
Abstract
Hearing loss frequently occurs in Noonan syndrome and related RASopathies (NS-RAS), with conductive hearing loss being common. However, the genotypic and phenotypic features of sensorineural hearing loss (SNHL) in NS-RAS, as well as genotype-phenotype correlations, remain unknown. Leveraging in-house database for syndromic deafness (N = 1666), we analyzed the genomic landscape and clinical phenotypes of 94 NS-RAS families with genetically confirmed via targeted panel sequencing. In particular, we explored the genetic signature of SNHL in NS-RAS and provided a detailed description of the auditory characteristics of SNHL, including its natural progression and outcomes of audiological rehabilitation. Additionally, molecular modeling and functional assays were conducted to explore how PTPN11 variants with distinct auditory phenotypes affect downstream signaling pathways. Resultantly, eighteen (19.1%) exhibited SNHL, predominantly with PTPN11 (88.9%) and RAF1 (11.1%) variants. Of these patients, the majority exhibited severe-to-profound SNHL with congenital onset, and cochlear implantation yielded favorable auditory outcomes. The remaining patients maintained normal hearing throughout the follow-up periods. The PTPN11 variants linked to SNHL compromise autoinhibition between the N-SH2 and PTP domains or disrupts the interaction between the C-SH2 domain and phosphorylated tyrosine (pTyr), showing a gain-of-function effect on the RAS/ERK cascade. Specifically, those variants associated with high penetrance and severe expressivity significantly enhanced ERK phosphorylation more than the variants associated with low penetrance and milder expressivity. This is the first cohort study on NS-RAS in South Korea, elucidating the gene signatures and phenotypic characteristics of SNHL and suggesting genotype-auditory phenotype correlations that inform clinical practice.
Similar content being viewed by others


Introduction
Noonan syndrome and related RASopathies (NS-RAS) is a clinically heterogeneous disorder characterized by short stature, craniofacial malformations, cardiac abnormalities, short and/or webbed neck, and hearing loss. The incidence is reported to occur in 1 in 1,5001. The genetic disorders known as RASopathies, which encompass both Noonan syndrome and/or Noonan syndrome with multiple lentigines (NSML), result from gain-of-function variants in genes involved in the RAS-Mitogen-Activated Protein Kinase (RAS-MAPK) signaling pathway2. The advances of high-throughput next-generation sequencing (NGS) technologies have enhanced the understanding of NS-RAS and expanded our knowledge of the associated genomic landscape3. It has been concluded that PTPN11 (~ 50%), SOS1 (10–15%), RAF1 (~ 5%), RIT1 (~ 5%), KRAS (~ 5%), BRAF, NRAS, SHOC2, and MAP2K (< 5%) are causative genes for NS-RAS, with PTPN11 comprising the largest proportion4,5,6,7,8,9,10,11.
Hearing loss has been described in 40% of the patients with Noonan syndrome, primarily due to conductive hearing loss resulting from recurrent otitis media12. While previous studies have mainly focused on the conductive component, often caused by recurrent middle ear effusion and related craniofacial anomalies, the incidence and associations of sensorineural hearing loss (SNHL) in NS-RAS remain poorly understood. Gao et al. elucidated the mechanism by which overexpression of mutant PTPN11 induces SNHL, leading to a significant decrease in hair and supporting cells in a zebrafish model10. The PTPN11 gene, which encodes Src homology-containing protein tyrosine phosphatase 2 (SHP2), is expressed in mouse cochlea and plays a crucial role for the normal morphogenesis and maintenance of cochlear hair cells and supporting cells in the neuromast of zebrafish10. This highlights the potential value and significance of identifying SNHL in NS-RAS, as without timely identification and intervention, prolonged SNHL can significantly impact language development during the pediatric period. Nevertheless, there is a lack of research on SNHL and associated auditory phenotypes in NS-RAS (Supplementary Table 1), which hinders the understanding of the genomic landscape and the genotype-phenotype correlations in these conditions. Only a few case series have been reported in the literature, hindering an accurate estimation of the incidence of Noonan syndrome. Moreover, despite evidence supporting genotype–phenotype correlations for various clinical features of NS-RAS, limited attention have been directed toward SNHL and related auditory rehabilitation outcomes. Additionally, difference in methodologies and study designs resulted in heterogeneous incidences of SNHL and genetic profiles in NS-RAS. Consequently, a cohort study involving a large number of NS-RAS patients with a specific focus on SNHL is necessary.
In this study, we comprehensively decomposed the genomic landscape and clinical phenotypes of 94 unrelated Korean families with genetically confirmed NS-RAS from a single tertiary rare disease center. Through this, we elucidated the genetic signature of SNHL in NS-RAS and detailed the auditory characteristics of SNHL, including its natural course and audiological rehabilitation. Additionally, we suggested a genotype-phenotype correlation by investigating the functional consequences of PTPN11 gain-of-function variants, which exhibit distinct penetrance rates of SNHL, on downstream signaling pathways.
