
Abstract
In recent years, evidence for the anti-cancer activity of lichen secondary metabolites has been rapidly increasing. In this study, we synthesised analogues of diffractaic acid, a lichen secondary metabolite, and evaluated their ability to suppress colorectal cancer stem potential. Among the 10 compounds after H/CH₃/benzylation of the diffractaic acid structure or modifications in an aromatic hydrophobic domain, TU3 has a more inhibition effect on the stem potential of colorectal cancer compared to other compounds. The compound TU3 targets ALDH1 and suppresses key signalling pathways such as WNT, STAT3, NF-κB, Hedgehog, and AP-1. Inhibition of these signalling pathways by TU3 contribute to attenuate the survival mechanisms of colorectal cancer stem cell and thus inhibit cancer progression.
Similar content being viewed by others

Introduction
Globally, colorectal cancer (CRC) is the second most common cause of cancer-related death and the third most common cancer1. Multiple treatments, including surgery, radiotherapy, chemotherapy, and immunotherapy, have been designed to reduce the risk of colorectal cancer onset and recurrence. However, even with these advancements, the metastasis rate remains between 10 and 25% in patients following primary tumor resection2,3. Consequently, creating therapies that specifically target cancer stem cells (CSCs) holds significant promise. Cancer stem cells are recognized for their self-renewal capabilities and their potential to initiate the growth of diverse cancer cell populations. CSCs play a crucial role in cancer development and spread4. Rising evidence shows that cancer stem cells are a key factor in chemoresistance, which hinders the efficacy of anticancer therapies5,6.
The term lichen refers to a stable symbiotic association between fungi and algae or cyanobacteria. Despite their symbiosis with many different species, lichens function as a truly unified organism, possessing unique qualities that are not part of a single fungus, algae, or cyanobacterium7,8. Lichens have been extensively studied for their potential as pharmaceuticals. Lichens are currently used for a wide range of medicinal purposes across the globe. Lichens can be applied externally as a wound dressing as a disinfectant or to stop bleeding. Companies worldwide offer a variety of products based on lichen compounds. The use of lichen compounds to make various remedies is common in folk medicine, and screening studies with lichens have indicated the presence of various metabolites with antibiotic, antimycobacterial, antiviral, antipyretic, analgesic, anti-inflammatory, antioxidant, cytotoxic and anticancer7,9,10,11,12. Most of these compounds are derivatives of orsellinic acid, which is a chromophore comprised of ortho-hydroxy-carbonyl units. In these compounds, two or three orcinol or β-orcinol phenolic groups have been linked by an ester (depsides, Di-depsides, Trideptides) and by an ether (diphenyl ethers, depsidones, dibenzofurans) or by a C(O)C bond (depsones)13,14. There are several groups of compounds that belong to this group of aromatic compounds, but depsides and depsidones are the most studied groups, and they have a wide range of therapeutic applications particularly in reducing oxidative stress. The antioxidant properties of depside may be due to their phenolic structure15,16. The carboxyl group of one molecule is esterified with the phenolic hydroxyl group of another molecule in these types of compounds, which are formed by the condensation of two or more hydroxybenzoic acids. Due to their phenolic chemical structures, these molecules are useful for evaluating their effects on cancer cells since a major class of inhibitors also contains an aromatic hydroxyl ring17,18.
A lichen-derived secondary metabolite is diffractaic acid (DA), which has a distinct chemical structure that includes a benzene ring replaced with different hydroxyl and methoxy groups18. DA has considerable anti-cancer capabilities, reducing the proliferation of several cancer cell lines and hence regulating tumor growth. It causes cancer cells to undergo apoptosis, or programmed cell death, which is necessary for the removal of malignant cells and the reduction of tumor burden. Budak`s works that looked into DA’s impact on lung, liver, breast and cervical cancer cells discovered that it inhibits the thioredoxin reductase 1 (TrxR1) enzyme19,20,21,22. DA also possesses anti-inflammatory properties, which can help it fight cancer by lowering inflammation related with the disease’s progression19,23,24. Furthermore, according to DA has developed into a mosquito-borne antiviral agent with a suggested target at viral replication for its selectivity indices and DA exhibited anti-respiratory syncytial virus properties25,26. Barbatic acid (BA), which is molecularly similar and an analogue of DA, has anti-cancer, anthelminthic, ultrastructural, schistosomicidal and diuretic activity27,28,29,30. To the best of our knowledge, although BA has been used in many cytotoxic studies28,31, its molecular mechanism on anticancer effects has not been adequately addressed in previous studies. In this study, we synthesized analogues of diffractaic acid, a depside molecule, and evaluated how it responds to the colorectal cancer stem cell (CCSC) and how it changes structurally based biological activity.
Results and discussion
Structure-based modifications
Despite advances in colonoscopy screening and treatment, which have led to decreased incidence and mortality rates of CRC in some highly developed countries, many developing countries are still experiencing an upward trend in both incidence and mortality rates32. Surgical treatment and chemotherapy are the primary methods used to treat colorectal cancer. Adjuvant chemotherapy, which typically includes drugs such as 5-fluorouracil, leucovorin, doxorubicin or capecitabine are several of the most effective treatment options for colorectal cancer patients.33,34,35. Thus, there is an urgent need to develop new treatment strategies or therapeutic agents for colorectal cancer to improve clinical outcomes. Increasingly, investigations are focusing on the biosafety and prolonged use of natural molecules or extracts, as well as on therapies that engage multiple pathways and reveal the molecular basis of their activity32,36,37,38. Several secondary metabolites derived from lichens have demonstrated anti-cancer activity such as usnic acid, lecanoric acid, norstictic acid, stictic acid, gyrophoric acid, atranorin, physciosporin, parietin (or Physcion), salazinic acid, protolichesterinic acid, evernic acid, fumarprotocetraric acid, orsellinic acid and etc.39,40,41,42,43,44,45,46,47. These metabolites offer significant potential for developing new cancer treatments.
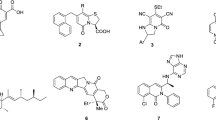
The structures of naturally occurring bioactive depsides consist of the ester bond between two phenolic acids (Fig. 1). In our ongoing efforts to discover novel chemotype anticancer agents, we attempted to synthesize derivatives based on the 2,4-dihydroxybenzoic acid skeleton of depsides as a starting point for medicinal chemistry. In particular, the symmetrical and dimeric structural features of depsides attracted our interest in the biological evaluation of structurally similar derivatives. Therefore, we fixed the 2,4-dihydroxybenzoic acid skeleton of the depside and synthesized various analogues through modification of the phenolic acids on the other side (Table 1), and compared their inhibition ability on the stemness of colorectal cancer.

Structures of naturally occurring bioactive depsides.
In vitro efficacy study
In our previous studies, other lichen secondary metabolites tumidulin, physciosporin, laboric acid and atraric acid have regulatory effects on stem cells48,49,50,51. Therefore, we subjected the synthesized compounds to spheroid formation experiment in order to create a treatment option targeting stem cells that play a role in cancer prognosis with diffractaic acid and its analogs. We evaluated various diffractaic acid analogs, identifying TU-1 (diffractaic acid), TU-3 (barbatic acid), and TU-4 as the most potent compounds. These compounds feature an acid moiety and a free hydroxy group, which enhance their binding affinity to target proteins. Notably, TU-3 demonstrated superior efficacy in promoting spheroid formation in CSC221 and CaCo2 compared to TU-1. This enhanced activity is attributed to the presence of a free hydroxy group at the ortho position in TU-3, whereas TU-1 has an ortho-methylation.
TU-2, in contrast, has its acid moiety protected by a benzyl group, resulting in reduced binding affinity relative to TU-1 and TU-3. The esterification of carboxylic acid groups in TU-2 diminishes its biological activity. TU1 itself was assessed for its spheroid-forming ability across CSC221 and CaCo2 cell line (Fig. 2). Furthermore, we investigated cell viability. The results are shown in Supplementary Fig. S1. In addition to suppressing stem cell potential, TU3 has a higher ability to inhibit cell viability compared to TU1. TU1 (diffractaic acid) shown that large scare cytoxicity against UACC-62 (melanoma cell), B16-F10 (melanoma cell), MCF-7 (hormone sensitive breast cancer cell), MDA-MB-453 (triple negative breast cancer cell), A549 (lung cancer cell), NCI-H460 (lung cancer cell), HeLa (cervical cancer cell), PRCC (papillary renal cell carcinoma), U87MG (malignant glioma), AGS (gastric cancer cell) and human lymphocytes cells19,20,22,52,53,54,55,56. On the other hand, TU3 (barbatic acid) shown cytotoxic effect on several cancer cells such as HeLa (cervical cancer cell), A549 (lung cancer), MCF-7 (breast cancer cell), DU-145 (prostate cancer), NCI-H292 (lung mucoepidermoid carcinoma)28,31. Overall, TU3 shows cytotoxic effects on both CSC221 and CaCo2 cell lines.

