
Abstract
Viral codon fitness (VCF) of the host and the VCF shifting has seldom been studied under quantitative measurements, although they could be concepts vital to understand pathogen epidemiology. This study demonstrates that the relative synonymous codon usage (RSCU) of virus genomes together with other genomic properties are predictive of virus host codon fitness through tree-based machine learning. Statistical analysis on the RSCU data matrix also revealed that the wobble position of the virus codons is critically important for the host codon fitness distinction. As the trained models can well characterise the host codon fitness of the viruses, the frequency and other details stored at the leaf nodes of these models can be reliably translated into human virus codon fitness score (HVCF score) as a readout of codon fitness of any virus infecting human. Specifically, we evaluated and compared HVCF of virus genome sequences from human sources and others and evaluated HVCF of SARS-CoV-2 genome sequences from NCBI virus database, where we found no obvious shifting trend in host codon fitness towards human-non-infectious. We also developed a bioinformatics tool to simulate codon-based virus fitness shifting using codon compositions of the viruses, and we found that Tylonycteris bat coronavirus HKU4 related viruses may have close relationship with SARS-CoV-2 in terms of human codon fitness. The finding of abundant synonymous mutations in the predicted codon fitness shifting path also provides new insights for evolution research and virus monitoring in environmental surveillance.
Similar content being viewed by others


Introduction
The COVID-19 pandemic outbreak at the end of 2019 has made global impacts on human society causing over multi-million deaths so far and countless economic loss. With the development of sequencing technology and incredible efforts of scientists, massive virus genome sequencing data were generated from environmental sampling to identify critical mutations and to monitor the evolution of SARS-CoV-2 during the pandemic1, especially by small-size sequencing equipment such as Nanopore MinION sequencer allowing scientists to sequence virus genome on-site directly after sample harvest2,3. An important question after sample collection is often that whether the virus infects human, or what is the virus host ranges for the on-site scientists to identify the virus’ potential threat. The virus host range is defined as a group of host species where the pathogen can proliferate. It is one of the most important concepts helping understand pathogen epidemiology and evolution. A pathogen’s host range is difficult to characterise due to the lack of quantitative measurements, which leads to ineffective early precaution predictions for early precaution alerts. Host range shifting is a chain of changes in host range (i.e. non-human to human) of a virus. Currently, there is no effective methodology to predict characteristics of host shifting between different virus strains (i.e., critical mutation of significant host range change); instead, the virus evolutions are mainly studied through constructing phylogenetic trees using the viruses’ genomes after a host range shifting occurred4,5,6,7,8,9. Historically, many harmful virus outbreaks are attributed to their unknown host range shifting, especially towards human, such as the MERS-CoV epidemic (2012, over 800 deaths) from bats or dromedary camels10,11, H1N1 influenza virus pandemic (2009, over 280,000 deaths) from swine12,13, SARS-CoV epidemic (2003, over 700 deaths) from horseshoe bats or palm civets14,15,16. Thus, lacking quantitative measurements in studying virus host range may be the stumbling block of virus evolution study. Moreover, there are studies trying to predict host ranges in specific group of viruses17,18, but limited studies were found to predict general host range.
There are various potential determining factors in virus host range such as codon fitness to hosts19, mechanism entering hosts20,21, immune evasion mechanisms22 et al. Because translations of viral genes rely dramatically on host translational machinery, and codon fitness is a correlation between virus codon usage biases and host tRNA pool, thus incompatibilities in codon fitness will eventually lead to inefficiency in virus proteins translation and failure in virus proliferation23,24. Thus, virus codon fitness (VCF) is one of the most vital determining factors to virus host range, which has huge potential in virus host range prediction. Host tRNA pool is dramatically affected by the host genotypes while it is still difficult to represent it at the species level consisting of different host genotypes with significant variety. Although we may set a reference genome with certain individual, the reference genotype may be insusceptible to certain human virus but not to the majority population. Thus, we propose to study virus host codon fitness with virus codon usage bias directly from viral genomes and virus host range label for generalisation.
Virus codon usage bias, as a major metric for host translational adaptions, is the key property of coding sequences to decide intracellular translation efficiency25,26,27, and the intracellular translational efficiency of viral proteins directly determines the efficiency of virus replications28,29,30. We hypothesise that the virus codon biases would have relation to the host translational mechanism (i.e., tRNA pool), and generally reflect the adaptation level if studied by machine learning, which therefore could be used to predict viral host codon fitness. There are many metrics to study virus codon biases such as Relative Synonymous Codon Usage (RSCU)31,32, Codon Adaptation Index (CAI)32, tRNA adaptation index (tAI)33, et al. However, most of them required gene expression level of host genes as reference, which may lead to extra biases in species-scale representation and in later prediction. Relative synonymous codon usage, or RSCU, is a statistical propensity parameter representing essential biases of the codon usages in a coding sequence31,32, which is purely computed from coding sequences without computational loss. RSCU preferences have been studied in individual viruses including SARS-CoV-234, Flaviviridae Virus35, Zika virus36, and Transmissible Gastroenteritis Virus37. However, most of these studies are only focused on the statistical analysis of the RSCU contents of the individual virus in an aim to find RSCU correlations between the individual viruses and their host labels35,38,39,40,41. The integrative RSCU contents and preferences about the collection of all the viruses have never been systematically examined, and there is no study aiming to bridge the gap between the virus codon biases and the viral host codon fitness. Although the micro-environments of virus-host interactions are extremely complicated and they should be clearly distinct between different species of viruses42,43, it is possible through competent machine learning algorithms44 to discover previously unknown rules underlining the association between codon usage biases and the viral codon fitness in hosts.