Results
Genotypes
A total of 94 patients (53 males, 41 females) were included in the study, and the mean age at diagnosis of NS-RAS was 67.6 ± 62.6 months. A flow diagram on our cohort of 94 families with NS-RAS was illustrated (Fig. 1A). In our cohort, the PTPN11 (54.3%) variants constituted the largest number for the genomic landscape of NS-RAS, followed by BRAF (6.4%), KRAS (1.1%), LZTR1 (1.1%), MAP2K1 (2.1%), NRAS (4.3%), RAF1 (9.6%), RIT1 (7.5%), SHOC2 (8.5%), and SOS1 (5.3%) variants (Fig. 1B). Among the total of 95 mutant alleles identified from 94 unrelated families, 93 were classified as missense variants, one as an in-frame variant, and one as a nonsense variant. Ninety-three families demonstrated patterns of inheritance that were either autosomal dominant (N = 18, 19.1%) or de novo traits (N = 75, 79.8%). Meanwhile, one proband harbored compound heterozygous variants (LZTR1 p.Tyr433His and p.Tyr155*), segregated as an autosomal recessive trait (Fig. 1C). When collapsed, 57 disease-causing variants across 10 NS-RAS genes were identified. We further expanded genotypic spectrum including three novel variants (PTPN11 p.Tyr62His, NRAS p.Arg68Ile, RAF1 p.Asn262dup). Detailed information about the mutational landscape and a conserved map of the corresponding residues are depicted in Supplementary Fig. 1. When focusing on PTPN11, the most frequent variant was p.Asn308Asp (N = 9), followed by p.Tyr279Cys (N = 5), p.Glu139Asp (N = 4), p.Gly503Arg (N = 3), and p.Met504Val (N = 3) (Supplementary Fig. 2). PTPN11 consists of N-SH2 (aa 3-104), C-SH2 (aa 112–216), and PTP (aa 221–524) domains. The distribution and residue of the identified variants showed conservation among orthologs across various species (Supplementary Fig. 2). Accordingly, all these variants met the criteria for pathogenic or likely pathogenic based on ACMG/AMP guideline (Supplementary Table 2).

A flow chart depicting Noonan syndrome and related RASopathies (NS-RAS), and percentages and inheritance patterns of each gene are illustrated. (A) A flow chart illustrating the diagnostic process of clinically diagnosed and genetically confirmed NS-RAS patients. (B) The percentages of 10 causative genes for NS-RAS identified in our study cohort are illustrated. The PTPN11 variant accounts for the largest proportion (54.3%). (C) The inheritance patterns for each gene and the number of patients corresponding to each inheritance pattern among individuals with NS-RAS included in our study are delineated. The most frequent inheritance pattern observed was autosomal dominant de novo (79.8%), followed by autosomal dominant (19.1%), and autosomal recessive (1.1%).
Clinical phenotypes
The most distinct clinical features of NS-RAS, including cardiac defects, craniofacial malformations (i.e. dysmorphic facial features), and abnormal skeletal appearances (i.e. short statures), were documented in 81 (86.2%), 80 (85.1%), and 76 (80.1%) patients, respectively. Other features including skin/hair anomaly, multiple lentigines, intellectual disabilities, cryptorchidism, and coagulation defects were relatively rare in our cohort, which were 26 (27.7%), 22 (23.4%), 20 (21.3%), 16 (30.2%), and 11 (11.7%) patients, respectively. Other malignancies were found in only 2 patients.
NSML, previously known as LEOPARD syndrome, has similar and overlapping clinical features with Noonan syndrome but with the additional hallmark of multiple lentigines. NSML and Noonan syndrome commonly share pathogenic variants in PTPN11 gene. Additionally, Noonan syndrome-like with loose anagen hair (NSLAH) overlaps clinically with Noonan syndrome, including facial dysmorphism, short stature, and cardiac defects, but can be distinguished by its characteristic thin, curly, and sparsely growing hair that is easily pluckable13. Thus, the distinguishing feature between NSLAH and Noonan syndrome lies in the presence of loose anagen hair and the typical involvement of the SHOC2 gene. Conclusively, 21 patients in our cohort were diagnosed with NSML, 6 with NSLAH, and the remaining 67 were diagnosed with Noonan syndrome (Table 1).
Clinical profiles of the PTPN11 variant cohort between those with hearing loss (hearing loss group) and without hearing loss (normal hearing group) as well as within the overall cohort were compared (Fig. 2 and Supplementary Fig. 3, respectively). Among the total cohort, 18 (19.1%) exhibited hearing loss, predominantly with PTPN11 (N = 16, 88.9%) and RAF1 (N = 2, 11.1%) variants (Fig. 2A). Among patients with PTPN11 variants, the prevalence of skeletal anomalies, multiple lentigines, and coagulation disorders significantly differed between the two groups. The hearing loss group showed higher rates of multiple lentigines and coagulation disorders, while skeletal anomalies were more prevalent in the normal hearing group (p-value = 0.044, p-value = 0.016, p-value = 0.044, respectively; Chi-square test) (Fig. 2G, K and C). No significant differences were observed in craniofacial anomaly, cardiac defects, pectus deformity, skin/hair anomaly, intellectual disability, cryptorchidism in male, and malignancy between the two groups (Fig. 2B, D, E, F, H, I and J).

Clinical profiles of the PTPN11 variant cohort between those with hearing loss (hearing loss group) and without hearing loss (normal hearing group) as well as within the overall cohort were compared. (A) Among the total cohort, 18 (19.1%) exhibited hearing loss, predominantly with PTPN11 (N = 16, 88.9%) and RAF1 (N = 2, 11.1%) variants. (B–K) A comparative analysis of the prevalence of clinical phenotypes (i.e. craniofacial anomaly, skeletal anomaly, cardiac defects, pectus deformity, skin/hair anomaly, multiple lentigines, intellectual disability, cryptorchidism in male, malignancy, and coagulation disorder) between patients with and without hearing loss using an in-house database are illustrated within the PTPN11 cohort.
In the entire cohort, qualitative analysis showed that the prevalence of coagulation disorder was significantly greater in the hearing loss group compared to that of the normal hearing group (p-value = 0.001; Chi-square test) (Supplementary Fig. 3J). The prevalence of craniofacial anomaly, skeletal anomaly, cardiac defects, pectus deformity, skin/hair anomaly, multiple lentigines, intellectual disability, cryptorchidism in male, and malignancy were not significantly different between the two groups (Supplementary Fig. 3A-I).