Evaluation of Spheroid Formation in CSC Cell Lines Treated with Diffractaic Acid and Its Analogs. Representative images of spheroid formation by CSC221 (A) and CaCo2 (B) cells treated with diffractaic acid and its analogs at 10 μM concentration for 10–14 days, and quantitative analysis of the number of spheroids formed following each treatment. Data represent the mean ± SD, *p < 0.05; **p < 0.01; ***p < 0.001.
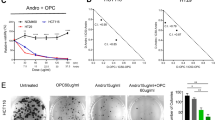
Therefore, we decided to evaluate the potential of TU3 for future experiments. First, to test its dose-dependent specificity, we reconfirmed the effect of CSC221 and CaCo2 on spheroid formation of cells by testing them at concentrations of 2.5, 5, and 10 μM. Our results revealed a dose-dependent inhibition effect of TU3 on spheroid formation (Fig. 3A,B). Then to check whether TU3 arrests the cell cycle or not. While TU3 triggers the S and G2 M population to accumulate in the G1 phase in a dose-dependent manner on human colorectal adenocarcinoma-enriched CSC (CSC221), it also increases the Sub G1 population in a dose-dependent manner. In the CaCo2 cell line, although it cannot establish a clear pattern in the dose-dependent G1, S and G2 M phases, Sub G1 shows a dose-dependent increase (Fig. 3C,D). Additionally, we checked the PARP and Bax protein level. Cleavage PARP tended to be induced in a dose-dependent manner, whereas Bax levels were unchanged (Supplementary Fig. S2). Important biological functions that PARP actively participates in include transcription, cell cycle regulation, DNA damage response, apoptosis, and genomic integrity preservation. Cleaved PARP is one of the most widely utilised indicators for apoptosis detection57.

Compound TU3 suppress spheroid formation in CSC221 and CaCo2 cell lines in a dose-dependent manner. Representative images of spheroid formation by CSC221 (A) and CaCo2 (B) cells treated with TU3 at 10 μM concentration for 10–14 days, and quantitative analysis of the number of spheroids formed following each treatment. Flow cytometric analysis of cell cycle distribution. CSC221 (C) and CaCo2 (D) cells were treated with indicated concentrations of TU3 for 48 h incubation. Data represent the mean ± SD, *p < 0.05; **p < 0.01; ***p < 0.001.
Furthermore characterization, we checked the cancer stemness markers, transcriptional regulators, and inflammatory related proteins. The self-renewal, proliferation, survival and differentiation properties of CSCs are regulated by many signaling pathways, and most of these pathways are non-linear58. The detoxifying enzyme aldehyde dehydrogenase (ALDH) is in charge of oxidising intracellular aldehydes. Through both in vitro and in vivo tests, it has been shown that cells with enhanced ALDH1 activity possess stem cell characteristics59. The protein level of ALDH1 was inhibited after TU3 treatment (2.5, 5, and 10 μM) for 48 h in CSC221 and CaCo2 cells (Fig. 4). The hedgehog signaling pathway is activated when sonic hedgehog ligand (SHH) binds to its receptor, patched (PTCH1), unleashes the transmembrane protein smoothened (SMO), which in turn actives several members of the GLI family of transcription factors. There has been a strong correlation found in the expression of aldehyde dehydrogenase family 1 member A1 (ALDH1A1)6,60,61. TU3 treatment inhibited Gli1 protein level in both cell lines in a dose-dependent manner, showing the same pattern as the ALDH1 protein level (Fig. 4).

TU3 targets colorectal cancer stem cells by blocking cellular signaling. Western blot analysis of ALDH1, Gli1, STAT3, NF-κB, β-catenin, c-Myc and Cyclin-D1 protein levels in CSC221 (A) and CaCo2 (B) cells treated with indicated concentrations of TU3 and incubated 48 h. (C–D) Quantitative analysis of protein expression. Data represent the mean ± SD, *p < 0.05; **p < 0.01; ***p < 0.001; difference compared with DMSO-treated.
STAT3 plays an active role in the development and spread of tumours62. The signal transduction molecule STAT3 is crucial for controlling stem cell characteristics as well as cell development, proliferation, and survival63. In certain situations, STAT3, NF-kB and AP-1 can cooperate and work together64. NF-κB signalling makes sense in the cells that initiate tumours, considering the links between the NF-B pathway and the initial oncogenesis processes. The expression of NF-κB signalling is closely linked to stemness and is essential for the upkeep and survival of cancer stem cells (CSCs). Increased invasion, metastasis, proliferation, and self-renewal are linked to CSCs that have activated NF-κB. The expression of genes linked to stem cells is regulated by NF-κB, which is frequently activated by cytokines present in the tumour microenvironment. Targeting CSCs with NF-κB inhibition has showed potential as a therapeutic approach65,66. Our results showed that TU3 downregulated the protein levels of STAT3 and NF-κB (Fig. 4). In addition, STAT3 has an important function in increasing ALDH1A3 expression. Aberrant STAT3 activation increases the expression of ALDH1A3, which is critical for the maintenance of cancer stem cells in non-small cell lung cancer67,68. Moreover, the STAT3-NF kB/DDIT3/CEBPβ axis regulates ALDH1A3 expression in chemo resistant cell subpopulations. Briefly, ALDH + expression is influenced by STAT3 and NF-κB, which helps maintain cancer stem cells and withstand chemotherapy69.
Moreover, WNT/β-catenin signaling may play an important role in the regulation of cancer stem cells (CSCs) and may regulate the expression of ALDH1 and influence the properties of cancer stem cells. When WNT signaling is activated, β-catenin accumulates intracellularly and passes into the nucleus. In the nucleus, β-catenin interacts with transcription factors that affect the transcription of target genes70,71,72. TU3 also inhibits the protein levels of WNT and AP-1 common target genes c-Myc and Cyclin-D1, including β-catenin (Fig. 4). Cyclin D1 is most well-known for its activity in the nucleus as a cell cycle regulator. Cyclin D1 controls the transition from G1 to S phase by acting as an allosteric regulator of cyclin-dependent kinase 4 (CDK) and CDK6. Furthermore, Cyclin-D1 regulates cellular adhesion and invasion, which are key processes in tumor growth and metastasis73. Another transcription factors, c-Myc, is emphasized because of its important functions in stem cell function and carcinogenic pathways. c-Myc is a transcription factor that controls genes related to cell proliferation, differentiation, and apoptosis. Its overexpression is connected to the preservation of cancer stem cell (CSC) features such as self-renewal and chemoresistance74,75.
We also characterized WNT and JAK2/STAT signaling to further characterize GSK3B and JAK2 levels in CSC221 cells (Supplementary Fig. S3). TU3 down-regulated GKS3B expression but did not cause significant changes for JAK2. Co-activation of β-catenin and STAT3 is linked to tumour growth, cellular survival, and proliferation. STAT3 is activated by WNT signalling through β-catenin. WNT1 overexpression causes β-catenin to stabilise, which raises STAT3 phosphorylation at Tyr705 in the process. WNT1 overexpression enhances STAT3’s transcriptional activity and encourages its nuclear localization76,77. Additionally, one study shown that JAK/STAT3 inhibition decreased intestinal tumour growth in mice and blocked Wnt/β-catenin signalling78. Therefore, inhibition of JAK/STAT and WNT/GSK3β/β-catenin together with small molecules is important.
In conclusion, we evaluated the inhibition ability of analogues and derivatives derived from diffractaic acid on bioactivity-mediated colorectal cancer. We discovered the existence of TU3, which can well suppress cancer stemness, and characterized its effect. TU3 potently inhibits ALDH1, an important stemness marker by blockade of the several signaling pathways (Fig. 5).

Schematic showing how the TU3 supresses the colorectal cancer stem cell. TU3 strongly suppresses the ALDH1 expression by regulated the STAT, NF-κB, AP-1, WNT, and HH signaling pathways.
Experimental section
For diffractaic acid analogs, 1H spectra were obtained from JEOL 600 MHz NMR spectrometer and LC–MS related data was obtained from HPLC (Agilent, 1260 series) with DAD (diode array detector) and single quadrupole mass (Agilent, 6100 series) (Supporting Information File). All compounds are > 95% pure by HPLC analysis.
Preparation of monomer for diffractaic analogs
The synthetic procedure of A–P monomer compounds (Fig. 6).

Synthetic procedure of monomer for diffractaic analogs. (i) Na, BnOH, refluxed, 24 h; (ii) K2CO3, Alkyl halide, DMF, 50 °C, 10 h; (iii) KOH, MeOH, rt, 12 h; (iv) SO2Cl2, Diethyl ether, 0 °C, 15 min and 37 °C, 30 min.
Benzyl 2,4-dihydroxy-3,6-dimethylbenzoate

To a solution of Methyl-2,4-dihydroxy-3,6-dimethylbenzoate (1 g, 1 eq) and Na (123.32 mg, 1.1 eq.) in Benzyl alcohol (10 mL) and was reflux for 24 h. The mixture was extracted with ethyl acetate, washed with brine, and dried over anhydrous Na2SO4, concentrated under a high-pressure vacuum and purified by column chromatography, to give a white solid as a product, yield (743 mg, 60%). Rf 0.25 (10% EA: HEX). 1HNMR (400 MHz, CDCl3) δ 12.05 (s, 1H), 7.39 (br s, 5H),6.19 (s, 1H), 5.38 (s, 2H), 2.45 (s, 3H), 2.11 (s, 3H).
Methyl-2,4-dihydroxy-3,6-dimethylbenzoate