In this study, we propose to use tree-based machine learning algorithms such as random forest (RF) to establish accurate models predictive to the probability of virus host codon fitness with RSCU of virus genomes and other virus genome composition properties as input data. This classification technique, as empowered by entropy or information gain dichotomy, is specially used by this study due to their advantages in dealing with non-linear features such as RSCU, which the RF model is a committee of different Decision Tree models making the prediction by voting. Additional important features of the input data include coding sequences (CDS) length profiles, and virus taxonomy classifications. The tree-based algorithms are a branch of supervised machine learning technique, where each tree is a dichotomy hierarchy structure of true/false decision-making rules for deciding the output classification according to the input feature values. Here, we propose using the predicted probability from trained RF model as representative readout score for virus codon fitness (VCF) in certain host range (i.e. human). In this study, the human virus codon fitness score, or HVCF score, predicted from the trained RF model for the human host is further explored for virus genomes sequence data from different sources, and for monitoring of SARS-CoV-2 human VCF shifting during COIVD-19 pandemic. Moreover, we attempted to simulate codon-based mutation process of SARS-CoV-2 from other Betacoronavirus through examining changes in HVCF when applied mutations. We have found that the virus codon biases, and machine learning models can serve as measurements in defining the boundaries of virus host codon fitness and can make predictions for virus host codon fitness shifting.
Results
Distinct codon usage biases observed in viruses which have different host codon fitness
To reveal the distinctness in codon usage biases of virus genomes that have different host ranges, we first analysed RSCU compositions, readout metrics of codon usage biases, and constructed their visualisations via Uniform Manifold Approximation and Projection (UMAP) dimensional reduction algorithm (Fig. 1A)45. The result shows that bacteriophages have distinct distributions compared to other viruses with different host ranges. When bacteriophages are excluded, the other host-ranged viruses still showed similar but slightly different patterns of distributions. Therefore, we aim to train machine learning models using RSCU features to predict codon fitness probabilities in specific host labels.

RSCU characteristics of virus genomes. (A) UMAP Dimensional reduction for the RSCU of virus genomes with different host ranges. The RSCU data was first normalised with Z-score normalisation then lossless compressed with Principal component analysis (PCA). (B) UMAP Dimensional reduction for the virus genome RSCUs on different codons’ wobble nucleotides (others in supplemental Fig. 1A). The RSCU data was first normalised with Z-score normalisation then lossless compressed with PCA. (C) Independent T-test on the virus genome RSCU of human and bacteria. Results of other hosts could be found in supplemental Fig. 1B.
We also transposed the RSCU data matrix to study the general patterns of the codon behaviours among the virus genomes. See Fig. 1B for the analysis result and visualisations (via UMAP dimensional reduction algorithm). The third nucleotide indicates robust clustering patterns, where two major clusters are identified with either A/U-ended codons or G/C-ended codons (Fig. 1B), while no obvious patterns in the first and second nucleotides (Supplementary Fig. 1A). Within the two major clusters, the A-ended codons and the U-ended codons have separate minor clusters, the same as the G-ended codons and C-ended codons. Interestingly, each of the A/U/G/C-ended codons have certain levels of individual clustering patterns suggesting that they are also distinct to each other. This direct evidence strongly supports that the wobble position of virus codons is vital. Surprisingly, two exceptional codons are observed, where the G-ended UUG (Leu) and AGG (Arg) are instead clustered with the A/U-ended codon. UUG is clustered with U-ended codons, and AGG is more clustered with A-ended. This finding highlights the potential important roles of both the UUG and AGG codons in the virus genomes.
To further compare codon biases within a specific host, independent T-test was performed on the RSCU data to find significantly varying codons in the context of a specific host codon fitness (Fig. 1C, supplementary Fig. 1B). Generally speaking, the RSCUs of the A/U-ended codons are significantly higher compared to the G/C-ended codons in the human viruses. This finding indicates that A/U-ended codons are more abundant in the human viruses compared to G/C-ended codons. However, exceptions were also spotted in the human-infectious viruses. For example, the RSCUs of the G/C-ended codons AGG (Arg), GGG (Gly) and CCC (Pro) are significantly abundant unlike the other G/C-ended codons, while the A/U-ended codons CGU (Arg), GGU (Gly) and CGA (Arg) are conversely less preferred. This finding implies a distinct behaviour of Arginine- and Glycine-encoding codons and their potentially different biological roles in the codon usage selection. Similar pattern was observed in the other hosts including vertebrates, invertebrates, and land plants, where the preference of the A/U-ended codons is clearly exceeding the G/C-ended codons with only a few exceptions (Supplementary Fig. 1B). Consistent to the above UMAP analysis, the bacteriophage has unique RSCU compositions compared to the other viruses with the general preferences of the G/C-ended codons over the A/U-ended codons.
Accurate range prediction for the hosts of virus through machine learning with RSCU and other virus genome characteristics as machine learning features
We then applied tree-based machine learning algorithms to use the RSCU datasets of virus genomes in predicting whether a viral genome has or does not have strong potential to infect a certain host due to affinised VCF. The class labels of these datasets are binary: host vs. non-host (i.e. human vs. not-human). To overcome sample imbalance and to achieve higher classification accuracy for the test data, we resampled the data to make the training data class-balanced by the SMOTE method46. The datasets with pure RSCU features are denoted by DR (or DRSCU). These datasets and the algorithms successfully trained accurate Random Forest (RF) models to predict VCF in different host labels including human, vertebrates, invertebrates, land plants and bacteria. Different train-test-split ratios show increasing accuracy of predictions with increasing training data sizes (Fig. 2A). Even with extremely low train data ratio of 0.05, the accuracies are all better than blind guessing (0.5 in accuracy), suggesting the use of RSCU data to predict virus host is reliable.
