res. Commun. 338(1), 311–317. https://doi.org/10.1016/j.bbrc.2005.08.162 (2005).
Gondouin, B. et al. Indolic uremic solutes increase tissue factor production in endothelial cells by the aryl hydrocarbon receptor pathway. Kidney Int. 84(4), 733–744. https://doi.org/10.1038/ki.2013.133 (2013).
Banoglu, E., Jha, G. G. & King, R. S. Hepatic microsomal metabolism of indole to indoxyl, a precursor of indoxyl sulfate. Eur. J. Drug Metab. Pharmacokinet. 26(4), 235–240 (2001).
Banoglu, E. & King, R. S. Sulfation of indoxyl by human and rat aryl (phenol) sulfotransferases to form indoxyl sulfate. Eur. J. Drug Metab. Pharmacokinet. 27(2), 135–140. https://doi.org/10.1007/BF03190428 (2002).
Ma, Q., Zhang, X. & Qu, Y. Biodegradation and biotransformation of indole: advances and perspectives. Front. Microbiol. 9,12. https://www.frontiersin.org/articles/, https://doi.org/10.3389/fmicb.2018.02625 (2018).
Niwa, T. Indoxyl sulfate is a nephro-vascular toxin. J. Ren. Nutr. 20(5), S2–S6. https://doi.org/10.1053/j.jrn.2010.05.002 (2010).
Kamiński, T. W., Pawlak, K., Karbowska, M., Myśliwiec, M. & Pawlak, D. Indoxyl sulfate—the uremic toxin linking hemostatic system disturbances with the prevalence of cardiovascular disease in patients with chronic kidney disease. BMC Nephrol. 18(1), 35. https://doi.org/10.1186/s12882-017-0457-1 (2017).
Lano, G., Burtey, S. & Sallée, M. Indoxyl sulfate, a uremic endotheliotoxin. Toxins 12(4), 4. https://doi.org/10.3390/toxins12040229 (2020).
Santana Machado, T. et al. Indoxyl sulfate upregulates liver P-glycoprotein expression and activity through aryl hydrocarbon receptor signaling. J. Am. Soc. Nephrol. JASN 29(3), 906–918. https://doi.org/10.1681/ASN.2017030361 (2018).
Dreisbach, A. W. & Lertora, J. J. The effect of chronic renal failure on drug metabolism and transport. Expert Opin. Drug Metab. Toxicol. 4(8), 1065–1074. https://doi.org/10.1517/17425255.4.8.1065 (2008).
Schmidt, J. V., Su, G. H., Reddy, J. K., Simon, M. C. & Bradfield, C. A. Characterization of a murine Ahr null allele: involvement of the Ah receptor in hepatic growth and development. Proc. Natl. Acad. Sci. 93(13), 6731–6736. https://doi.org/10.1073/pnas.93.13.6731 (1996).
du Sert, N. P. et al. Reporting animal research: explanation and elaboration for the ARRIVE guidelines 2.0. PLOS Biol. 18(7), e3000411. https://doi.org/10.1371/journal.pbio.3000411 (2020).
Calaf, R. et al. Determination of uremic solutes in biological fluids of chronic kidney disease patients by HPLC assay. J. Chromatogr. B Analyt. Technol. Biomed. Life. Sci. 879(23), 2281–2286. https://doi.org/10.1016/j.jchromb.2011.06.014 (2011).
‘Willekens et al. A universal anti-Xa assay for rivaroxaban, apixaba.pdf. (2021, accessed 24 Jun 2024). https://zlmsg.ch/wp-content/uploads/2021/05/2021-Br-J-Haematol_Willekens.pdf.
Livak, K. J. & Schmittgen, T. D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods San Diego Calif 25(4), 402–408. https://doi.org/10.1006/meth.2001.1262 (2001).
Smadja, D. M. et al. Increased VEGFR2 expression during human late endothelial progenitor cells expansion enhances in vitro angiogenesis with up-regulation of integrin alpha(6). J. Cell. Mol. Med. 11(5), 1149–1161. https://doi.org/10.1111/j.1582-4934.2007.00090.x (2007).