To a solution of methyl 3,6-dimethyl-2,4-dioxocyclohexane-1-carboxylate (100 mg, 1 eq.) dissolved in acetonitrile, was added copper bromide (112 mg, 1 eq.) and calcium chloride (55 mg, 1 eq.) under N2 at rt. Keep string at 50 °C for 4 h. Monitoring reaction progress by checking TLC. After completion of the reaction, the mixture was extracted with ethyl acetate, washed with brine, and dried over anhydrous Na2SO4, concentrated under a high-pressure vacuum and purified by column chromatography, to give a white solid as a product, yield (90 mg, 91%). Rf 0.25 (10% EA: HEX). 1HNMR (400 MHz, CDCl3) δ 12.02 (s, 1H), 6.21 (s, 1H), 5.07 (s, 1H), 3.92 (s, 3H), 2.46 (s, 3H), 2.10 (s, 3H).
Methyl 2,4-dimethoxy-3,6-dimethylbenzoate

To a solution of methyl 2, 4-dihydroxy-3, 6-dimethyl benzoate (250 mg, 1 eq.) in DMF (3 mL) was added K2CO3 (1.4 gm, 8 eq.) and methyl iodide (317 µL, 4 eq.) at the room temperature under N2. After being stirred at 50 °C for 9 h, the reaction mixture was filtered through a pad of celite. The filtrate was diluted with EtOAc and acidified with 3 M HCl. The organic layer was separated, and the aqueous layer was extracted twice with ethyl acetate, washed with brine, and dried over anhydrous MgSO4, concentrated under a high-pressure vacuum and purified by column chromatography, to give a white solid as a product, yield (251 mg, 88%). Rf 0.15 (10% MeOH: MC). 1HNMR (400 MHz, CDCl3) δ 6.45 (s, 1H), 3.89 (s, 3H), 3.80 (s, 3H), 3.74 (s, 3H), 2.29 (s, 3H), 2.10 (s, 3H).
2,4-dimethoxy-3,6-dimethylbenzoic acid

To a solution of 2,4-dimethoxy-3,6-dimethyl benzoate (1) (200 mg, 1 eq.) and KOH (134.4 mg 3 eq.) in DMSO (4 mL) and H2O (1 mL) was reflux for 24 h. Then the solution was acidified with 1 N HCl (20 mL) and extracted with DCM. The organic layer washed with brine, and dried over anhydrous Na2SO4, concentrated under a high-pressure vacuum and purified by column chromatography, to give a white solid as a product, yield (100 mg, 80%). Rf 0.25 (10% MeOH: MC). 1HNMR (400 MHz, CDCl3) δ 6.45 (s, 1H), 3.80 (s, 3H), 3.74 (s, 3H), 2.29 (s, 3H), 2.10 (s, 3H).
4-(benzyloxy)-2-methoxy-3,6-dimethylbenzoic acid

To a solution of Methyl 4-(benzyloxy)-2-methoxy-3,6-dimethylbenzoate (170 mg, 1 eq.) and KOH (202.8 mg 8 eq.) in MeOH (50 mL) was reflux for 24 h. Then the solution was extracted with EA. The organic layer was separated, and the aqueous layer was extracted twice with ethyl acetate, washed with brine, and dried over anhydrous Na2SO4, concentrated under a high-pressure vacuum and purified by column chromatography, to give a white solid as a product, yield (82 mg, 50%). Rf 0.15 (5% MeOH: MC). 1HNMR (400 MHz, CDCl3) δ 11.85 (d, J = 0.9 Hz, 1H), 7.40–7.38 (m, 5H), 6.45 (s, 1H), 5.10 (s, 2H), 3.87 (s, 3H), 2.38 (s, 3H), 2.16 (s, 3H).
Methyl 4-(benzyloxy)-2-methoxy-3,6-dimethylbenzoate

Methyl 4-(benzyloxy)-2-hydroxy-3,6-dimethylbenzoate; To a solution of Methyl-2,4-dihydroxy-3,6-dimethylbenzoate (500 mg, 1 eq.) in Acetone (7 mL) were added K2CO3 (1.05 g, 3 eq.) and benzyl bromide (908 µL, 3 eq.) at rt under N2 and stirred at 50 °C for 9 h. After completion of the reaction, the mixture was extracted with ethyl acetate, washed with brine, and dried over anhydrous Na2SO4, concentrated under a high-pressure vacuum and purified by column chromatography, to give a white solid as a product, yield (604 mg, 63%). Rf 0.75 (10% EA: HEX). 1HNMR (400 MHz, CDCl3) δ 11.85 (d, J = 0.9 Hz, 1H), 7.45–7.33 (m, 5H), 6.35 (s, 1H), 5.12 (s, 2H), 3.93 (s, 3H), 2.51 (s, 3H), 2.15 (s, 3H).
Methyl 4-(benzyloxy)-2-methoxy-3,6-dimethylbenzoate; To a solution of Methyl 4-(benzyloxy)-2-hydroxy-3, 6-dimethylbenzoate (700 mg, 1 eq.) in DMF (5 mL) were added K2CO3 (845 mg, 2.5 eq.) and methyl iodide (316 µL, 2 eq.) at the room temperature under N2 and was reflux at 50 °C for 10 h. The mixture was extracted with ethyl acetate, washed with brine, and dried over anhydrous Na2SO4, concentrated under a high-pressure vacuum and purified by column chromatography, to give a colorless oil as a product, yield (518 mg, 71%). Rf 0.45 (10% EA: Hex). 1HNMR (400 MHz, CDCl3) δ 7.42–7.40 (m, 5H), 6.54 (s, 1H), 5.07 (s, 2H), 3.90 (s, 3H), 3.77 (s, 3H), 2.29 (s, 3H), 2.17 (s, 3H).
Methyl 2-hydroxy-4-methoxy-3,6-dimethylbenzoate

To a solution of Methyl 2, 4-dihydroxy-3, 6-dimethyl benzoate (1 g, 1 eq.) in DMF (5 mL) were added K2CO3 (774 mg, 1.1 eq.) and methyl iodide (317 µL, 1 eq.) at the room temperature under N2. After being stirred at 50 °C for 10 h, the reaction mixture was filtered through a pad of celite. The filtrate was diluted with ethyl acetate and acidified with 3 M HCl. The organic layer was separated, and the aqueous layer was extracted twice with ethyl acetate, washed with brine, and dried over anhydrous MgSO4, concentrated under a high-pressure vacuum and purified by column chromatography, to give a white solid as a product, yield (941 mg, 88%). Rf 0.75 (50% Hex: MC). 1HNMR (400 MHz, CDCl3) δ 11.82 (s, 1H), 6.27 (s, 1H), 3.92 (s, 3H),3.85 (s, 3H), 2.52 (s, 3H), 2.07 (s, 3H).
2-Hydroxy-4-methoxy-3,6-dimethylbenzoic acid

To a solution of Methyl 2-hydroxy-4-methoxy-3,6-dimethylbenzoate (200 mg, 1 eq.) and KOH (534 mg 10 eq.) in MeOH (10 mL) was reflux for 48 h. Then the solution was acidified with 1 N HCl (20 mL) and extracted with DCM. The organic layer washed with brine, and dried over anhydrous Na2SO4, concentrated under a high-pressure vacuum and purified by column chromatography, to give a white solid as a product, yield (147 mg, 78%). Rf 0.25 (5% MeOH: MC). 1HNMR (400 MHz, DMSO-d6) δ 6.46 (s, 1H), 3.83 (s, 3H), 2.51 (S, 3H), 1.96 (s,3H).
Methyl 2-hydroxy-3,6-dimethyl-4-((4-methylbenzyl)oxy)benzoate

To a solution of Methyl-2,4-dihydroxy-3,6-dimethylbenzoate (500 mg, 1 eq.) in DMF (3 mL) were added K2CO3 (704 mg, 2 eq.) and 1-(bromomethyl)-4-methylbenzene (565 mg, 1.2 eq.) at rt under N2. After being stirred at 50 °C for 9 h, the reaction mixture was filtered through a pad of celite. The filtrate was diluted with ethyl acetate and acidified with 3 M HCl. The organic layer was separated, and the aqueous layer was extracted twice with ethyl acetate, washed with brine, and dried over anhydrous Na2SO4, concentrated under a high-pressure vacuum and purified by column chromatography, to give a white solid as a product, yield (370 mg, 48%). Rf 0.61 (10% EA: HEX). 1HNMR (400 MHz, CDCl3) δ 11.89–11.81 (m, 1H), 7.32 (d, J = 8.2 Hz, 2H), 7.21–7.15 (m, 2H), 6.37 (d, J = 14.6 Hz, 1H), 5.17–5.07 (m, 2H), 4.00–3.89 (m, 3H), 2.67–2.47 (m, 3H), 2.45–2.33 (m, 4H), 2.21–2.09 (m, 3H), 1.57 (t, J = 14.6 Hz, 1H).
2-Hydroxy-3,6-dimethyl-4-((4-methylbenzyl)oxy)benzoic acid

To a solution of Methyl 4-(benzyloxy)-2-methoxy-3,6-dimethylbenzoate (100 mg, 1 eq.) and KOH (149.6 mg 8 eq.) in MeOH (50 mL) was reflux for 24 h. Then the solution was acidified with 1 N HCl (20 mL) and extracted with DCM. The organic layer was separated, and the aqueous layer was extracted twice with ethyl acetate, washed with brine, and dried over anhydrous Na2SO4, concentrated under a high-pressure vacuum and purified by column chromatography, to give a white solid as a product, yield (50 mg, 53%). Rf 0.15 (10% MeOH: MC). 1HNMR (400 MHz, DMSO-d6) δ 7.27 (dd, J = 52.4, 8.0 Hz, 4H), 6.57 (s, 1H), 5.12 (s, 2H), 2.48 (s, 3H), 2.31 (s, 3H), 1.98 (s, 3H), -0.00 (s, 4H).
Methyl 2,4-dihydroxy-6-methyl benzoate