To achieve higher accuracy in training models, extra features including Taxonomy dataset and CDS Length dataset of viruses are included in addition to RSCU dataset (Supplementary Fig. 2). The combination of RSCU dataset and Taxonomy dataset are denoted as DRT (or DRSCU−Taxonomy); when CDS length are further included, the datasets are denoted as DRTC (or DRSCU−Taxonomy−CDS Length). The model performances including excellent ROC-AUC and F1 performance of these classification models (train data ratio = 0.9) are showed at Fig. 2B. These highly accurate models confirm the facts that differently host-ranged viruses do have their distinct codon usage biases. Thus, we propose to use the predicted probability of trained RF models as the readout of VCF with regards to the various hosts.

Performances of trained random forest models to predict different hosts based on different datasets. (A) The balanced accuracy of models trained with DRSCU with different train-test-split ratios, which are better than blind guessing (0.5 accuracy) even with extremely low train data ratio of 0.05. (B) The model performances (Balanced accuracy and F1 score) and ROC curve of models trained with different datasets: DR (DRSCU), DRT (DRSCU−Taxonomy), DRTC (DRSCU−Taxonomy−CDS Length). The ROC-AUC scores are shown.
To further verify the feasibility of creating practical tool in predicting human virus codon fitness score (HVCF score), the Leave-One-Out (LOO) train-test-split method is carried out to predict one sample each time with model trained with all other samples. Both balanced accuracy and recall score are optimised in hyper-parameters tuning. The performances including accuracy, recall score and ROC curve are shown in Fig. 3A, where DRTC generate the best performance compared to other datasets. No significant differences are observed between the recall optimisation and balanced accuracy optimisation, but the recall-optimised LOO training show slightly better performance. Moreover, the LOO performances could be better for hyper-parameters tunning with more computation (Supplementary Fig. 3A). The prediction performances to different virus families are later examined in Fig. 3B. Several important virus families are highlighted including Coronaviridae, Filoviridae, Flaviviridae, Orthomyxoviriade, Paramyxoviridae, Poxviridae, Retroviridae, where prediction towards all the important virus families show very low false negative rates and high accuracy. Additionally, all the viruses causing previous pandemics are predicted correctly in LOO predictions except ‘SARS coronavirus Tor2’, which has HVCF very close to correct ones (=0.482) (Supplementary Fig. 3B). These results indicate it is reliable to use all the samples for training a general RF model to predict host codon fitness of unknown viruses, and we use the DRTC-trained Recall-optimised RF model for the following applications (RSCU feature importance in Supplementary Fig. 4).

Leave-One-Out train-test-split method to prove possibility of generating predictive tool to VCF. The optimising score in hyper-parameters tuning is set either to balanced accuracy or recall scores. (A) Performances of all models trained by Leave-One-Out methods, including balanced accuracy and Recall score of different Datasets: DR, DRT, DRTC. The ROC curve with ROC-AUC scores, and the boxplot of predict probabilities are also shown. (B) The prediction performances including accuracy and false negative percentages (%) towards important virus families.
Host codon fitness shifting of SARS-CoV-2 in the COVID-19 pandemic
We considered HVCF score derived from the viruses’ RSCU and other features by the tree classification models as an indexing readout of VCF in human host, which we mainly used the HVCF score derived from the DRTC-trained Recall-optimised RF model to analyse the virus genome sequences.

Using DRTC-trained moel to predict HVCF scores of virus genome sequence data from environmental source. (A) Predict HVCF of virus genome sequence data that was harvested from human or non-human sources. All the data points are shown in supplemental Fig. 6A. (B) Predicted HVCF scores of SARS-CoV-2 in USA across timeline from April 2020 to December 2023.
Firstly, the HVCF scores of virus genome sequence data harvested from environmental sources of either human or not-human isolation host show no obvious differences in predicted labels between human-sourced or not-human-sourced virus genome sequence data. The model predicts non-human-sourced virus genome sequences as human-infecting. However, the predicted HVCF scores were lowered for the virus genome sequences from not-human source than human source for viruses including MERS-CoV, Zaire Ebolavirus, Zika virus, Influenza A virus, and Henipavirus (Fig. 4A). Similar outcomes were also observed with different sources species taxonomy (Supplementary Fig. 5A).
We also calculated and ranked the HVCF scores of SARS-CoV-2 genomes sequenced in the USA throughout the pandemic timeline (Fig. 5B, supplemental Fig. 6A). The first or the reference genome of SARS-CoV-2 (NC_04551247) receives a HVCF score 0.992, which is a very high probability. Complete SARS-CoV-2 genomes sequenced after the pandemic outbreak generally have a lower HVCF score. The lowest score of the HVCF is 0.740 (Jan 2021) and the highest one is 1.000 (Nov 2021), while the HVCF scores fluctuate approximately around 0.953 (Fig. 4B). The overall result shows that there does not exist an obvious host-shifting trend towards human-non-infectious in the evolution of the SARS-COV-2 virus during pandemic era. Interestingly, an increase in mean HVCF score is spotted between August and Nov 2021. After that, the mean HVCF score is gradually decreasing, and an obvious decline is spotted in December 2021. The mean HVCF is fluctuating after December 2021. To identify potential threating strain of viruses, we also ranked the predicted mean HVCF scores of different pango lineages, where top 20 pango lineages are showed in supplemental Fig. 6B. BF.5. recorded the highest mean HVCF of 0.992, followed by AY.49 with 0.987. Infectiousness probabilities of the virus binding to other hosts are also investigated (Supplemental Fig. 6D). SARS-CoV-2 has consistent and significantly high VCF to human and vertebrates while other hosts maintain low infectiousness probabilities (< 0.138), suggesting potential risks to infect other vertebrate species but not significant risks to other hosts. That is, the VCF of SARS-CoV-2 has been remaining in the range of human and vertebrates throughout the pandemic without a noticeable host-shifting trend.