Auditory phenotypes and CI outcomes
Eighteen (19.1%) exhibited SNHL, predominantly with PTPN11 (N = 16, 88.9%) and RAF1 (N = 2, 11.1%) variants. All patients diagnosed with SNHL in our cohort study (N = 19) underwent targeted panel sequencing of 246 SNHL-related genes (Supplementary Table 3). As a result, no second hits were identified based on ACMG guidelines (pathogenic/likely pathogenic variants) and the ClinVar database (pathogenic/likely pathogenic variants). Among them, majority with PTPN11 variants had severe-to-profound SNHL with congenital onset. The mean age at ascertainment for Noonan syndrome within those with SNHL was 38.7 ± 53.4 month. Specifically, among the patients with the PTPN11 variant (N = 51), 16 (31.4%) had SNHL, with most exhibiting bilateral profound SNHL (N = 14, 87.5%) and only two exhibiting single-sided deafness (12.5%). Conversely, within the non-PTPN11 variants (N = 43), only two patients (4.7%) showed SNHL phenotypes: one with bilateral mixed hearing loss (RAF1 p.Ser257Leu) and another with bilateral moderate SNHL (RAF1 p.Ser259Phe). Importantly, pediatric patients who passed newborn hearing screening (NHS) or had normal audiological tests at initial evaluation did not experience progressive hearing loss. In this context, the hearing loss phenotype in pediatric patients with NS-RAS, excluding cases of conductive hearing loss, appears to manifest an “all-or-nothing” pattern. This indicates that their hearing is distinct either normal or severely impaired from birth.
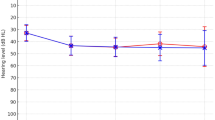
Among 14 bilateral profound SNHL patients with PTPN11 variants, 9 patients were identified as Noonan syndrome and the remainder 5 with NSML. With respect to the development of speech performances, CAP scores gradually increased over time in all patients, reaching up to a maximum score of 7 points in most of the cases at postoperative 3-year period (Fig. 3A). Receptive and expressive language scores, as evaluated by SELSI results, markedly improved after postoperative 1-year period (Fig. 3B). Throughout 5-year period, language development, as measured by PRES scores, also gradually improved in most of the patients. However, the overall language development status did not surpass the developmental level of normal children with equivalent age (50th percentile), although 4 patients (4/14, 28.6%) exceeded the normal development standard (Fig. 3C) in terms of the receptive scores. The audiological profiles in patients with hearing loss and CI profiles for those with bilateral SNHL are detailed in Supplementary Tables 4 and 5, respectively.

Graphs representing the outcomes of cochlear implantation from patient 1 to 14 are illustrated. (A) Left: A graph representing the development of the average Category of Auditory Performance (CAP) scores at each test period pre- and postoperatively. Right: CAP scores of each patient at each test period pre- and postoperatively. (B) A graph representing the development of Sequenced Language Scale for Infants (SELSI) receptive and expressive scores from preoperative period to postoperative 1-year period. (C) A graph representing the development of Preschool Receptive-Expressive Language Scale (PRES) receptive and expressive scores since postoperative 1-year period.
No significant differences in post-CI speech outcomes (i.e. receptive and expressive scores) were observed between patients diagnosed with Noonan syndrome and those with NSML over the 5-year postoperative period (Mann-Whitney U tests, p-value < 0.05).
Genotype-auditory phenotype correlation
The genomic landscape of PTPN11 variants linked to SNHL is presented in Fig. 4A. Among the PTPN11 variants identified, the four variants recurred in three or more individuals and were categorized into two groups according to the penetrance and expressivity of SNHL. Specifically, the two variants, p.Glu139Asp and p.Tyr279Cys, were associated with high penetrance (100%) and severe expressivity. All patients carrying either the p.Glu139Asp or p.Tyr279Cys variants underwent CI due to bilateral profound SNHL. In contrast, the other two variants, p.Asn308Asp and p.Gly503Arg, were associated with low penetrance (11%) and milder expressivity. None of the patients with the p.Asn308Asp or p.Gly503Arg variants manifested a hearing loss phenotype, except for one patient with SNHL (Fig. 4B). Other clinical phenotypes have also been analyzed whether there were significant differences in penetrance between those showing high and low penetrance on SNHL, and the results showed that penetrance of other phenotypes did not differ (Supplementary Table 6; Chi-square test).

Distinct auditory phenotypes in PTPN11 variants. (A) The figure illustrates the distribution of patients with SNHL and normal hearing for all variants of PTPN11 (B) The penetrance and expressivity of SNHL for four variants of PTPN11 (p.Glu139Asp, p.Tyr279Cys, p.Asn308Asp, and p.Gly503Arg).
Subcellular localization
We proposed a hypothesis that the genotype-phenotype correlation of hearing loss in NS-RAS might be associated with molecular consequences of PTPN11 variants. In particular, four variants (p.Glu139Asp, p.Tyr279Cys, p.Asn308Asp, and p.Gly503Arg), which were characterized by distinct levels of expressivity and penetrance on hearing loss, were chosen. We first investigated the subcellular localization of the four aforementioned PTPN11 variants to determine whether they were localized, which is a prerequisite for downstream signaling. Our findings revealed that all mutants, regardless of their localized domain, were effectively localized to the membrane (Fig. 5A). This was demonstrated by the overlapping signals of concanavalin A (red) with the SHP2 protein encoded by PTPN11 (green) in both HEK293T (Supplementary Fig. 4A) and HEI-OC1 cells (Supplementary Fig. 4B). These results were further confirmed by the graphs showcasing signal peaks of concanavalin A (a membrane marker, red) and Myc (SHP2, green) (Supplementary Fig. 4).

The subcellular localization and protein expression of wild-type and four missense variants of PTPN11 were investigated in HEI-OC1 cells. (A) Immunofluorescence of HEI-OC1 cells transfected with C-terminally Myc-DDK-tagged PTPN11 wild-type, p.Glu139Asp, p.Tyr279Cys, p.Asn308Asp, and p.Gly503Arg. Cells were immunostained with anti-Myc (green) and anti-Concanavalin A (red) antibodies. The four mutants and wild-type of PTPN11 are well localized to the membrane. (B) Expression levels of pERK/ERK of PTPN11 wild-type and mutants were detected time dependently by western blotting in HEI-OC1 cells upon 30 ng/ml EGF stimulation. In comparison to the PTPN11 wild-type, the four mutants exhibited an elevation in expression level of pERK/ERK. The original immunoblots (uncropped, membrane edges visible, and standard protein size markers and expected molecular weight labeled) matched to the cropped blots (this figure) were all provided in Supplementary Fig. 5. (C) When EGF treatment was prolonged to 30 min, there was a significant difference in the pERK levels between the variant group with high penetrance (p.Glu139Asp, p.Tyr279Cys) and the variant group with low penetrance (p.Asn308Asp, and p.Gly503Arg). The band intensity was quantified by Image J. Intensity data was presented as means standard deviations from 3 independent plots.