A solution of methyl acetoacetate (2.78 mL, 25.8 mmol) in 30 mL dry tetrahydrofuran was added slowly to a stirred suspension of NaH (1.5 g, 30 mmol) in 10 mL dry THF at 0 °C. After the gas evaluation had closed, the reaction mixture was cooled to − 78 °C and n-BuLi (10.4 ml, 2.6 mmol) was added drop by drop. The reaction mixture could warm at room temperature and be stirred overnight and then refluxed for 26 h, monitoring the reaction progress by checking the TLC. After completion of the reaction, the dark red suspension was cooled to 0 °C, quenched with 2N HCl up to pH 2–3 and extracted with ethyl acetate, washed with brine, and dried over anhydrous Na2SO4, concentrated under a high-pressure vacuum and purified by column chromatography, yield (4.69 g, 10.2%). 1HNMR (400 MHz, CDCl3) δ 11.73 (s, 1H), 6.25 (dd, J = 20.1, 2.7 Hz, 2H), 5.30 (s, 1H), 3.92 (s, 3H), 2.49 (s, 3H).
Methyl 3,5-dichloro-2,4-dihydroxy-6-methyl benzoate

To a solution of Methyl 2,4-dihydroxy-6-methylbenzoate (0.5 g, 2.74 mmol) was dissolved in diethyl ether (10 mL) and stirred for 15 min. at 0 °C after that SO2Cl2 (0.3 mL) was added drop by drop. As SO2Cl2 was added colors of the solution has become changed. After stirring for 10 min. at 0 °C, the resulting mixture was heated at 37 °C for 10 min. and the random progress was monitored by TLC. After 10 min. water was added to the reaction mixture at RT. An additional 5 mL of NaHCO3 was added to the mixture and shacked well. With the addition of NaHCO3, the reaction color turned black. Further, the product was extracted with EtOAc, dried over anhydrous Na2SO4, and concentrated on rotavapor to give a yellow solid (0.622 g, 90.8%). This crude compound was used for the further reaction without purification. 1HNMR (400 MHz, CDCl3) δ 12.07 (s, 1H), 3.96 (s, 3H), 2.60 (s, 3H).
Methyl 2,4-bis(benzyloxy)-3,5-dichloro-6-methylbenzoate

A solution of benzyl bromide (0.48 mL, 4.03 mmol), Methyl 3,5-dichloro-2,4-dihydroxy-6-methylbenzoate (0.336 g, 1.34 mmol) and potassium carbonate (0.557 g, 4.03 mmol) in acetone (30 mL) was heated to reflux for 12 h. After completion of reaction the solution was diluted with EtOAc, washed with water (30 mL) three times and brine respectively. The organic phase was dried over anhydrous sodium sulfate and concentrated under vacuum. Further, purification by using column chromatography to give a pale yellow solid as a product, yield (0.429 g, 74.6%). Rf 0.30 (EA: HEX-1:9%). 1HNMR (400 MHz, CDCl3) δ 7.58 (d, J = 6.4 Hz, 2H), 7.46 (d, J = 1.4 Hz, 2H), 7.37–7.42 (m, 4H), 7.35 (d, J = 4.1 Hz, 2H), 5.06 (d, J = 2.7 Hz, 4H), 3.83 (s, 3H), 2.34 (s, 3H).
2,4-Bis(benzyloxy)-3,5-dichloro-6-methylbenzoic acid

To a solution of Methyl 2,4-bis(benzyloxy)-3,5-dichloro-6-methylbenzoate (400 mg, 0.93 mmol, 1 eq.) and KOH (156.4 mg, 2.7 mmol) in DMSO (4 mL) and H2O (1 mL) was reflux for 24 h. after the reaction completion the solution was acidified with 1 N HCl (10 mL) and extracted with DCM. The organic phase was washed with brine (20 mL) and dried over anhydrous sodium sulfate; filter the solvent and concentrated with the help of a rotary. The residue was applied to chromatography to give a white solid as a product, yield(160 mg, 41.3%). Rf 0.1 (5% MeOH/DCM). 1HNMR (400 MHz, DMSO-d6) δ 7.55 (dd, J = 7.8, 1.4 Hz, 2H), 7.45–7.48 (m, 2H), 7.37–7.45 (m, 6H), 5.02 (d, J = 7.3 Hz, 4H), 2.53 (s, 1H), 2.32 (s, 3H).
The synthetic procedure of TU-01 to TU-10 compounds
See Fig. 7.

Synthetic procedure of depside ester, TU-01, and TU-02.
Synthesis of 3-hydroxy-4-(methoxycarbonyl)-2,5-dimethylphenyl 2,4-dimethoxy-3,6-dimethylbenzoate (depside ester, Fig. 7)
To a solution of 2, 4-dimethoxy -3,6-dimethyl benzoic acid (1.0 eq.) in anhydrous toluene (3 mL) was added TFFA (5.0 eq.) by dropwise addition at 0 °C. After completing of additions transfer the reaction mixture to rt. String the reaction mixture at room temperature for 2 h. Evaporate the reaction mixture and keep over column chromatography. 1H-NMR (400 MHz, DMSO-d6) δ 6.78 (s, 1H), 6.60 (s, 1H), 3.92 (s, 3H), 3.84 (s, 3H), 3.76 (s, 3H), 2.50 (t, J = 1.6 Hz, 3H), 2.39 (s, 3H), 2.07 (d, J = 11.4 Hz, 6H). 13C-NMR (400 MHz, DMSO-d6): δ 173.66, 166.06, 162.01, 159.99, 156.88, 152.64, 139.63, 135.29, 119.86, 116.92, 116.48, 116.07, 112.11, 108.98, 62.33, 56.36, 23.39, 20.0225, 9.5006, 9.31.
Synthesis of 4-((2,4-dimethoxy-3,6-dimethylbenzoyl) oxy)-2-hydroxy-3,6dimethylbenzoic acid (TU-01, Fig. 7)
To a solution of 3-hydroxy-4-(methoxycarbonyl)-2,5-dimethylphenyl 2,4-dimethoxy-3,6-dimethylbenzoate (200 mg, 1 eq.) and KOH (134.4 mg, 3 eq.) in DMSO (4 mL) and H2O (1 mL) was reflux for 2 h. Then the solution was cooled at room temperature, acidified with 1 N HCl (20 mL), and extracted with DCM. The organic phase was washed with brine (20 mL) and dried over anhydrous sodium sulfate; the filtrate was concentrated with the help of a rotary. The residue was applied to chromatography over silica gel. 1H-NMR (400 MHz, DMSO-d6) δ 6.74 (s, 1H), 6.56 (s, 1H), 3.80 (s, 3H), 3.71 (s, 3H), 2.35 (s, 3H), 2.04 (s, 3H), 2.01 (s, 3H). 13C-NMR (400 MHz, DMSO-d6): δ 173.66, 166.06, 162.02, 160.00, 156.88, 152.64, 139.62, 135.29, 119.87, 116.92, 116.48, 116.08, 112.11, 108.99, 62.33, 56.36, 23.40, 20.03, 9.50, 9.32. MS (ESI + , m/z): 375 [M + H] (Figs. S4 and S5).
Synthesis of benzyl 4-((2,4-dimethoxy-3,6-dimethylbenzoyl) oxy)-2-hydroxy-3,6-dimethylbenzoate (TU-02, Fig. 7)
To a solution of 4-((2,4-dimethoxy-3,6-dimethylbenzoyl) oxy)-2-hydroxy-3,6-dimethylbenzoic acid (50 mg, 1 eq.) in dry DMF was added KHCO3 (1.1 eq.) and BnBr (1.1 eq.). The mixture was stirred at RT for 2 h. Monitor the reaction by using TLC, after consumption of starting material stop the reaction. The solution was diluted with EtOAc (3 mL). The organic phase was washed with water and brine, respectively, and was then dried over anhydrous sodium sulfate and concentrated. The residue was purified by silica gel column chromatography. 1HNMR (400 MHz, CDCl3) δ 11.87 (s, 1H), 7.35–7.44 (m, 5H), 6.55 (s, 1H), 6.52 (s, 1H), 5.41 (s, 2H), 3.85 (s, 3H), 3.83 (d, J = 3.2 Hz, 3H), 2.52 (s, 3H), 2.45 (s, 3H), 2.16 (s, 3H), 2.15 (s, 3H). 13C-NMR (400 MHz, CDCl3): δ 171.77, 166.28, 163.11, 160.08, 157.28, 153.46, 139.67, 135.47, 135.25, 128.79, 128.62, 128.54, 119.81, 117.62, 117.33, 116.49, 109.74, 108.18, 67.48, 62.16, 55.79, 29.79, 24.45, 20.24, 9.20, 8.94. MS (ESI + , m/z): 465 [M + H] (Figs. S6 and S7).
A solution of 2-hydroxy-4-methoxy-3,6-dimethylbenzoic acid (144 mg, 0.735 mmol, 1 eq.) in dichloromethane (6 mL) with stirring DMAP (44 mg, 0.36 mmol, 0.5 eq.) was added as a solid and then resulting clear solution was stirred for 10 min. at RT followed by the addition of benzyl 2,4-dihydroxy-3,6-dimethylbenzoate (200 mg, 0.735 mmol, 1 eq.), and the resulting solution was stirred for 10 min. before DIC (115.9 µL, 0.735 mmol, 1 eq.) was added by syringe. The mixture was stirred for 12 h. At room temperature reaction was monitored by checking TLC. After completion of the reaction in a mixture add excess DCM for the quenching of the reaction and 1 N HCl (5 mL) was added, extracted with dichloromethane, dried in the solution with anhydrous sodium sulfate, and the solvent was evaporated in a vacuum. The residue was purified by column chromatography (hexane/EtOAc 5–10%) as a white powder. Rf = 0.4(hex/EA 20%).
The depside ester i.e., (TU-04) (97 mg, 0.215 mm) was dissolved in ethyl acetate (5 mL) containing 10% palladium on carbon (10 mg), and the suspension was stirred in an atmosphere of hydrogen for 2 h. After completion of the reaction, the catalyst was filtered using a celite pad, and the solvent evaporated. The residue was purified by column chromatography. Rf = 0.4 (hex/EA 20%). The obtained product was a white crystal. The structural data is as follows.
Synthesis of 2-hydroxy-4-((2-hydroxy-4-methoxy-3,6-dimethylbenzoyloxy)-3,6-dimethylbenzoic acid (TU-03, barbatic acid, Fig. 8)