Simulation of SARS-CoV-2 mutation process starting from other betacoronavirus through HVCF gradients
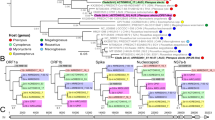
To unveil the unknown genetic links between SARS-CoV-2 and human-non-infectious Betacoronavirus, we used HVCF readouts as gradient scores to simulate codon-mutation-driven (including codon substitutions, codon addition, codon deletion) evolution-like process between two viruses for False-to-True VCF jump (i.e., human-non-infectious to human-infectious jump). In the simulation, each of the human-non-infectious Betacoronavirus is taken to perform a step-by-step codon-mutation to screen efficient codon-mutations evolving till SARS-CoV-2’s HVCF score (see more details in the method section). At each step, it is required to generate a mutated codon profiles such that it has a possibly highest HVCF score and the best correlation to SARS-CoV-2’s RSCU matrix (Forward Mutation Path). Similarly on the other hand, SARS-CoV-2 is also taken to screen efficient mutations to evolve into a target Betacoronavirus, then the mutation path is reversed after to generate a False-to-True result (Backward Mutation Path). These paths are shown in Fig. 5A.

Prediction and analysis of SARS-CoV-2 codon fitness simulation processes using codon mutations from other Betacoronavirus. (A) Predicted SARS-CoV-2 codon fitness simulation processes using codon mutations from other Betacoronavirus. (B) Simulation efficiencies in both HVCF changes and correlation coefficient changes of different Betacoronavirus are shown. (C) Analysis of codon mutations in codon fitness simulation processes from Tylonycteris bat coronavirus HKU4 to SARS-CoV-2 with both abundant codon mutations and codon abundancy changes (another figure format in Supplemental Fig. 7).
From the construction of these putative simulation processes, we can see that the Tylonycteris bat coronavirus HKU4 (NC_009019) has the highest level of efficiency to ‘evolve’ into SARS-CoV-2 equivalent VCF according to the HVCF score changes per mutation compared to other Betacoronavirus reference genomes (Fig. 5B). More importantly, the compositions of both the forward and backward mutation path are also studied, we found that UUA(Leu)-to-CUC(Leu) and GAU(Asp)-to-GAC(Asp) mutations are significantly abundant in forward mutation path, while UAU(Tyr)-to-UAC(Tyr) and GCU(Ala)-to-GCC(Ala) are significantly abundant in reverse mutation path (Fig. 5C). Besides, GGU(Gly)-to-GGA(Gly) is abundant in both paths. Based on the simulation results, it is predicted that significant changes in codon usage for the amino acids Leu, Asp, Tyr, Ala, and Gly may be spotted in the intermediate strain of the viruses, if SARS-CoV-2 is evolved from intermediate viruses related to Tylonycteris bat coronavirus HKU4. Interestingly, the third nucleotide mutations are spotted in all those mutations, especially U-to-C mutation, where most of those mutations are synonymous mutations. Similar results are observed in codon usage changes which multiple U-ended codons are significantly decreased in abundance including GCU(Ala), UAU(Tyr), GGU(Gly), CGU(Arg), CCU(Pro) in both paths, while multiple C-ended codons are significantly increased in abundance like CUC(Leu), GCC(Ala), UAC(Tyr) besides GGA(Gly). This finding provides clues in virus evolution for searching human infectious intermediate viruses from environmental sampling.
Discussion
Our study showed that the viral genomes with different host ranges have distinct codon usage biases, especially the A/U-ended codons (3rd nt) that have distinct characteristics compared to the G/C-ended codons excepts UUG (Leu) and AGG (Arg). This evidence supports our finding that the wobble position of codons is significantly important in virus host codon fitness and host ranges although the underlying mechanism is largely unknown. In addition, the significant abundancy of the A/U-ended codons, especially the A-ended codons implies that the transcripts from viruses infecting human are potentially more susceptible to A-to-I editing on the wobble position, but the real outcomes need further investigation. This study verifies our hypothesis that machine learning can detect distinguishable boundaries of codon usage biases from virus genomes having different host ranges, and codon usage biases have predictive power to virus codon fitness in host ranges and the underlying probabilities of infectiousness.
This modelling methodology has an advantage in its generalisability because it purely relies on the codon usage biases of viral genomes and other general genomic characteristics regardless the diversity in virus-host interactions of different viruses such as expression regulation, protein interactions, cellular immunity, tRNA pool regulation et al. It overcomes the dilemma that the real micro-environment of virus-host interactions are complicated, and the significant diversity in different individuals from the same host type. Incomplete virus genome sequence data can also generate codon usage biases for sub-optimal prediction, expanding the range of application scenarios. This new way of predicting virus host codon fitness provides new insight into how we understand virus host ranges complementing the current major research focus on host entry of virus (i.e. Spike-membrane protein interaction)48,49,50. Moreover, data mining of using codon usage biases to represent coding sequences is significantly more computationally efficient compared to other methods such as natural language processing (NLP)51. The sample quantity limitation and imbalance need improvement when using only virus reference genomes, especially the imbalances in virus sample amount of different host ranges (i.e. human virus vs. not-human virus). This may be possible to overcome with sample synthetic algorithms or generative deep learning networks to simulate virus genomes. Additionally, the representation of virus genome through summing codons counts within all gene CDS may not be biased towards the gene CDS of longer length. This may be improved through other embedding algorithms or through derivatives like Transformer model.