Türei, D., Korcsmáros, T. & Saez-Rodriguez, J. OmniPath: guidelines and gateway for literature-curated signaling pathway resources. Nat. Methods 13(12), 966–967. https://doi.org/10.1038/nmeth.4077 (2016).
Välikangas, T., Suomi, T. & Elo, L. L. A systematic evaluation of normalization methods in quantitative label-free proteomics. Brief. Bioinform. 19(1), 1–11. https://doi.org/10.1093/bib/bbw095 (2016).
Phipson, B. et al. Robust hyperparameter estimation protects against hypervariable genes and improves power to detect differential expression. Ann. Appl. Stat. 10(2), 946–963. https://doi.org/10.1214/16-AOAS920 (2016).
Badia-i-Mompel, P. et al. DecoupleR: ensemble of computational methods to infer biological activities from omics data. Bioinforma Adv. 2(1), vbac016. https://doi.org/10.1093/bioadv/vbac016 (2022).
Liberzon, A. et al. The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst. 1(6), 417–425. https://doi.org/10.1016/j.cels.2015.12.004 (2015).
Jansen, J. et al. Remote sensing and signaling in kidney proximal tubules stimulates gut microbiome-derived organic anion secretion. Proc. Natl. Acad. Sci. 116(32), 16105–16110. https://doi.org/10.1073/pnas.1821809116 (2019).
König, C. et al. Thirty mouse strain survey of voluntary physical activity and energy expenditure: influence of strain, sex and day–night variation. Front. Neurosci. 14, 896. https://doi.org/10.3389/fnins.2020.00531 (2020).
Hylek, E. M. Apixaban for end-stage kidney disease. Circulation 138(15), 1534–1536. https://doi.org/10.1161/CIRCULATIONAHA.118.036449 (2018).
Zhang, D. et al. Comparative metabolism of 14 C-labeled apixaban in mice, rats, rabbits, dogs, and humans. Drug Metab. Dispos. Biol. Fate Chem. 37(8), 1738–1748. https://doi.org/10.1124/dmd.108.025981 (2009).
Bushi, D. et al. Apixaban decreases brain thrombin activity in a male mouse model of acute ischemic stroke. J. Neurosci. Res. 96(8), 1406–1411. https://doi.org/10.1002/jnr.24253 (2018).
Beery, A. K. Inclusion of females does not increase variability in rodent research studies. Curr. Opin. Behav. Sci. 23, 143–149. https://doi.org/10.1016/j.cobeha.2018.06.016 (2018).
Bierer, B. E., Meloney, L. G., Ahmed, H. R. & White, S. A. Advancing the inclusion of underrepresented women in clinical research. Cell. Rep. Med. 3(4), 100553. https://doi.org/10.1016/j.xcrm.2022.100553 (2022).
Alahamadi, Z. et al. Race, sex, and kidney disease trial participation. Kidney Med. 5, 3. https://doi.org/10.1016/j.xkme.2022.100594 (2023).
Heerspink Hiddo, J. L. et al. Dapagliflozin in patients with chronic kidney disease. N. Engl. J. Med. 383(15), 1436–1446 https://doi.org/10.1056/NEJMoa2024816 (2020).
Vinson, A. J., Collister, D., Ahmed, S. & Tennankore, K. Underrepresentation of women in recent landmark kidney trials: the gender gap prevails. Kidney Int. Rep. 7(11), 2526–2529. https://doi.org/10.1016/j.ekir.2022.08.022 (2022).
Granger Christopher, B. et al. Apixaban versus warfarin in patients with atrial fibrillation. N. Engl. J. Med. 365(11), 981–992. https://doi.org/10.1056/NEJMoa1107039 (2011).
Santema, B. T. et al. Identifying optimal doses of heart failure medications in men compared with women: a prospective, observational, cohort study’, The Lancet 394(10205), 1254–1263. https://doi.org/10.1016/S0140-6736(19)31792-1 (2019).