Synthetic procedure of TU-03, and TU-04.
1H-NMR (400 MHz, DMSO-d6) δ 10.70 (s, 1H), 6.63 (s, 1H), 6.57 (s, 1H), 3.82 (s, 3H), 2.52 (s, 3H), 2.44 (s, 3H), 1.96 (s, 3H), 1.95 (s, 3H). 13C-NMR (400 MHz, DMSO- d6): δ 173.64, 169.17, 162.02, 161.61, 159.94, 152.22, 139.47, 116.32, 116.19, 112.21, 110.53, 107.63, 106.87, 56.31, 23.61, 23.38, 9.66, 8.62. MS (ESI + , m/z): 361 [M + H] (Fig. S8 and S9).
Synthesis of benzyl 2-hydroxy-4-((2-hydroxy-4-methoxy-3, 6dimethylbenzoyl) oxy)-3, 6-dimethylbenzoate (TU-04, Fig. 8)
1H-NMR (400 MHz, CDCl3) δ 11.90 (s, 1H), 11.49 (s, 1H), 7.37–7.44 (m, 5H), 6.50 (s, 1H), 6.36 (s, 1H), 5.42 (s, 2H), 3.89 (s, 3H), 2.67 (s, 3H), 2.51 (s, 3H), 2.08 (d, J = 4.1 Hz, 6H). 13C-NMR (400 MHz, CDCl3): δ 171.70, 170.27, 163.07, 162.34, 152.71, 140.82, 139.85, 135.18, 128.81, 128.68, 128.57, 117.11, 116.51, 111.39, 109.99, 106.51, 104.43, 67.57, 55.66, 25.16, 24.41, 9.41, 7.90. MS (ESI + , m/z): 449[M-H] (Figs. S10 and S11).
Synthesis of benzyl 2-hydroxy-4-((2-hydroxy-3, 6-dimethyl-4-((4-methylbenzyl) oxy) benzoyl) oxy)-3, 6-dimethylbenzoate (TU-05, Fig. 9)

Synthetic procedure of TU-05.
A solution of 2-hydroxy-3,6-dimethyl-4-((4-methylbenzyl)oxy)benzoic acid (210 mg, 0.735 mmol, 1 eq.) in dichloromethane (6 mL) with stirring DMAP (44 mg, 0.36 mmol, 0.5 eq.) was added as a solid and then resulting clear solution was stirred for 10 min. at RT followed by the addition of benzyl 2,4-dihydroxy-3,6-dimethylbenzoate (200 mg, 0.735 mmol, 1 eq.), and the resulting solution was stirred for 10 min. before DIC (115.9 µL, 0.735 mmol, 1 eq.) was added by syringe. The mixture was stirred for 12 h. At room temperature reaction was monitored by checking TLC. After completion of the reaction in a mixture add excess DCM for the quenching of the reaction and 1 N HCl (5 mL) was added, extracted with dichloromethane, dried in the solution with anhydrous sodium sulfate, and the solvent was evaporated in a vacuum. The residue was purified by column chromatography (hexane/EtOAc 5–10%) as a white powder. 1H-NMR (400 MHz, CDCl3) δ 11.92 (s, 1H), 11.52 (s, 1H), 7.35–7.44 (m, 5H), 7.32 (d, J = 7.8 Hz, 2H), 7.20 (d, J = 8.2 Hz, 2H), 6.49 (s, 1H), 6.43 (s, 1H), 5.42 (s, 2H), 5.11 (s, 2H), 2.65 (s, 3H), 2.51 (s, 3H), 2.37 (s, 3H), 2.14 (s, 3H), 2.07 (s, 3H). 13C-NMR (400 MHz, CDCl3): δ 171.71, 170.28, 163.22, 163.08, 161.60, 152.68, 140.71, 139.87, 137.93, 135.16, 133.65, 129.40, 128.82, 128.68, 128.57, 127.32, 117.11, 116.50, 111.86, 109.98, 107.78, 104.52, 70.03, 67.58, 25.21, 24.45, 21.33, 9.43, 8.17. MS (ESI + , m/z): 541[M + H] (Figs. S12 and S13).
Synthesis of 3-hydroxy-4-(methoxy carbonyl)-2, 5-dimethylphenyl 2-hydroxy-3, 6-dimethyl-4-((4-methylbenzyl) oxy) benzoate (TU-06, Fig. 10)

Synthetic procedure of TU-06.
A solution of 2-hydroxy-3,6-dimethyl-4-((4-methylbenzyl)oxy)benzoic acid (210 mg, 0.735 mmol, 1 eq.) in dichloromethane (6 mL) with stirring DMAP (44 mg, 0.36 mmol, 0.5 eq.) was added as a solid and then resulting clear solution was stirred for 10 min. at RT followed by the addition of methyl 2,4-dihydroxy-3,6-dimethylbenzoate (144 mg, 0.735 mmol, 1 eq.), and the resulting solution was stirred for 10 min. before DIC (115.9 µL, 0.735 mmol, 1 eq.) was added by syringe. The mixture was stirred for 12 h. At room temperature reaction was monitored by checking TLC. After completion of the reaction in a mixture add excess DCM for the quenching of the reaction and 1 N HCl (5 mL) was added, extracted with dichloromethane, dried in the solution with anhydrous sodium sulfate, and the solvent was evaporated in a vacuum. The residue was purified by column chromatography (hexane/EtOAc 5–10%) as a white powder.
1H-NMR (400 MHz, CDCl3) δ 11.92 (s, 1H), 11.52 (s, 1H), 7.32 (d, J = 7.8 Hz, 2H), 7.21 (d, J = 7.8 Hz, 2H), 6.50 (s, 1H), 6.43 (s, 1H), 5.11 (s, 2H), 3.96 (s, 3H), 2.65 (s, 3H), 2.52 (s, 3H), 2.37 (s, 3H), 2.14 (s, 3H), 2.07 (s, 3H). 13C-NMR (400 MHz, CDCl3): δ 172.39, 170.29, 163.22, 162.94, 161.60, 152.61, 140.72, 139.77, 137.93, 133.66, 129.40, 127.32, 117.02, 116.43, 111.87, 110.02, 107.78, 104.53, 70.03, 52.37, 29.81, 25.22, 24.15, 21.33, 9.43, 8.18. MS (ESI + , m/z): 463 [M-H] (Figs. S14 and S15).
Synthesis of 3-hydroxy-4-(methoxy carbonyl)-2, 5-dimethylphenyl furan-2-carboxylate (TU-07, Fig. 11)

Synthetic procedure of TU-07.
An oven-dried flask (25 mL) equipped with a stir bar was charged with methyl 2,4-dihydroxy-3,6-dimethylbenzoate (0.5 mmol, 1.0 equiv), dimethylaminopyridine (typically, 0.005 equiv), triethylamine (typically, 1.2 equiv), and dichloromethane (7 mL), placed under a positive pressure of argon, Acyl chloride (typically, 1.0 equiv) was added dropwise to the reaction mixture with vigorous stirring at 0 °C, and the reaction mixture was stirred 12 h at room temperature. After the indicated time, the reaction mixture was diluted with ethyl acetate (30 mL), and washed with 1 M HCl (20 mL), H2O (20 mL), and brine (20 mL). Then the organic layer was dried by Na2SO4, filtrated, and concentrated. The residue was purified by column chromatography (hexane/EtOAc 5–10%) as a white powder to give an analytically pure product.
1H-NMR (400 MHz, CDCl3) δ 11.90 (s, 1H), 7.68 (d, J = 0.9 Hz, 1H), 7.39 (d, J = 3.2 Hz, 1H), 6.60 (q, J = 1.7 Hz, 1H), 6.56 (s, 1H), 3.96 (s, 3H), 2.51 (s, 3H), 2.08 (s, 3H). 13C-NMR (400 MHz, CDCl3): δ 172.43, 162.84, 156.19, 152.46, 147.49, 143.67, 139.64, 119.84, 116.98, 116.42, 112.36, 109.99, 52.33, 24.15, 9.16. MS (ESI + , m/z): 291 [M + H] (Figurs. S16 and S17).
Synthesis of benzyl 4-((4-benzoyloxy-2-methoxy-3, 6dimethylbenzoyl) oxy)-2-hydroxy-3,6-dimethylbenzoate (TU-08, Fig. 12)