Protein expression
We then examined the protein expression level of ERK phosphorylation (pERK) relative to ERK. The mutants (p.Glu139Asp, p.Tyr279Cys, p.Asn308Asp, and p.Gly503Arg) in HEI-OC1 cells showed a significantly enhanced pERK expression and pERK/ERK ratio compared to wild-type protein, upon 30 ng/ml EGF stimulation, supporting the evidence for their pathogenicity by gain-of-function variants upon RAS/ERK cascade (Fig. 5B). The original immunoblots (uncropped, membrane edges visible, and standard protein size markers and expected molecular weight labeled) matched to the cropped blots were provided in Supplementary Fig. 5.
However, treatment with EGF for 30 min resulted in decreased pERK levels for both p.Asn308Asp and p.Gly503Arg mutants compared to treatment with EGF for 15 min. (Fig. 5C). Furthermore, the expression levels of pERK upon the RAS/ERK cascade of the two mutants (p.Asn308Asp and p.Gly503Arg), exhibiting a milder phenotype and low penetrance of SNHL, were notably lower compared to those of the other mutants (p.Glu139Asp and p.Tyr279Cys) with more severe phenotype and higher penetrance. Thus, different PTPN11 variants can induce two distinct SNHL phenotypes, suggesting a genotype-phenotype correlation that would guide medical implications.
Structural analysis
The structural model of human PTPN11 (SHP-2) was analyzed, focusing on the three key domains: N-SH2 (light cyan), C-SH2 (brown), and PTP domain (mustard), with variants localized within these regions (Supplementary Fig. 6). In particular, the Glu139Asp variant in the C-SH2 domain, located near the pTyr interaction interface, was examined through structural alignment with a pTyr-bound RAS complex (PDB ID: 6PXC). This variant shortens the side chain at position 139, weakening its interaction with backbone of G115-H116 loop, which destabilizes the surrounding loop and affects the configuration of Arg138, a critical residue for pTyr binding. As a result, the altered conformation of Arg138 disrupts the C-SH2-pTyr interaction, potentially impairing substrate recognition and leading to pathological consequences. These findings support the evidence of C-SH2’s functional importance on pTyr binding, a role initially underestimated14 but increasingly acknowledged in recent studies15,16.
Discussion
This is the first cohort study of NS-RAS in South Korea, specifically focusing on the genetic signature and associated phenotypes. Similar to previous studies3,5,17, the PTPN11 variants represented the largest proportion for the genomic landscape of NS-RAS in South Korea, and PTPN11 was identified as a gene signature linked to SNHL. Although the frequency of SNHL among NS-RAS patients was below 20%, most cases had bilateral profound SNHL with congenital onset, requiring CI. In contrast, the prevalence of mild-to-moderate SNHL phenotype was extremely rare, and most patients without SNHL who passed NHS or had documented normal hearing at their initial evaluation maintained normal hearing throughout their pediatric years. Through this, our results highlight the importance of NHS in NS-RAS patients. That is, patients who do not pass NHS are highly likely to have congenital profound SNHL, making timely intervention and rehabilitation crucial. Moreover, it is essential to ensure that treatment should not be delayed due to other comorbidities, thereby providing care within the appropriate time window.
Different PTPN11 variants induce two distinct SNHL phenotypes, suggesting a genotype-phenotype correlation that would guide medical implications. The p.Glu139Asp and p.Tyr279Cys variants of PTPN11 were the most prevalent, showing 100% penetrance and severe-to-profound SNHL. Meanwhile, the p.Asn308Asp and p.Gly503Arg variants displayed relatively lower penetrance and were associated with mostly normal hearing and a non-progressive nature. In NS-RAS patients, significant comorbidities (e.g., cardiac defects) other than hearing loss can lead to missed opportunities for timely hearing evaluations. Prolonged hearing deficits may lead to the negative maturation of central auditory pathways, followed by degradation of central neurological and higher neurocognitive functioning, and diminished speech performances18,19. In this regard, a growing body of evidences highlights the importance of early detection of SNHL and early-stage auditory rehabilitation to preserve neural auditory cortical structures and maximize language development in children with bilateral congenital SNHL20,21. Based on the experience from this study, at least in South Korea, patients with the p.Glu139Asp and p.Tyr279Cys variants need vigilant hearing assessments. The genotype-phenotype correlations could have significant medical implications for the prognosis of hearing status and therapeutic approaches in patients with NS-RAS.