Nolin, T. D. A synopsis of clinical pharmacokinetic alterations in advanced CKD. Semin. Dial. 28(4), 325–329. https://doi.org/10.1111/sdi.12374 (2015).
Jalal, D., Chonchol, M. & Targher, G. Disorders of hemostasis associated with chronic kidney disease. Semin. Thromb. Hemost. 36(1), 034–040. https://doi.org/10.1055/s-0030-1248722 (2010).
Dou, L. et al. Aryl hydrocarbon receptor is activated in patients and mice with chronic kidney disease. Kidney Int. 93(4), 986–999. https://doi.org/10.1016/j.kint.2017.11.010 (2018).
Frost, C. E. et al. Effects of age and sex on the single-dose pharmacokinetics and pharmacodynamics of apixaban. Clin. Pharmacokinet. 54(6), 651–662. https://doi.org/10.1007/s40262-014-0228-0 (2015).
Zhang, D. et al. Characterization of efflux transporters involved in distribution and disposition of apixaban. Drug Metab. Dispos. Biol. Fate Chem. 41(4), 827–835. https://doi.org/10.1124/dmd.112.050260 (2013).
Kim, H. et al. ABCG2 gene polymorphisms may affect the bleeding risk in patients on apixaban and rivaroxaban. Drug Des. Devel. Ther. 17, 2513–2522. https://doi.org/10.2147/DDDT.S417096 (2023).
Attelind, S. et al. Genetic determinants of apixaban plasma levels and their relationship to bleeding and thromboembolic events. Front. Genet. 13, 89. https://doi.org/10.3389/fgene.2022.982955 (2022).
Faber, M. S., Jetter, A. & Fuhr, U. Assessment of CYP1A2 activity in clinical practice: why, how, and when? Basic. Clin. Pharmacol. Toxicol. 97(3), 125–134. https://doi.org/10.1111/j.1742-7843.2005.pto_973160.x (2005).
Krekels, E. H. J., Rower, J. E., Constance, J. E., Knibbe, C. A. J. & Sherwin, C. M. T. Chapter 8—hepatic drug metabolism in Pediatric patients. In Drug Metabolism in Diseases (ed. Xie, W.) 181–206 (Academic, 2017). https://doi.org/10.1016/B978-0-12-802949-7.00008-0.
Sindhu, R. K. & Vaziri, N. D. Upregulation of cytochrome P450 1A2 in chronic renal failure: does oxidized tryptophan play a role? Adv. Exp. Med. Biol. 527, 401–407. https://doi.org/10.1007/978-1-4615-0135-0_47 (2003).
Xiong, L. et al. Direct androgen receptor regulation of sexually dimorphic gene expression in the mammalian kidney. BioRxiv Prepr. Serv. Biol. 2023, 539585. https://doi.org/10.1101/2023.05.06.539585 (2023).
Breljak, D., Brzica, H., Sweet, D. H., Anzai, N. & Sabolic, I. Sex-dependent expression of Oat3 (Slc22a8) and Oat1 (Slc22a6) proteins in murine kidneys. Am J. Physiol. Ren. Physiol. 304(8), F1114–F1126. https://doi.org/10.1152/ajprenal.00201.2012 (2013).
Sakurai, Y. et al. Expression levels of renal organic anion transporters (OATs) and their correlation with anionic drug excretion in patients with renal diseases. Pharm. Res. 21(1), 61–67. https://doi.org/10.1023/B:PHAM.0000012153.71993.cb (2004).
Zhang, J., Wang, H., Fan, Y., Yu, Z. & You, G. Regulation of organic anion transporters: role in physiology, pathophysiology, and drug elimination. Pharmacol. Ther. 217, 107647. https://doi.org/10.1016/j.pharmthera.2020.107647 (2021).
Deguchi, T. et al. Characterization of uremic toxin transport by organic anion transporters in the kidney. Kidney Int. 65(1), 162–174. https://doi.org/10.1111/j.1523-1755.2004.00354.x (2004).