Synthetic procedure of TU-08.
A solution of 4-(benzyloxy)-2-methoxy-3,6-dimethylbenzoic acid (210 mg, 0.735 mmol, 1 eq.) in dichloromethane (6 mL) with stirring DMAP (44 mg, 0.36 mmol, 0.5 eq.) was added as a solid and then resulting clear solution was stirred for 10 min. at RT followed by the addition of benzyl 2,4-dihydroxy-3,6-dimethylbenzoate (199 mg, 0.735 mmol, 1 eq.), and the resulting solution was stirred for 10 min. before DIC (115.9 µL, 0.735 mmol, 1 eq.) was added by syringe. The mixture was stirred for 12 h. At room temperature reaction was monitored by checking TLC. After completion of the reaction in a mixture add excess DCM for the quenching of the reaction and 1 N HCl (5 mL) was added, extracted with dichloromethane, dried in the solution with anhydrous sodium sulfate, and the solvent was evaporated in a vacuum. The residue was purified by column chromatography (hexane/EtOAc 5–10%) as a white powder. 1H-NMR (400 MHz, CDCl3) δ 11.88 (s, 1H), 7.35–7.45 (m, 10H), 6.60 (s, 1H), 6.55 (s, 1H), 5.42 (s, 2H), 5.10 (s, 2H), 3.84 (s, 3H), 2.52 (s, 3H), 2.44 (s, 3H), 2.22 (s, 3H), 2.17 (s, 3H). 13C-NMR (400 MHz, CDCl3): δ 171.76, 166.25, 163.11, 159.23, 157.38, 153.45, 139.68, 136.91, 135.40, 135.25, 128.78, 128.69, 128.63, 128.54, 128.06, 127.20, 120.15, 118.10, 117.32, 116.48, 109.76, 109.56, 70.31, 67.47, 62.17, 29.78, 24.43, 20.21, 9.19. MS (ESI + , m/z): 539[M-H] (Figus. S18 and S19).
Synthesis of benzyl 4-((4-bromobenzoyloxy-2-hydroxy -3, 6-dimethylbenzoate (TU-09, Fig. 13)

Synthetic procedure of TU-09.
An oven-dried flask (25 mL) equipped with a stir bar was charged with methyl 2,4-dihydroxy-3,6-dimethylbenzoate (0.5 mmol, 1.0 equiv), dimethylaminopyridine (typically, 0.005 equiv), triethylamine (typically, 1.2 equiv), and dichloromethane (7 mL), placed under a positive pressure of argon, Acyl chloride (typically, 1.0 equiv) was added dropwise to the reaction mixture with vigorous stirring at 0 °C, and the reaction mixture was stirred 12 h at room temperature. After the indicated time, the reaction mixture was diluted with ethyl acetate (30 mL), and washed with 1 M HCl (20 mL), H2O (20 mL), and brine (20 mL). Then the organic layer was dried by Na2SO4, filtrated, and concentrated. The residue was purified by column chromatography (hexane/EtOAc 5–10%) as a white powder to give an analytically pure product.
1H-NMR (400 MHz, CDCl3) δ 11.91 (s, 1H), 8.05 (dd, J = 6.9, 1.8 Hz, 2H), 7.65 (dd, J = 6.9, 1.8 Hz, 2H), 7.37–7.45 (m, 4H), 6.53 (s, 1H), 5.41 (s, 2H), 2.50 (s, 3H), 2.07 (s, 3H). 13C-NMR (400 MHz, CDCl3): δ 171.76, 163.79, 163.03, 153.17, 139.80, 135.17, 132.16, 131.78, 129.22, 128.82, 128.69, 128.61, 128.03, 116.94, 116.45, 109.89, 67.57, 24.47, 9.18. MS (ESI + , m/z): 454 [M-H] (Figs. S20 and S21).
Synthesis of 3-hydroxy-4-(methoxy carbonyl)-2, 5-dimethylphenyl 2, 4bis (benzyloxy)-3,5-dichloro-6-methylbenzoate (TU-10, Fig. 14)