Based on Muller’s morph definitions, gain-of-function variants can be categorized into hypermorph (hyper-; enhanced function), antimorph (anti-; opposing function), or neomorph (neo-; completely novel function). From this perspective, the PTPN11 variants tested in our study (p.Glu139Asp, p.Tyr279Cys, p.Asn308Asp, and p.Gly503Arg) can all be classified as hypermorph variants, acting as gain-of-function variants by enhancing ERK phosphorylation activity compared to the wild type. Although gain-of-function variants that result in enhanced or novel functions are generally rare, they are frequently observed in genes that follow an autosomal dominant inheritance pattern. Additionally, most of the dominant variants, including PTPN11 missense variants associated with NS-RAS, can be characterized as gain-of-function variants through dominant-negative mechanisms. PTPN11 encodes SHP2, which consists of two SH2 domains (N-SH2 and C-SH2) and one PTP domain22. In this study, structural modeling and biochemical assays provided evidence for the gain-of-function nature of these variants. As shown in Supplementary Fig. 6, crystal structure alignment (PDB IDs: 6PXC) revealed that p.Glu139Asp destabilizes Arg138, a critical residue for pTyr binding, disrupting the C-SH2-pTyr interaction and enhancing SHP2 activation. Qiu et al.23 also demonstrated that these conformational changes of Arg138 disrupts the C-SH2-pTyr interaction, potentially impairing substrate recognition and leading to pathological consequences. Similarly, p.Tyr279Cys altered intramolecular interactions, weakening autoinhibition and increasing catalytic activity. Qiu et al. also showed the loss of interaction between Tyr279 and Tyr62, where –OH group of Tyr62 interacts with the π-electrons of the Tyr279’s aromatic ring, decreases the intramolecular binding strength between the PTP and N-SH2 domains, leading to diminished autoinhibition. The p.Asn308Asp and p.Gly503Arg variants localized in PTP domain altered intramolecular interactions that form strong hydrogen bonds with adjacent residues, thereby compromising the autoinhibition between the N-SH2 and PTP domains. These all mutants were properly localized to the membrane, and immunoblot analyses in HEI-OC1 cells specific to the inner ear demonstrated elevated pERK/ERK ratios in all mutants compared to wild-type SHP2. Notably, the p.Glu139Asp and p.Tyr279Cys variants exhibited the most pronounced effects (see Fig. 5). These findings suggest correlations between genotype and auditory phenotype at the molecular level, as demonstrated through clinical genetic analyses. The gain-of-function effects of PTPN11 variants observed in our study are supported by previous reports. For example, Fragale et al.6 and Niihori et al.24 demonstrated that PTPN11 variants associated with RASopathies exhibit increased enzymatic activity and sustained RAS/ERK signaling. Collectively, the PTPN11 variants linked to SNHL compromise autoinhibition between the N-SH2 and PTP domains or disrupts the interaction between the C-SH2 domain and phosphorylated tyrosine (pTyr), showing a gain-of-function effect on the RAS/ERK cascade.
We have demonstrated a range of clinical phenotypes of NS-RAS based on causative genes. Despite SNHL being distinct, there is a clear overlap with other phenotypes. Nonetheless, a side-by-side comparison of clinical phenotypes between NS-RAS patients with and without hearing loss has not been previously conducted in the literature. Gao et al. summarized the incidence rates of several clinical phenotypes, including hearing loss, in 11 patients with Noonan syndrome (all with PTPN11 variants)10 and found that all patients exhibited dysmorphic facial features such as ocular hypertelorism, while nine patients exhibited with multiple lentigines, four with pectus deformity, three with skin and hair abnormalities, one male patient with cryptorchidism, and none with cardiac abnormalities, albeit of statistical significances due to the small sample size. Similarly, Han et al. elucidated that cardiac abnormalities (6/11), facial dysmorphism (7/11), skin pigmentation (4/11), growth problems (5/11), and sensorineural hearing loss (2/11) have been observed in patients with the PTPN11 p.Arg498Trp variant25. However, these two studies were unable to evaluate the prevalence rates due to the small sample sizes. In this study, we analyzed the prevalence of different clinical phenotypes depending on the presence of SNHL. This approach benefits from statistically analyzing the prevalence rates of various clinical phenotypes in a large cohort, offering valuable insights. We observed that patients with SNHL in NS-RAS are at increased risk for coagulation disorders and multiple lentigines but exhibit fewer skeletal anomalies. Reflecting on our results, it provides clues to predict the occurrence of other hidden clinical phenotypes of NS-RAS (e.g., mimics), especially when genetically confirmed NS-RAS patients first present with SNHL.
In conclusion, this is the first cohort study of NS-RAS in South Korea, specifically elucidating the genetic signature and associated phenotypic attributes of SNHL. Notably, the clinical characteristics of SNHL in NS-RAS patients include a congenital onset, severe-to-profound deafness, and a non-progressive course. These findings provide clinicians with a solid foundation for developing evidence-based treatment guidelines for NS-RAS–associated SNHL. Moreover, we propose that PTPN11 variants produce distinct SNHL phenotypes in terms of both expressivity and penetrance, suggesting genotype–phenotype correlations. In particular, highly penetrant and severe SNHL appears to be associated with gain-of-function effects of PTPN11 variants on the RAS/ERK cascade (e.g., ERK phosphorylation). Future research should aim to replicate these findings in additional cohorts and further elucidate the pathogenic mechanisms by which PTPN11 variants cause SNHL.
Our study has some limitations that should be addressed in future research. First, our sample size, comprising a total of 94 patients, was relatively small. Larger sample sizes and more diverse patient populations are needed in future studies to validate our findings and minimize potential biases in the correlation between NS-RAS and auditory phenotypes. Secondly, a more extensive functional analysis is required to clarify the involvement of SHP2 in the auditory function of the mammalian inner ear, and other downstream pathways need to be studied. Such analysis will contribute to a more precise understanding of the genotype-phenotype correlation for hearing loss in NS-RAS. Third, given the retrospective nature of this study, we acknowledge that certain biases may have arisen. Specifically, selection bias is a pervasive issue, as the study population is typically not randomly selected but rather defined by pre-existing diagnosis, conditions or outcomes, potentially limiting the generalizability of the results. Recall bias also poses a challenge, often leading to inaccuracies in collected data. To address these potential limitations, multicenter prospective studies that include diverse ethnic populations are necessary.
Methods
Cohort description and study subjects
This study utilized a retrospective design and focused on patients with NS-RAS attending the Hereditary Hearing Loss Clinic at the Center for Rare Diseases at the tertiary medical center. From our in-house database for syndromic deafness (N = 1666) using the van der Burgt criteria (suggestive facial features + 2 major or 3 minor criteria), we identified 138 patients (8.3% of the database) with clinically-suspected NS-RAS (International Classification of Diseases (ICD) code Q87.1). This encompasses genetic disorders that exhibit similar clinical characteristics to NS-RAS, including cardio-facio-cutaneous syndrome (CFCS), Noonan syndrome with multiple lentigines (NSML), and Noonan syndrome-like with loose anagen hair (NSLAH)26. Next, targeted panel sequencing for NS-RAS was performed, and 44 patients (44/138, 31.9%) who remained undiagnosed or lacked causative genes were excluded. Consequently, 94 patients (94/138, 68.1%) with genetically confirmed NS-RAS were included for the analysis (Fig. 1A, flow chart).