The concept of the human virus codon fitness score (HVCF score), sourced from the decision tree models, has the potential of monitoring the dynamics of virus host codon fitness shifting, which could help assess the potential host codon fitness and host ranges of emerging viruses which may cause disease outbreaks or even pandemic. However, there is still no evidence supporting that this readout of VCF is correlating to virus lethality to host or virus infection outcomes. The accuracy of predicting different types of viruses may be different because the limitation and imbalance of training data. This results with HVCF scores of human-sourced and not-human-sourced viruses in different viruses suggest that this modelling method has potential to development accurate prediction tools to monitor virus host codon fitness shifting accordingly. In the SARS-CoV-2 pandemic analysis, the HVCF score remains in the similar level suggesting that the current attenuation in COVID-19 mortality rate is less likely leading to gradual vanish, but it remains a long persisting disease52,53. Besides, this method could also identify the potential threating viruses with routine virus genome sequencing of environmental sampling (e.g., bats, mice, rats et al.). The deficiency of this method is the difficulty in acquiring new samples to build models in species-specific scope (i.e. cats, dogs et al.) because it is unethical and dangerous if infecting various species with various specific viruses.
More importantly, this study proposes an innovative method to simulate mutational process between two viruses (original virus and target virus). Comparisons among different simulation processes could help identify the relations of VCF between the two viruses. SARS-CoV-2 and other Betacoronavirus are taken as example by this study, where Tylonycteris bat coronavirus HKU4 stood out closely relating to SARS-CoV-2 in terms of VCF. Further studies on the simulation processes conclude that codon-related mutation signatures have significant abundancy in synonymous mutations, especially with U-to-C mutations in wobble position, of the Leu, Asp, Tyr, Ala, Gly. Moreover, this finding of abundant synonymous mutations in the simulation also demonstrates the importance of synonymous mutations in virus evolution. This method provides guidelines for searching evolutional relations between viruses and guidance for virus traceability research. The predicted probabilities generated from the RF models are discontinuous due to the nature of the algorithm leading to inefficiency and inaccuracy in predicting impacts of different codon-related mutations, which may be overcome with deep learning algorithms in the future work. Additionally, RF modelling may have limitations in integrating diverse data types, predicting values outside the range of the training data, and producing discontinuous predicted probabilities, among other challenges. Deep learning methods often outperform traditional models in accuracy due to their ability to capture complex patterns and relationships in diverse datasets, while deep learning also excels at integrating heterogeneous types of data, which is particularly relevant for predicting virus-host interactions.
Methods
Acquisition of virus genome reference sequences
The accession IDs of all the virus genome reference sequences (RefSeq) and their corresponding host range label (under label ‘Host’) are acquired from the ‘Viral genome browser’ of National Center for Biotechnology Information (NCBI)54. The accession IDs was used to download coding sequences later through Biopython toolkit. Viruses with limited sample count in host ranges are ignored in later studies except ‘Human’, ‘Vertebrates’, ‘Invertebrates’, ‘Land plants’, and ‘Bacteria’, but they are remained in the dataset as negative samples throughout the study. The incomplete viral genome sequences (labelled as ‘Incomplete’ in ‘RefSeq type’) were discarded throughout the study. The multi-partite virus which has multiple NCBI accession IDs for multiple genome segments are summarised as the same virus, which all the genes in different genome segments are considered in the same virus. Total 10,820 samples were retrieved with 488 Human samples, 1758 Vertebrates samples, 1851 Invertebrates samples, 1763 Land plants samples, and 4041 Bacteria samples.
Acquisition of other virus genome sequences
For testing the trained RF model, complete virus genome sequence data of MERS-CoV, Zaire Ebolavirus, West Nile virus, Zika virus, Orthohantavirus, Influenza A virus, Henipavirus, Lyssavirus Rabies, and SARS-CoV-2 were acquired from NCBI database. The accession IDs of all those virus genomes used by this study are acquired from the NCBI Virus database55, which other information such as ‘Host’, ‘Pangolin’ et al. were also downloaded there (incomplete genomes were discarded), and only the viruses has ‘Host’ label were downloaded. The accession IDs was used to download coding sequences through Biopython toolkit, and the viruses are separated into two groups based on whether they are labelled as ‘Homo Sapiens’ in ‘Host’. Total 639 genome sequences of MERS-CoV (256 human-sourced, 383 non-human-sourced), 563 genome sequences of Zaire Ebolavirus (435 human-sourced, 128 non-human-sourced), 1823 genome sequences of West Nile virus (137 human-sourced, 1686 non-human-sourced), 240 genome sequences of Zika virus (208 human-sourced, 32 non-human-sourced), 826 genome sequences of Orthohantavirus (142 human-sourced, 684 non-human-sourced), 614 genome sequences of Influenza A virus (131 human-sourced, 483 non-human-sourced), 55 genome sequences of Henipavirus (35 human-sourced, 20 non-human-sourced), 1862 genome sequences of Lyssavirus Rabies (30 human-sourced, 1832 non-human-sourced), and 755151 genome sequences of SARS-CoV-2 (all human-sourced) were retrieved. The WHO Name information related to SARS-CoV-2 was acquired from ‘cov-lineages.org’ database, which is assigned to the samples according to their pangolin labels56.
Calculation for RSCU
The Relative Synonymous Codon Usages (RSCU) of the virus genome, as readouts of codon usage biases, are calculated based on codon counts and amino acid counts of coding sequences according to their definition proposed in previous publication31,32. All the coding sequences, or CDS, in a virus genomes (either mono-partite or multi-partite) were converted into counts of each codon and counts of each amino acid. The counts of the same codons from all CDS in a virus genome were summed to represent codon counts for the whole genome, which same method was applied to the counts of amino acids. RSCU of each codon were generated based on each codon count and respective amino acid count. The codons for 1-box amino acids, UGG (Try) and AUG (Met) are ignored due to unchanged values (= 1). The stop codons UAA, UAG and UGA are also ignored because they were not relevant to translation efficiency. Thus, the RSCU dataset (or DR) consists of total 59 codon features. As the start and stop codons are discarded, our analysis was purely focused on the codon usage biases of the coding sequences in translation efficiency.