Synthetic procedure of TU-10.
A solution of 2,4-bis(benzyloxy)-3,5-dichloro-6-methylbenzoic acid (306 mg, 0.735 mmol, 1 eq.) in dichloromethane (10 mL) with stirring DMAP (44 mg, 0.36 mmol, 0.5 eq.) was added as a solid and then resulting clear solution was stirred for 10 min. at RT followed by the addition of methyl 2,4-dihydroxy-3,6-dimethylbenzoate (144 mg, 0.735 mmol, 1 eq.), and the resulting solution was stirred for 10 min. before DIC (115.9 µL, 0.735 mmol, 1 eq.) was added by syringe. The mixture was stirred for 12 h. At room temperature reaction was monitored by checking TLC. After completion of the reaction in a mixture add excess DCM for the quenching of the reaction and 1 N HCl (5 mL) was added, extracted with dichloromethane, dried in the solution with anhydrous sodium sulfate, and the solvent was evaporated in a vacuum. The residue was purified by column chromatography (hexane/EtOAc 5–10%) as a white powder. 1H-NMR (400 MHz, CDCl3.): δ 11.89 (s, 1H), 7.58–7.60 (m, 2H), 7.49 (dd, J = 7.3, 1.8 Hz, 2H), 7.36–7.43 (m, 6H), 6.32 (s, 1H), 5.12 (s, 2H), 5.08 (s, 2H), 3.95 (s, 3H), 2.53 (s, 3H), 2.38 (s, 3H), 2.03 (s, 3H). 13C-NMR (400 MHz, CDCl3): δ 172.37, 164.65, 162.90, 153.47, 152.62, 151.52, 139.71, 136.07, 133.74, 128.65, 126.91, 126.61, 122.13, 116.98, 116.13, 110.09, 52.35, 29.81, 23.99, 17.84, 9.23. MS (ESI + , m/z): 594 [M-H] (Figure S22 and S23).
Cell culture
Human colorectal adenocarcinoma-enriched cancer stem cells (CSC221) and CaCo2 cells (microsatellite stable with wild-type KRAS, BRAF, and PIK3CA) were cultured in DMEM (GenDepot, Katy, TX, USA) with the addition of 10% fetal bovine serum (FBS) and 1% penicillin–streptomycin solution. CSC221 and CaCo2 cells were purchased from BioMedicure (San Diego, CA, USA) and the American Type Culture Collection (ATCC, Manassas, VA, USA), respectively. The cells were maintained at 37 °C in a humidified incubator with a 5% CO2.
Cell viability
Cells were seeded at an appropriate density in a 96-well plate and allowed to adhere overnight. Then, the cells were treated with test compounds in desired concentrations and incubated for 48 h. After treatment, 15 µL of MTT solution (5 mg/mL in PBS) was added to each well to give a final concentration of 0.5 mg/mL. The plate was then incubated at 37 °C for 4 h in the dark to allow the viable cells to reduce MTT into formazan crystals. The medium was carefully removed, and 150 µL of dimethyl sulfoxide (DMSO) was added to dissolve the crystals. After incubation at 37 °C in a non-CO2 incubator for 10 min, the absorbance was measured at 570 nm using a microplate reader.
Spheroid assay
Cells were trypsinized and then washed with N2-supplemented DMEM/F12 (Invitrogen, Carlsbad, CA, USA). Human basic fibroblast growth factor (hbFGF; Invitrogen) and human recombinant epidermal growth factor (hrEGF; Biovision, Atlanta, GA, USA) were added to the N2-supplemented DMEM/F12 medium. Cells were seeded at a density of 5–7 × 103 number per well in ultra-low attachment 24-well plates. After 10–14 of day incubation period, sphere formation was quantified using an inverted phase contrast microscope. The relative sphere formation ability was determined using IMT iSolution software (IMT iSolution Inc., Northampton, NJ, USA) by measuring the pixel intensity of the sphere areas randomly selected from each plate.
Western blotting
CSC221 and CaCo2 cells were treated with TU3 for 48 h and then washed twice with ice-cold phosphate-buffered saline (PBS). Protein extraction was performed using lysis buffer, and the extracted proteins were separated by SDS-PAGE. The band density was analyzed using Multi Gauge 3.0 software (Fujifilm, Tokyo, Japan), and the relative density of the bands was calculated by comparing them to the density of the control bands in each sample. Antibodies against ALDH1 (sc-166362; Santa Cruz Biotechnology, Dallas, TX, USA), Gli1 (sc-20687; Santa Cruz, Dallas, TX, USA), STAT3 (Cell signaling, #12640), NF-ΚB (Cell signaling, #8242), β-catenin (Cell signaling, #9562), c-Myc (Santa Cruz Biotech., sc-40, 9E10), Cyclin-D1 (DCS-6, Merck, Kenilworth, NJ, USA), PARP (Cell signaling, #9542), BAX (Cell signaling, #2772), and β-Actin (sc-47778; Santa Cruz) were used as a loading control. Full-length uncropped blots are shown in Fig. S24.
qRT-PCR
Total RNA was extracted from CSC221 cells using RNAiso Plus (TaKaRa,Otsu, Japan) properly to the producer’s instructions. 1 µg of RNA was used for reverse transcription to create cDNA, which was then diluted for qPCR. SYBR Green reagents (Enzynomics, Seoul, Korea) were utilized to assess the relative expression of genes. Gene expression levels were normalized to housekeeping genes such as ACTIN. CFX instrument (Bio-Rad, Hercules, CA, USA) was used to perform the analysis.
Cell cycle analysis by flow cytometry
CSC221 and CaCo2 cells were seeded in 6-well plates at the density of 2 × 105 cells/well, incubated overnight, and treated with DMSO or TU3 (2.5, 5, and 10 μM) for 48 h. Cells were harvested and washed with FACS washing buffer, then incubated with trypsin solution and RNase A for 10 min at room temperature. Cells were centrifuged, and the pellets were collected and stained with 100 μL of 4 mg/mL PI (Sigma-Aldrich, St. Louis, MO, USA) for 2 h in the dark at 4 °C. A CytoFLEX device was used for cell cycle analysis.
Statistical analysis
Data are expressed as means ± standard deviation. All statistical analyses were performed using the Sigma Plot software. The statistically significant between two groups was compared using the student’s t test.
Data availability
All data generated or analysed during this study are included in this published article and its supplementary information files.
References
Sung, H. et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 Cancers in 185 countries. CA Cancer J. Clin. 71, 209–249 (2021).
Pellino, G. et al. Comparison of western and asian guidelines concerning the management of colon cancer. Dis. Colon Rectum. 61, 250–259 (2018).
Shin, J. M., Lim, E., Cho, Y. S. & Nho, C. W. Cancer-preventive effect of phenethyl isothiocyanate through tumor microenvironment regulation in a colorectal cancer stem cell xenograft model. Phytomedicine 84, 153493 (2021).
Najafi, M., Farhood, B. & Mortezaee, K. Cancer stem cells (CSCs) in cancer progression and therapy. J. Cell Physiol. 234, 8381–8395 (2019).
Batlle, E. & Clevers, H. Cancer stem cells revisited. Nat. Med. 23, 1124–1134 (2017).
Varlı, M. et al. 1′-O-methyl-averantin isolated from the endolichenic fungus Jackrogersella sp. EL001672 suppresses colorectal cancer stemness via sonic Hedgehog and Notch signaling. Sci. Rep. https://doi.org/10.1038/s41598-023-28773-z (2023).
Kumar, K. C. S. & Müller, K. Lichen metabolites. 1. Inhibitory action against leukotriene B4 biosynthesis by a non-redox mechanism. J. Nat. Prod. 62, 817–820 (1999).
Yang, J. et al. Continental scale comparison of mycobiomes in Parmelia and Peltigera lichens from Turkey and South Korea. BMC Microbiol. 24, 1–10 (2024).
Tsukamoto, S. et al. Halenaquinone inhibits RANKL-induced osteoclastogenesis. Bioorg. Med. Chem. Lett. 24, 5315–5317 (2014).
Neamati, N. et al. Depsides and depsidones as inhibitors of HIV-1 integrase: Discovery of novel inhibitors through 3D database searching. J. Med. Chem. 40, 942–951 (1997).
Solárová, Z. et al. Anticancer potential of lichens’ secondary metabolites. Biomolecules 10, 87 (2020).
Açıkgöz, B., Gökalsın, B., Karaltı, İ, Çobanoğlu, G. & Sesal, C. Interactions of PKS gene expression and antimicrobial and antibiofilm activity levels in the lichen-forming fungus Hypogymnia tubulosa due to light and heat stress. Mycol. Progr. 24, 1–16 (2025).
Boustie, J. & Grube, M. Lichens—a promising source of bioactive secondary metabolites. Plant Genet. Resour. 3, 273–287 (2005).
Zhou, R. et al. The depside derivative pericodepside inhibits cancer cell metastasis and proliferation by suppressing epithelial-mesenchymal transition. ACS Omega 9, 6828–6836 (2024).
AlQranei, M. S., Aljohani, H., Majumdar, S., Senbanjo, L. T. & Chellaiah, M. A. C-phycocyanin attenuates RANKL-induced osteoclastogenesis and bone resorption in vitro through inhibiting ROS levels, NFATc1 and NF-κB activation. Sci. Rep. 10, 1–13 (2020).
Ibrahim, S. R. M., Sirwi, A., Eid, B. G., Mohamed, S. G. A. & Mohamed, G. A. Fungal depsides—naturally inspiring molecules: Biosynthesis, structural characterization, and biological activities. Metabolites 11, 683 (2021).
Stoeckli-Evans, H. & Blaser, D. Structure of the methyl esters of barbatic and evernic acids: natural para-depsides. Acta Crystallograph. Sect. C Crystal Struct. Commun. 47, 2620–2624 (1991).
Shameera Ahamed, T. K., Rajan, V. K., Sabira, K. & Muraleedharan, K. DFT and QTAIM based investigation on the structure and antioxidant behavior of lichen substances atranorin, evernic acid and diffractaic acid. Comput. Biol. Chem. 80, 66–78 (2019).
Günaydın, Ş, Sulukoğlu, E. K., Kalın, ŞN., Altay, A. & Budak, H. Diffractaic acid exhibits thioredoxin reductase 1 inhibition in lung cancer A549 cells. J. Appl. Toxicol. 43, 1676–1685 (2023).
Kalın, ŞN., Altay, A. & Budak, H. Diffractaic acid, a novel TrxR1 inhibitor, induces cytotoxicity, apoptosis, and antimigration in human breast cancer cells. Chem. Biol. Interact. 361, 109984 (2022).
Sulukoğlu, E. K., Günaydın, Ş, Kalın, ŞN., Altay, A. & Budak, H. Diffractaic acid exerts anti-cancer effects on hepatocellular carcinoma HepG2 cells by inducing apoptosis and suppressing migration through targeting thioredoxin reductase 1. Naunyn. Schmiedebergs Arch. Pharmacol. https://doi.org/10.1007/S00210-024-02980-5/FIGURES/4 (2024).
Budak, B., Kalın, ŞN. & Yapça, Ö. E. Antiproliferative, antimigratory, and apoptotic effects of diffractaic and vulpinic acids as thioredoxin reductase 1 inhibitors on cervical cancer. Naunyn Schmiedebergs Arch. Pharmacol. 397, 1525–1535 (2024).
Bayir, Y. et al. The inhibition of gastric mucosal lesion, oxidative stress and neutrophil-infiltration in rats by the lichen constituent diffractaic acid. Phytomedicine 13, 584–590 (2006).
Okuyama, E., Umeyama, K., Yamazaki, M., Kinoshita, Y. & Yamamoto, Y. Usnic acid and diffractaic acid as analgesic and antipyretic components of Usnea diffracta. Planta Med. 61, 113–115 (1995).