The clinical phenotypes of NS-RAS were comprehensively retrieved from electronic medical records, and a comparative analysis was conducted to illustrate the phenotypic differences between individuals with hearing loss and those with normal hearing. The study was approved by the Institutional Review Boards (IRB-H-0905-041-281 and IRB-2201-040-1288). Written informed consents were obtained from all participants or their legal guardians, in accordance with the Declaration of Helsinki.
Audiological and speech evaluations
Patients who visited the outpatient clinic of otorhinolaryngology department all underwent endoscopic examinations as a standard procedure to confirm the presence of conductive component including middle ear effusion. Detailed audiological examinations including click-evoked and bone-conduction auditory brainstem response (ABR) tests and auditory steady-state response (ASSR) tests within four different octave frequencies (500, 1000, 2000, and 4000 Hz) were performed. Conductive hearing loss was determined if there was a threshold difference between click-evoked and bone-conduction ABR, along with type B configuration on tympanometry27. Otherwise, SNHL was defined if there was no threshold difference between click-evoked and bone-conduction ABR, compatible with a normal configuration on tympanometry. Degree of SNHL was classified into normal hearing (lower than 25 dB HL), mild (26 to 40 dB HL), moderate (41 to 55 dB HL), moderately severe (56 to 70 dB), severe (71 to 90 dB HL), and profound (more than 90 dB HL) degree according to ABR threshold and average threshold of four different octave frequencies of ASSR.
For those who were diagnosed with bilateral SNHL in our cohort study (N = 14), all cases were of congenital onset, as evidenced by failure of newborn hearing screening test. They underwent initial auditory rehabilitations with bilateral hearing aids if speech development was poorer than normal range or hearing threshold surpassed over 40 dB HL. When their hearing threshold exceeded over 90 dB or exhibited no response at the ABR tests, CI was performed. Given this, those who were diagnosed with SNHL were less likely to have acquired deafness by drugs or trauma.
For speech evaluations, we analyzed pre- and post-cochlear implantation (CI) outcomes using categories of speech perception tests, including Sequenced Language Scale for Infants (SELSI) and Preschool Receptive-Expressive Language Scale (PRES). The SELSI and PRES tests are diagnostic tools for the evaluation of receptive and expressive language and vocabulary development in native Korean users under 2 years old and from 2 to 6 years old, respectively28,29. These two tests have the advantages of measuring in forms of raw scores and percentiles, which are comparable with the flow of chronological ages. In addition, Category of Auditory Performance (CAP) score was used to evaluate children’s auditory performance in daily life30. All the speech evaluation tests were implemented by professional speech and language pathologists at Seoul National University Hospital during each pre- and post-CI session.
Molecular genetic testing
Genomic DNA was extracted from peripheral blood using a standard procedure, and subjected to target panel sequencing for patients with NS-RAS. The target regions were captured using a SureSelect DNA targeted sequencing panel (ACTB, ACTG1, BRAF, CBL, CCNK, CDC42, EPHB4, FGD1, HRAS, KAT6B, KRAS, LZTR1, MAP2K1, MAP2K2, MAP3K8, MAPK1, MRAS, NF1, NF2, NRAS, NSUN2, PPP1CB, PTPN11, RAF1, RASA1, RASA2, RIT1, RRAS, RRAS2, SASH1, SHOC2, SMARCB1, SOS1, SOS2, SPRED1, SPRED2, STAMBP, and SYNGAP1). The library was paired-end sequenced with a NovaSeq 6000 sequencing system (Illumina, San Diego, CA, USA). Sequence reads were aligned to the human reference genome GRCh37 (hg19) and processed according to the Genome Analysis Toolkit (GATK) best-practice pipeline to call SNVs and indels. The ANNOVAR program was used for variant annotation, such as the RefSeq gene set and the Genome Aggregation Database (gnomAD, https://gnomad.broadinstitute.org/). Rare non-silent variants were selected as candidates, including nonsynonymous SNVs, coding indels, and splicing variants. We also used the KRGDB (http://coda.nih.go.kr/coda/KRGDB/index.jsp) and KOVA2 (https://www.kobic.re.kr/kova/) databases for further filtration of ethnic specific variants. In addition, LitVar2, ClinVar, and HGMD databases were screened to check whether candidate variants were previously identified in other patients. The pathogenicity of all variants was classified in accordance with the ACMG/AMP guideline31.
Plasmids, cell culture, and transfection
A human PTPN11 cDNA clone (SC112718) was purchased from Origene. The PTPN11 variant plasmids, including pCMV6-Myc-DDK entry, pCMV6-PTPN11 WT-Myc-DDK, pCMV6-PTPN11 Glu139Asp-Myc-DDK, pCMV6- PTPN11 Tyr279Cys-Myc-DDK, pCMV6-PTPN11 Asn308Asp Myc-DDK, pCMV6-PTPN11 Gly503Arg-Myc-DDK were generated utilizing the QuickChange mutagenesis method. HEI-OC1 cells were cultured in DMEM (LM001-05, Welgene) supplemented with 10% fetal bovine serum (12483-020, Gibco), 100 units/mL penicillin/streptomycin (LS015-01, Welgene), and 2 mM L-glutamine (LS002-01, Welgene). HEI-OC1 cells were maintained in a humidified atmosphere containing 10% CO2 at 33℃32,33. For transient overexpression, cells were transfected with 0.5–1 µg of total plasmid DNA in a 12-well culture plate (> 95% confluent or at a density of 106 cells/well) for 24 h using jetPRIME reagent (101000015, Polyplus), in accordance with the manufacturer’s guidelines.