Dimensionality reduction of the RSCU data matrix
The raw RSCU data matrix, which virus genomes as samples and codons as features, was first normalised through the Z-score Normalisation method. The normalised RSCU data matrix was later compressed by Principal Component Analysis (PCA) method, where the cut-off threshold was set as 1.0 (no loss in explained variance). This method can compress the normalised data without loss of the variances by removing the redundant features. Dimensional reduction analysis was performed on the normalised and compressed data using the Uniform Manifold Approximation and Projection (UMAP) algorithm45. The raw RSCU data matrix was then transposed to study the viral codon behaviours, which codon labels become samples and all the virus genomes become features. The transposed RSCU data matrix was applied with the same dimensional reduction pipeline as above.
Independent T-test
The independent T-test was performed through the python package Scipy. The RSCU of each codon was analysed by comparing host and non-host virus genomes (i.e. human virus vs. not-human virus), which results with p-value smaller than 0.05 will be considered as significantly different.
Random forest machine learning
The train datasets were resampled by the Synthetic Minority Oversampling Technique (SMOTE)46 to overcome sample imbalance. The Random Forest models were trained with SMOTE-resampled train datasets with Scikit-learn57, when Balance accuracy, F1 score, Recall scores and ROC-AUC scores were used as the standard of the model performance due to sample imbalances. The open-source OPTUNA framework was used for hyper-parameters tunning with specific trials for different scenarios (20 or 50 ‘n_trials’)58. For parameters suggested in OPTUNA, ‘n_estimators’ was set 2 ~ 300; ‘criterion’ was set between ‘entropy’ and ‘gini’; ‘min_samples_split’ was set 2 ~ 20; ‘min_samples_leaf’ was set 1 ~ 10; ‘max_features’ was set between ‘sqrt’ and ‘log2’. The ‘class_weight’ was set as ‘balanced’, and 20-fold cross validation was applied for hyper-parameters tunning. The target score in OPTUNA tuning was set as ‘balanced_accuracy’, which the mean of balanced accuracy in 20-fold cross validation was served as the standard to find the optimal set of hyper-parameters.
The predicted probabilities from the models’ predictions were considered as the readout of virus codon fitness (VCF) in a specific host, which is computed from embedded function of Scikit-learn. The feature importance metrics are also computed from embedded function of Scikit-learn.
Selection of additional features
Besides RSCUs, other feature datasets were used to achieve better machine learning prediction. Datasets ‘Codon%’ (DCodon%) and ‘AminoAcid%’ (DAminoAcid%) were simply calculated with percentages of different codons or amino acids in total codon count of amino acid count of virus genome. Stop codons were ignored in both DCodon% and DAminoAcid% (thus 61 features in DCodon% and 20 features in DAminoAcid%, stop codons were not included). Dataset ‘ATGC%’ (DATGC%) was simply calculated with frequency of each nucleotide (A%, U%, G%, C%, AU%, GC%), which AU% and GC% were calculated by summing A%/U% and G%/C%. Dataset ‘Start-Stop Codon%’ (DStartStopCodon%) is the calculated with frequency of the start codon (AUG%) in all the CDS of virus genomes, and the frequency of each stop codons (UAA%, UAG%, UGA%) in all the CDS of virus genomes. Dataset ‘CDS Length’ (DCDS Length) consists of features genome length, Concatenated CDS length, CDS count, and the mean and standard deviation of CDS length, which Concatenated CDS length is the sum of all CDS length. Dataset ‘HumanCorr’ (DHumanCorr) is correlation coefficients calculated between virus RSCU and Human reference RSCU, which is Human reference RSCU acquired from the CoCoPUTs database59. Dataset ‘HumanCorr(AA)’ (DHumanCorrAA) dataset is correlation coefficients calculated among between virus RSCU and Human standard RSCU from each Amino Acids. Met (M) and Trp (W) as well as Stop codons were ignored. Dataset ‘Partite’ (DPartite) consist of the virus classifications of either mono-partite or multi-partite. Dataset ‘Taxonomy’ (DTaxonomy) consist of tertiarily encoded data based on taxonomy information acquired from the NCBI Taxonomy database with Realm (or Clade), Kingdom, Phylum, Class, Order. The positive samples were labelled as ‘1’ and negative samples were labelled as ‘-1’, which unknown samples were labelled as ‘0’.
For searching additional features beneficial to model performance, above datasets were individually or combinatively added to RSCU dataset before training models in different conditions (i.e. different train-test ratio, different hyperparameter tuning strategies et al.), which the trained models were evaluated and compared with Balanced accuracy after parameters optimisation (Supplemental Fig. 2). 50 trials were set in OPTUNA hyper-parameters tuning. As consequences, Taxonomy dataset and CDS Length dataset additional to RSCU dataset increases Balanced accuracy of the trained models, and three datasets were generated: DR (DRSCU), DRT (combination of DRSCU and DTaxonomy), and DRTC (combination of DRSCU, DTaxonomy, and DCDS Length).
Leave-One-Out machine learning
To further confirm reliability of using RSCU and other features in predicting HVCF scores of different host-ranged viruses, the Leave-One-Out (LOO) method was carried out, which all other samples were used to train a RF model in predicting one test sample. The same process was carried out separately to all the samples, and only 5 trials were used in OPTUNA hyper-parameters tunning (other OPTUNA were identical as those from ‘Random forest machine learning’ in method section). Model performances such as Balanced accuracy and Recall score were generated with summary of total 10,820 predictions (correct/wrong predictions for 10820 samples or models). The models with wrong predictions were later re-trained with 50 trials setting in OPTUNA hyper-parameters tunning to see whether it will have correct predictions (Supplemental Fig. 3).