Loeanurit, N. et al. Lichen-derived diffractaic acid inhibited dengue virus replication in a cell-based system. Molecules 28, 974 (2023).
Filimonov, A. S. et al. Diffractaic acid and its ethers as anti-respiratory syncytial virus agents. Med. Chem. Res. 33, 677–686 (2024).
Yu, X. et al. Semisynthesis, biological evaluation and molecular docking studies of barbatic acid derivatives as novel diuretic candidates. Molecules 28, 4010 (2023).
Divya Reddy, S. et al. Comprehensive analysis of secondary metabolites in usnea longissima (Lichenized Ascomycetes, Parmeliaceae) using UPLC-ESI-QTOF-MS/MS and pro-apoptotic activity of barbatic acid. Molecules 24, 2270 (2019).
Silva, H. A. M. F. et al. Barbatic acid from Cladia aggregata (lichen): Cytotoxicity and in vitro schistosomicidal evaluation and ultrastructural analysis against adult worms of Schistosoma mansoni. Toxicol. Vitro 65, 104771 (2020).
Hager, A., Brunauer, G., Türk, R. & Stocker-Wörgötter, E. Production and bioactivity of common lichen metabolites as exemplified by Heterodea muelleri (Hampe) Nyl. J. Chem. Ecol. 34, 113–120 (2008).
Barroso Martins, M. C. et al. In vitro and in vivo antineoplastic activity of barbatic acid. Int. Arch. Med. https://doi.org/10.3823/1934 (2016).
Wang, D. et al. The role of the natural compound naringenin in AMPK-mitochondria modulation and colorectal cancer inhibition. Phytomedicine 131, 155786 (2024).
Iwashyna, T. J. & Lamont, E. B. Effectiveness of adjuvant fluorouracil in clinical practice: A population-based cohort study of elderly patients with stage III colon cancer. J. Clin. Oncol. 20, 3992–3998 (2002).
Zhang, M. et al. Edible ginger-derived nano-lipids loaded with doxorubicin as a novel drug-delivery approach for colon cancer therapy. Mol. Therapy 24, 1783–1796 (2016).
Zhao, R. et al. Targeting FGFR1 by β, β-dimethylacrylalkannin suppresses the proliferation of colorectal cancer in cellular and xenograft models. Phytomedicine 129, 155612 (2024).
Özenver, N. & Efferth, T. Small molecule inhibitors and stimulators of inducible nitric oxide synthase in cancer cells from natural origin (phytochemicals, marine compounds, antibiotics). Biochem. Pharmacol. 176, 113792 (2020).
Varlı, M. et al. An acetonic extract and secondary metabolites from the endolichenic fungus Nemania sp. EL006872 exhibit immune checkpoint inhibitory activity in lung cancer cell. Front. Pharmacol. 13, 986946 (2022).
He, X., Liao, Y., Liu, J. & Sun, S. Research progress of natural small-molecule compounds related to tumor differentiation. Molecules 27, 2128 (2022).
Jóhannsson, F., Cherek, P., Xu, M., Rolfsson, Ó. & Ögmundsdóttir, H. M. The anti-proliferative lichen-compound protolichesterinic acid inhibits oxidative phosphorylation and is processed via the mercapturic pathway in cancer cells. Planta Med. 88, 891 (2022).
Majchrzak-Celińska, A. et al. Lichen secondary metabolites inhibit the Wnt/β-catenin pathway in glioblastoma cells and improve the anticancer effects of temozolomide. Cells 11, 1084 (2022).
Murugesan, B. et al. Molecular insights of anticancer potential of usnic acid towards cervical cancer target proteins: An in silico validation for novel anti-cancer compound from lichens. J. Biomol. Struct. Dyn. https://doi.org/10.1080/07391102.2023.2252076 (2023).
Nugraha, A. S. et al. Chemistry, Biology and Pharmacology of Lichen 193–229 (Wiley, 2024). https://doi.org/10.1002/9781394190706.CH14.
Roser, L. A. et al. Lecanoric acid mediates anti-proliferative effects by an M phase arrest in colon cancer cells. Biomed. Pharmacother. 148, 112734 (2022).
Taş, İ et al. Physciosporin suppresses mitochondrial respiration, aerobic glycolysis, and tumorigenesis in breast cancer. Phytomedicine 91, 153674 (2021).
Tripathi, A. H. et al. A review of anti-cancer and related properties of lichen-extracts and metabolites. Anticancer Agents Med. Chem. 22, 115–142 (2021).
Varlı, M. et al. KITENIN promotes aerobic glycolysis through PKM2 induction by upregulating the c-Myc/hnRNPs axis in colorectal cancer. Cell Biosci. 13, 1–21 (2023).
Varlı, M. et al. Usnic acid targets 14-3-3 proteins and suppresses cancer progression by blocking substrate interaction. JACS Au. 4, 1521–1537 (2024).
Yang, Y. et al. Tumidulin, a lichen secondary metabolite, decreases the stemness potential of colorectal cancer cells. Molecules 23, 2968 (2018).
Yang, Y., Nguyen, T. T., Pereira, I., Hur, J. S. & Kim, H. Lichen secondary metabolite physciosporin decreases the stemness potential of colorectal cancer cells. Biomolecules 9, 797 (2019).
Pulat, S. et al. Atraric acid induces hair growth through the stimulation of sonic hedgehog/GLI1 in human dermal papilla cells. Stem Cell Rev. Rep. https://doi.org/10.1007/S12015-024-10798-0/METRICS (2024).
Pulat, S. et al. Lobaric acid suppresses the stemness potential of colorectal cancer cells through mTOR/AKT. BioFactors 51, e70002 (2025).
Brandão, L. F. G. et al. Cytotoxic evaluation of phenolic compounds from lichens against melanoma cells. Chem. Pharm. Bull. (Tokyo) 61, 176–183 (2013).
Truong, T. L., Nga, V. T., Huy, D. T., Chi, H. B. L. & Phung, N. K. P. A new depside from Usnea aciculifera growing in Vietnam. Nat. Prod. Commun. 9, 1179–1180 (2014).
Emsen, B., Aslan, A., Turkez, H., Joughi, A. & Kaya, A. The anti-cancer efficacies of diffractaic, lobaric, and usnic acid: In vitro inhibition of glioma. J. Cancer Res. Ther. 14, 941–951 (2018).
Demir, L. et al. The investigation of cytogenetic and oxidative effects of diffractaic acid on human lymphocyte cultures. Braz. Arch. Biol. Technol. 58, 75–81 (2015).
Klzll, H. E. & Aǧar, G. Antiproliferative and apoptotic effects of diffractaic acid in A549 and AGS cancer cells. In AIP Conf Proc, Vol 1833 (2017).
Vizetto-Duarte, C. et al. Isololiolide, a carotenoid metabolite isolated from the brown alga Cystoseira tamariscifolia, is cytotoxic and able to induce apoptosis in hepatocarcinoma cells through caspase-3 activation, decreased Bcl-2 levels, increased p53 expression and PARP cleavage. Phytomedicine 23, 550–557 (2016).
Matsui, W. H. Cancer stem cell signaling pathways. Medicine (United States) 95, S8–S19 (2016).
Ginestier, C. et al. ALDH1 is a marker of normal and malignant human mammary stem cells and a predictor of poor clinical outcome. Cell Stem Cell 1, 555–567 (2007).
Barnfield, P. C., Zhang, X., Thanabalasingham, V., Yoshida, M. & Hui, C. C. Negative regulation of Gli1 and Gli2 activator function by suppressor of fused through multiple mechanisms. Differentiation 73, 397–405 (2005).
Raz, G. et al. Hedgehog signaling pathway molecules and ALDH1A1 expression in early-stage non-small cell lung cancer. Lung Cancer 76, 191–196 (2012).
Garner, J. M. et al. Constitutive activation of signal transducer and activator of transcription 3 (STAT3) and nuclear factor κB signaling in glioblastoma cancer stem cells regulates the Notch pathway. J. Biol. Chem. 288, 26167–26176 (2013).
Lin, H. et al. STAT3-mediated gene expression in colorectal cancer cells-derived cancer stem-like tumorspheres. Adv. Dig. Med. 8, 224–233 (2021).
Ji, Z., He, L., Regev, A. & Struhl, K. Inflammatory regulatory network mediated by the joint action of NF-kB, STAT3, and AP-1 factors is involved in many human cancers. Proc. Natl. Acad. Sci. U. S. A. 116, 9453–9462 (2019).
Rajasekhar, V. K., Studer, L., Gerald, W., Socci, N. D. & Scher, H. I. Tumour-initiating stem-like cells in human prostate cancer exhibit increased NF-κB signalling. Nat. Commun. 2, 1–13 (2011).
Rinkenbaugh, A. L. & Baldwin, A. S. The NF-κB pathway and cancer stem cells. Cells 5, 16 (2016).
Poturnajova, M., Kozovska, Z. & Matuskova, M. Aldehyde dehydrogenase 1A1 and 1A3 isoforms—Mechanism of activation and regulation in cancer. Cell Signal 87, 110120 (2021).
Shao, C. et al. Essential role of aldehyde dehydrogenase 1A3 for the maintenance of non-small cell lung cancer stem cells is associated with the STAT3 pathway. Clin. Cancer Res. 20, 4154–4166 (2014).
Canino, C. et al. A STAT3-NFkB/DDIT3/CEBPβ axis modulates ALDH1A3 expression in chemoresistant cell subpopulations. Oncotarget 6, 12637 (2015).
Clevers, H. & Nusse, R. Wnt/β-catenin signaling and disease. Cell 149, 1192–1205 (2012).
Condello, S. et al. β-Catenin-regulated ALDH1A1 is a target in ovarian cancer spheroids. Oncogene 34, 2297–2308 (2015).
Singh, S. et al. ALDH1B1 is crucial for colon tumorigenesis by modulating Wnt/β-catenin, notch and PI3K/Akt signaling pathways. PLoS One 10, e0121648 (2015).
Montalto, F. I. & De Amicis, F. Cyclin D1 in cancer: A molecular connection for cell cycle control, adhesion and invasion in tumor and stroma. Cells 9, 2648 (2020).
Chu, P. Y. et al. IFITM3 promotes malignant progression, cancer stemness and chemoresistance of gastric cancer by targeting MET/AKT/FOXO3/c-MYC axis. Cell Biosci. 12, 1–16 (2022).
Zhang, H. L., Wang, P., Lu, M. Z., Zhang, S. D. & Zheng, L. c-Myc maintains the self-renewal and chemoresistance properties of colon cancer stem cells. Oncol. Lett. 17, 4487–4493 (2019).
Kim, S. J. et al. Crosstalk between WNT and STAT3 is mediated by galectin-3 in tumor progression. Gastric Cancer 24, 1050–1062 (2021).
Liu, S. C. et al. Isoorientin inhibits epithelial-to-mesenchymal properties and cancer stem-cell-like features in oral squamous cell carcinoma by blocking Wnt/β-catenin/STAT3 axis. Toxicol. Appl. Pharmacol. 424, 115581 (2021).
Phesse, T. J. et al. Partial inhibition of gp130-Jak-Stat3 signaling prevents Wnt-β-catenin-mediated intestinal tumor growth and regeneration. Sci. Signal 7, ra92 (2014).
Funding
This work was supported by the National Research Foundation of Korea grant (RS-2024-00413760) funded by the Korea government (MSIT).
Author information
Authors and Affiliations
Contributions
M.V. and H.K. conceived and designed the experiments. M.V., and Y.Y. performed the biological experiments. S.R.B., Y.H.Y and H.H.H synthesized compounds. H.K. was the project leader guiding bioassays. H.H.H. was the project leader guiding the chemical analysis experiments. M.V. and H.K. analyzed the data and wrote the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Varlı, M., Bhosle, S.R., Jo, E. et al. Development and synthesis of diffractaic acid analogs as potent inhibitors of colorectal cancer stem cell traits. Sci Rep 15, 6695 (2025). https://doi.org/10.1038/s41598-025-90552-9
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-90552-9