Western blotting
Proteins in whole cell lysates were separated by 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to 0.45 μm polyvinylidene difluoride (PVDF) membranes (IPVH00005, Millipore). The membranes were incubated with 5% skim milk at room temperature for 1 h and probed with the following primary antibodies: anti-Myc (2276 S, CST), anti-Erk (9102 S, CST), anti-phospho Erk, (9101 S, CST), anti-β-actin (sc-47778, Santa Cruz biotechnology). The membranes were incubated with a horseradish peroxidase conjugated anti-mouse IgG antibody (SA001-500, GenDEPOT) or anti-rabbit IgG antibody (SA002-500, GenDEPOT). The protein band were detected by chemiluminescence reagent (RPN2106, cytiva). The band intensity was measured by Image J software.
Immunocytochemistry
For immunofluorescence microscopy, cultured HEI-OC1 cells on a cover glass were transfected with 1–2 ug of total plasmid DNA for 24 h. After that, the cells were fixed with 4% paraformaldehyde for 20 min, permeabilized with 0.5% Triton X-100 in PBS for 20 min, and blocked with 1% BSA in PBS for 20 min. And then the cells were incubated with the Myc-Tag primary antibodies for overnight. Following incubation, the cells were washed 3-times for 10 min with PBS and incubated with Alexa Fluor-conjugated secondary antibody diluted in 1% BSA/PBS for 40 min at room temperature. The cells were then mounted with 4’,6’-diamidino-2-phenylindole (DAPI)-containing mounting medium (ab104139, abcam). Confocal images were captured by a laser scanning confocal microscope (Leica STELLARIS 8, Upright). The intensity of the signal of the peak graph was measured using LAS X software.
Structural analysis
A structural analysis of PTPN11 (SHP-2) was performed using its crystal structure from the Protein Data Bank (PDB ID: 2SHP) and aligned with RAS-pTyr complex structure (PDB ID: 6PXC) to examine the substrate-binding interface of the C-SH2 domain. The key variants of PTPN11 (p.Glu139Asp, p.Tyr279Cys, p.Asn308Asp, and p.Gly503Arg) were visualized and analyzed using PyMOL software (v. 2.6; PyMOL, Schrödinger Inc., New York, NY, USA).
Statistical analysis
All the data are presented as mean ± standard deviation. All statistical analyses were executed and depicted using the GraphPad Prism software (ver. 10.0.0; GraphPad Software Inc.). For the comparative analysis of clinical phenotypes between those with and without hearing loss, we employed the Chi square test. One-way ANOVA with Bonferroni’s correction was used to compare the differences of the subcellular localization of the four PTPN11 variants. Throughout the analyses, p-values less than 0.05 were considered to be significant.
Data availability
Sequence variations were submitted to Clinvar with the accession code (SCV005199862, SCV005199863, and SCV005199864; https://www.ncbi.nlm.nih.gov/clinvar/submitters/509667). The authors confirm that the data supporting the findings of this study are available within the article and its supplementary materials. The data that support the findings of this study are available from the corresponding author (S-Y.L, maru4843@hanamil.net) upon reasonable request. Some data may not be made available because of privacy or ethical restrictions.
References
Noonan, J. A. Noonan syndrome and related disorders: alterations in growth and puberty. Rev. Endocr. Metab. Disord. 7, 251–255. https://doi.org/10.1007/s11154-006-9021-1 (2006).
Zenker, M., Edouard, T., Blair, J. C. & Cappa, M. Noonan syndrome: improving recognition and diagnosis. Arch. Dis. Child. 107, 1073–1078. https://doi.org/10.1136/archdischild-2021-322858 (2022).
Li, X. et al. Molecular and phenotypic spectrum of Noonan syndrome in Chinese patients. Clin. Genet. 96, 290–299. https://doi.org/10.1111/cge.13588 (2019).
Jongmans, M. C. et al. Cancer risk in patients with Noonan syndrome carrying a PTPN11 mutation. Eur. J. Hum. Genet. 19, 870–874. https://doi.org/10.1038/ejhg.2011.37 (2011).
Lee, B. H. et al. Spectrum of mutations in Noonan syndrome and their correlation with phenotypes. J. Pediatr. 159, 1029–1035. https://doi.org/10.1016/j.jpeds.2011.05.024 (2011).
Fragale, A., Tartaglia, M., Wu, J. & Gelb, B. D. Noonan syndrome-associated SHP2/PTPN11 mutants cause EGF-dependent prolonged GAB1 binding and sustained ERK2/MAPK1 activation. Hum. Mutat. 23, 267–277. https://doi.org/10.1002/humu.20005 (2004).
Tartaglia, M. et al. PTPN11 mutations in Noonan syndrome: molecular spectrum, genotype-phenotype correlation, and phenotypic heterogeneity. Am. J. Hum. Genet. 70, 1555–1563. https://doi.org/10.1086/340847 (2002).
Nava, C. et al. Cardio-facio-cutaneous and Noonan syndromes due to mutations in the RAS/MAPK signalling pathway: genotype-phenotype relationships and overlap with Costello syndrome. J. Med. Genet. 44, 763–771. https://doi.org/10.1136/jmg.2007.050450 (2007).
Pandit, B. et al. Gain-of-function RAF1 mutations cause Noonan and LEOPARD syndromes with hypertrophic cardiomyopathy. Nat. Genet. 39, 1007–1012. https://doi.org/10.1038/ng2073 (2007).
Gao, X. et al. Congenital sensorineural hearing loss as the initial presentation of PTPN11-associated Noonan syndrome with multiple lentigines or Noonan syndrome: clinical features and underlying mechanisms. J. Med. Genet. 58, 465–474. https://doi.org/10.1136/jmedgenet-2020-106892 (2021).
van Trier, D. C. et al. External ear anomalies and hearing impairment in Noonan Syndrome. Int. J. Pediatr. Otorhinolaryngol. 79, 874–878. https://doi.org/10.1016/j.ijporl.2015.03.021 (2015).
Sharland, M., Burch, M., McKenna, W. M. & Paton, M. A. A clinical study of Noonan syndrome. Arch. Dis. Child. 67, 178–183. https://doi.org/10.1136/adc.67.2.178 (1992).