Simulation for SARS-CoV-2 mutation process
The predicted probability from the DRTC-trained Recall-optmised RF model trained with all 10,820 samples was considered as the readout of human virus codon fitness score (HVCF score) because it has best prediction performance. To simulate codon fitness mutations between two viruses, the start-point virus and the end-point virus were determined between SARS-CoV-2 and a target Betacoronavirus for either the Forward or Backward mutation simulation. The Forward mutation is from the target Betacoronavirus to SARS-CoV-2, while the Backward mutation is from SARS-CoV-2 to the target Betacoronavirus. At each mutation step in the simulation process, every possible mutation was applied to the codon count compositions of the virus, including all possible substitution (i.e. AAA→AAG), addition (i.e. +AAA), or deletion (i.e. −AAA) of codons. The DRSCU and DCDS Length of the updated virus codon compositions were re-calculated except for the DTaxonomy (remained as Coronaviridae). The new HVCF score was then predicted with the updated DRTC. Among new HVCF scores derived from all the possible mutations, the simulated mutation generating the lowest/highest HVCF score (depending on simulation direction, Forward or Backward) was selected. When multiple mutations have the same lowest/highest HVCF score, the additional analysis of correlation coefficient was calculated between updated DRSCU and DRSCU of the end-point virus (SARS-CoV-2 in the Forward path or the target Betacoronavirus in the Backward path). The simulated mutation generating the best correlation coefficient was selected.
Similar to the gradient descent, this process was step-by-step repeated until reaching the HVCF score of the end-point virus. In some cases, the simulation may reach a stagnation because of possibility of mutually contradictory mutations (i.e., AAA→AAG then AAG→AAA). To avoid such meaningless loop stagnation, mutually contradictory mutations were forbidden in the simulation process. For instance, if an ongoing simulation process has AAA→AAG, then mutation AAG→AAA must be excluded in the subsequent simulation.
Data availability
The accessions IDs of virus genomes RefSeq used for model training were downloaded from NCBI Virus Genomes Resource https://www.ncbi.nlm.nih.gov/genomes/GenomesGroup.cgi. The accessions IDs of other virus genomes were downloaded from NCBI Virus https://www.ncbi.nlm.nih.gov/labs/virus/vssi/#/ according to their virus search filter. The pango lineage classification of SARS-CoV-2 were downloaded from https://cov-lineages.org/lineage_list.html. The reference human codon usage was downloaded from https://dnahive.fda.gov/dna.cgi?cmd=codon_usage&id=537&mode=cocoputs.
References
Karthikeyan, S. et al. Wastewater sequencing reveals early cryptic SARS-CoV-2 variant transmission. Nature 609, 101–108. https://doi.org/10.1038/s41586-022-05049-6 (2022).
Kia, P. et al. Genomic characterization of SARS-CoV-2 from Uganda using minion nanopore sequencing. Sci. Rep. 13, 20507. https://doi.org/10.1038/s41598-023-47379-z (2023).
Barbe, L. et al. SARS-CoV-2 Whole-Genome sequencing using Oxford nanopore technology for variant monitoring in wastewaters. Front. Microbiol. 13, 889811. https://doi.org/10.3389/fmicb.2022.889811 (2022).
Lu, R. et al. Genomic characterisation and epidemiology of 2019 novel coronavirus: implications for virus origins and receptor binding. Lancet 395, 565–574. https://doi.org/10.1016/S0140-6736(20)30251-8 (2020).
Hadfield, J. et al. Nextstrain: real-time tracking of pathogen evolution. Bioinformatics 34, 4121–4123. https://doi.org/10.1093/bioinformatics/bty407 (2018).
Wang, T., Yu, Z. G. & Li, J. CGRWDL: alignment-free phylogeny reconstruction method for viruses based on chaos game representation weighted by dynamical Language model. Front. Microbiol. 15, 1339156 (2024).
Tang, R., Yu, Z., Li, J. & Kinn An alignment-free accurate phylogeny reconstruction method based on inner distance distributions of k-mer pairs in biological sequences. Mol. Phylogenet. Evol. 179, 107662 (2023).
Yang, W. F., Yu, Z. G. & Anh, V. Whole genome/proteome based phylogeny reconstruction for prokaryotes using higher order Markov model and chaos game representation. Mol. Phylogenet. Evol. 96, 102–111 (2016).
Xie, X. H., Yu, Z. G., Han, G. S., Yang, W. F. & Anh, V. Whole-proteome based phylogenetic tree construction with inter-amino-acid distances and the conditional geometric distribution profiles. Mol. Phylogenet. Evol. 89, 37–45 (2015).
Irving, A. T., Ahn, M., Goh, G., Anderson, D. E. & Wang, L. F. Lessons from the host defences of bats, a unique viral reservoir. Nature 589, 363–370. https://doi.org/10.1038/s41586-020-03128-0 (2021).
de Wit, E., van Doremalen, N., Falzarano, D. & Munster, V. J. SARS and MERS: recent insights into emerging coronaviruses. Nat. Rev. Microbiol. 14, 523–534. https://doi.org/10.1038/nrmicro.2016.81 (2016).
Zell, R. et al. Cocirculation of swine H1N1 influenza A virus lineages in Germany. Viruses 12, 762. https://doi.org/10.3390/v12070762 (2020).
Starick, E. et al. Reassorted pandemic (H1N1) 2009 influenza A virus discovered from pigs in Germany. J. Gen. Virol. 92, 1184–1188. https://doi.org/10.1099/vir.0.028662-0 (2011).
Wang, L. F. & Eaton, B. T. Bats, civets and the emergence of SARS. Curr. Top. Microbiol. Immunol. 315, 325–344. https://doi.org/10.1007/978-3-540-70962-6_13 (2007).
Graham, R. L. & Baric, R. S. Recombination, reservoirs, and the modular Spike: mechanisms of coronavirus cross-species transmission. J. Virol. 84, 3134–3146. https://doi.org/10.1128/JVI.01394-09 (2010).
Guan, Y. et al. Isolation and characterization of viruses related to the SARS coronavirus from animals in Southern China. Science 302, 276–278. https://doi.org/10.1126/science.1087139 (2003).