Ravid, J. D. & Chitalia, V. C. Molecular mechanisms underlying the cardiovascular toxicity of specific uremic solutes. Cells 9(9), 2020. https://doi.org/10.3390/cells9092024 (2024)
Lee, P. & Wu, X. Modifications of human serum albumin and their binding effect. Curr. Pharm. Des. 21(14), 1862–1865 (2015).
Yu, S. et al. Interaction of human serum albumin with uremic toxins: a thermodynamic study. RSC Adv. 7, 27913–27922. https://doi.org/10.1039/C7RA02838E (2017).
Wang, Q. et al. Determination of potential main sites of apixaban binding in human serum albumin by combined spectroscopic and docking investigations. RSC Adv. 5(99), 81696–81706. https://doi.org/10.1039/C5RA15430H (2015).
Devine, E., Krieter, D. H., Rüth, M., Jankovski, J. & Lemke, H. D. Binding affinity and capacity for the uremic toxin indoxyl sulfate. Toxins 6(2), 416–429. https://doi.org/10.3390/toxins6020416 (2014).
Sakai, T. et al. Interaction mechanism between indoxyl sulfate, a typical uremic toxin bound to site II, and ligands bound to site I of human serum albumin. Pharm. Res. 18(4), 520–524. https://doi.org/10.1023/A:1011014629551 (2001).
Su, L. et al. Abcb1a and Abcb1b genes function differentially in blood–testis barrier dynamics in the rat. Cell. Death Dis. 8(9), 9. https://doi.org/10.1038/cddis.2017.435 (2017).
Horsey, A. J., Cox, M. H., Sarwat, S. & Kerr, I. D. The multidrug transporter ABCG2: still more questions than answers. Biochem. Soc. Trans. 44(3), 824–830. https://doi.org/10.1042/BST20160014 (2016).
Grishanova, A. Y. & Perepechaeva, M. L. Aryl hydrocarbon receptor in oxidative stress as a double agent and its biological and therapeutic significance. Int. J. Mol. Sci. 23(12), 12. https://doi.org/10.3390/ijms23126719 (2022).
Wang, L. et al. In vitro assessment of metabolic drug-drug interaction potential of apixaban through cytochrome P450 phenotyping, inhibition, and induction studies. Drug Metab. Dispos. Biol. Fate Chem. 38(3), 448–458. https://doi.org/10.1124/dmd.109.029694 (2010).
Lu, C. L. et al. Indoxyl-sulfate-induced redox imbalance in chronic kidney disease. Antioxidants 10(6), 936. https://doi.org/10.3390/antiox10060936 (2021).
Sekine, T., Miyazaki, H. & Endou, H. Molecular physiology of renal organic anion transporters. Am. J. Physiol. -Ren. Physiol. 290(2), F251–F261. https://doi.org/10.1152/ajprenal.00439.2004 (2006).
Perez-Riverol, Y. et al. The PRIDE database resources in 2022: a hub for mass spectrometry-based proteomics evidences. Nucleic Acids Res. 50, D543–D552. https://doi.org/10.1093/nar/gkab1038 (2022).
Acknowledgements
This work was supported by the European Union’s Horizon 2020 research and innovation program (860329 Marie-Curie ITN “STRATEGY-CKD”) (SB, JSR) and the EU Horizon 2020 program Epic-XS (project 0000432) (BPB, SP, SB).
Author information
Authors and Affiliations
Contributions
B.P.B., S.B and S.P. wrote the main manuscript. B.P.B prepared all the figures. B.P.B., M.G. and S.P. performed animal experiments. D.D., J.P. and J.S.R. performed the bioinformatic analysis. Z.Z., D.P. and V.P. performed the proteomic analysis. B.P.B. and N.M. performed expression analysis. S.B. had the idea of the work. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Pina-Beltran, B., Dimitrov, D., McKay, N. et al. Unveiling the role of sex in the metabolism of indoxyl sulfate and apixaban. Sci Rep 15, 6075 (2025). https://doi.org/10.1038/s41598-025-90405-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-025-90405-5