Padhiyar, J., Mahajan, R. & Panda, M. RASopathies: Evolving concepts in pathogenetics, clinical features, and management. Indian Dermatol. Online J. 15, 392–404. https://doi.org/10.4103/idoj.idoj_594_23 (2024).
Barford, D. & Neel, B. G. Revealing mechanisms for SH2 domain mediated regulation of the protein tyrosine phosphatase SHP-2. Structure. 6, 249–254. https://doi.org/10.1016/s0969-2126(98)00027-6 (1998).
Liu, B. A., Engelmann, B. W. & Nash, P. D. The language of SH2 domain interactions defines phosphotyrosine-mediated signal transduction. FEBS Lett. 586, 2597–2605. https://doi.org/10.1016/j.febslet.2012.04.054 (2012).
Park, M. J. et al. SH2 domains serve as lipid-binding modules for ptyr-signaling proteins. Mol. Cell. 62, 7–20. https://doi.org/10.1016/j.molcel.2016.01.027 (2016).
Shoji, Y. et al. Genotype-phenotype correlation analysis in Japanese patients with Noonan syndrome. Endocr. J. 66, 983–994. https://doi.org/10.1507/endocrj.EJ18-0564 (2019).
Naik, A. N., Varadarajan, V. V. & Malhotra, P. S. Early pediatric cochlear implantation: an update. Laryngoscope Investig Otolaryngol. 6, 512–521. https://doi.org/10.1002/lio2.574 (2021).
Kral, A., Kronenberger, W. G., Pisoni, D. B. & O’Donoghue, G. M. Neurocognitive factors in sensory restoration of early deafness: a connectome model. Lancet Neurol. 15, 610–621. https://doi.org/10.1016/s1474-4422(16)00034-x (2016).
Yuan, D. et al. Early-stage use of hearing aids preserves auditory cortical structure in children with sensorineural hearing loss. Cereb. Cortex. 34 https://doi.org/10.1093/cercor/bhae145 (2024).
Lee, S. J. et al. Early postoperative benefits in receptive and expressive language development after cochlear implantation under 9 months of age in comparison to implantation at later ages. Clin. Exp. Otorhinolaryngol. 17, 46–55. https://doi.org/10.21053/ceo.2024.00011 (2024).
Tartaglia, M. et al. Mutations in PTPN11, encoding the protein tyrosine phosphatase SHP-2, cause Noonan syndrome. Nat. Genet. 29, 465–468. https://doi.org/10.1038/ng772 (2001).
Qiu, W. et al. Structural insights into Noonan/LEOPARD syndrome-related mutants of protein-tyrosine phosphatase SHP2 (PTPN11). BMC Struct. Biol. 14 https://doi.org/10.1186/1472-6807-14-10 (2014).
Niihori, T. et al. Functional analysis of PTPN11/SHP-2 mutants identified in Noonan syndrome and childhood leukemia. J. Hum. Genet. 50, 192–202. https://doi.org/10.1007/s10038-005-0239-7 (2005).
Han, J. Y. & Park, J. Paternally inherited Noonan syndrome caused by a PTPN11 variant may exhibit mild symptoms: a case report and literature review. Genes (Basel). 15 https://doi.org/10.3390/genes15040445 (2024).
Carcavilla, A. et al. Genotypic findings in Noonan and non-Noonan RASopathies and patient eligibility for growth hormone treatment. J. Clin. Med. 12 https://doi.org/10.3390/jcm12155003 (2023).
Reynard, P. et al. Contribution of bone conduction click-evoked auditory brainstem responses to diagnosis of hearing loss in infants in France. Eur. Ann. Otorhinolaryngol. Head Neck Dis. 138, 159–162. https://doi.org/10.1016/j.anorl.2020.09.007 (2021).
Yoon, J. A. et al. Correlation of language assessment batteries of toddlers with developmental language delay. Ann. Rehabil Med. 46, 256–262. https://doi.org/10.5535/arm.22045 (2022).
Byun, H. et al. Performance after timely cochlear implantation in prelingually deaf children with cerebral palsy. Int. J. Pediatr. Otorhinolaryngol. 77, 1013–1018. https://doi.org/10.1016/j.ijporl.2013.03.034 (2013).
Yoshida, H., Kanda, Y., Satoh, C., Kumai, Y. & Takahashi, H. Long-term speech perception performance in prelingually deafened adult cochlear implant recipients. Cochlear Implants Int. 24, 243–249. https://doi.org/10.1080/14670100.2023.2228031 (2023).
Richards, S. et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 17, 405–424. https://doi.org/10.1038/gim.2015.30 (2015).
Kalinec, G. M., Webster, P., Lim, D. J. & Kalinec, F. A cochlear cell line as an in vitro system for drug ototoxicity screening. Audiol. Neurootol. 8, 177–189. https://doi.org/10.1159/000071059 (2003).
Wijesinghe, P., Nunez, D. A. & Garnis, C. MicroRNA signature and cellular characterization of undifferentiated and differentiated house ear Institute-Organ of Corti 1 (HEI-OC1) cells. J. Assoc. Res. Otolaryngol. 23, 467–489. https://doi.org/10.1007/s10162-022-00850-6 (2022).
Acknowledgements
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Author information
Authors and Affiliations
Contributions
S.-Y.L. and M.K.P. designed the study. S.J.L. and S.L wrote the manuscript. S.J.L., S.J., S.L, and S.H.J. collected and analyzed data. S.J., S.H.J., and S.-Y.L. performed the functional study. M.-W.S., J.-J.S, J.H.L, J.M.K, M.K.P., and S.-Y.L. reviewed the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Lee, S.J., Jeong, S., Lee, S. et al. Gene signatures and genotype-phenotype correlations of sensorineural hearing loss in Noonan syndrome and related RASopathies. Sci Rep 15, 12102 (2025). https://doi.org/10.1038/s41598-025-90635-7
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-90635-7