Ruohan, W., Xianglilan, Z. & Jianping, W. Shuai Cheng, L. DeepHost: phage host prediction with convolutional neural network. Brief. Bioinform. 23, bbab385 (2022).
Bai, Z. et al. Identification of bacteriophage genome sequences with representation learning. Bioinformatics 38, 4264–4270 (2022).
Martinez, M. A., Jordan-Paiz, A., Franco, S. & Nevot, M. Synonymous virus genome recoding as a tool to impact viral fitness. Trends Microbiol. 24, 134–147. https://doi.org/10.1016/j.tim.2015.11.002 (2016).
Battles, M. B. & McLellan, J. S. Respiratory syncytial virus entry and how to block it. Nat. Rev. Microbiol. 17, 233–245. https://doi.org/10.1038/s41579-019-0149-x (2019).
Jackson, C. B., Farzan, M., Chen, B. & Choe, H. Mechanisms of SARS-CoV-2 entry into cells. Nat. Rev. Mol. Cell. Biol. 23, 3–20. https://doi.org/10.1038/s41580-021-00418-x (2022).
Minkoff, J. M. & tenOever, B. Innate immune evasion strategies of SARS-CoV-2. Nat. Rev. Microbiol. 21, 178–194. https://doi.org/10.1038/s41579-022-00839-1 (2023).
Chen, F. & Yang, J. R. Distinct codon usage bias evolutionary patterns between weakly and strongly virulent respiratory viruses. iScience 25, 103682. https://doi.org/10.1016/j.isci.2021.103682 (2022).
Chen, F. et al. Dissimilation of synonymous codon usage bias in virus-host Coevolution due to translational selection. Nat. Ecol. Evol. 4, 589–600. https://doi.org/10.1038/s41559-020-1124-7 (2020).
Yu, C. et al. Hepatitis B virus (HBV) codon adapts well to the gene expression profile of liver cancer: an evolutionary explanation for HBV’s oncogenic role. J. Microbiol. 60, 1106–1112. https://doi.org/10.1007/s12275-022-2371-x (2022).
Arella, D., Dilucca, M. & Giansanti, A. Codon usage bias and environmental adaptation in microbial organisms. Mol. Genet. Genom. 296, 751–762. https://doi.org/10.1007/s00438-021-01771-4 (2021).
Yang, S., Liu, Y., Wu, X., Cheng, X. & Wu, X. Synonymous codon pattern of Cowpea mild mottle virus sheds light on its host adaptation and genome evolution. Pathogens 11, 419. https://doi.org/10.3390/pathogens11040419 (2022).
Hernandez-Alias, X., Benisty, H., Schaefer, M. H. & Serrano, L. Translational adaptation of human viruses to the tissues they infect. Cell. Rep. 34, 108872. https://doi.org/10.1016/j.celrep.2021.108872 (2021).
Gale, M. Jr., Tan, S. L. & Katze, M. G. Translational control of viral gene expression in eukaryotes. Microbiol. Mol. Biol. Rev. 64, 239–280. https://doi.org/10.1128/MMBR.64.2.239-280.2000 (2000).
Balvay, L., Lopez Lastra, M., Sargueil, B., Darlix, J. L. & Ohlmann, T. Translational control of retroviruses. Nat. Rev. Microbiol. 5, 128–140. https://doi.org/10.1038/nrmicro1599 (2007).
Sharp, P. M., Tuohy, T. M. & Mosurski, K. R. Codon usage in yeast: cluster analysis clearly differentiates highly and lowly expressed genes. Nucleic Acids Res. 14, 5125–5143. https://doi.org/10.1093/nar/14.13.5125 (1986).
Puigbo, P., Bravo, I. G. & Garcia-Vallve, S. CAIcal: a combined set of tools to assess codon usage adaptation. Biol. Direct 3, 38. https://doi.org/10.1186/1745-6150-3-38 (2008).
dos Reis, M., Savva, R. & Wernisch, L. Solving the riddle of codon usage preferences: a test for translational selection. Nucleic Acids Res. 32, 5036–5044. https://doi.org/10.1093/nar/gkh834 (2004).
Ji, W., Wang, W., Zhao, X., Zai, J. & Li, X. Cross-species transmission of the newly identified coronavirus 2019-nCoV. J. Med. Virol. 92, 433–440. https://doi.org/10.1002/jmv.25682 (2020).
Yao, H., Chen, M. & Tang, Z. Analysis of synonymous codon usage bias in flaviviridae virus. Biomed. Res. Int. 2019, 5857285. https://doi.org/10.1155/2019/5857285 (2019).
Tao, J. & Yao, H. Comprehensive analysis of the codon usage patterns of polyprotein of Zika virus. Prog. Biophys. Mol. Biol. 150, 43–49. https://doi.org/10.1016/j.pbiomolbio.2019.05.001 (2020).
Cheng, S., Wu, H. & Chen, Z. Evolution of transmissible gastroenteritis virus (TGEV): A codon usage perspective. Int. J. Mol. Sci. 21, 898. https://doi.org/10.3390/ijms21217898 (2020).
Pinto, R. M. et al. Hepatitis A virus codon usage: implications for translation kinetics and capsid folding. Cold Spring Harb. Perspect. Med. 8, 781. https://doi.org/10.1101/cshperspect.a031781 (2018).
Deb, B., Uddin, A. & Chakraborty, S. Analysis of codon usage of horseshoe Bat hepatitis B virus and its host. Virology 561, 69–79. https://doi.org/10.1016/j.virol.2021.05.008 (2021).
Hou, W. Characterization of codon usage pattern in SARS-CoV-2. Virol. J. 17, 138. https://doi.org/10.1186/s12985-020-01395-x (2020).
Gu, H., Chu, D. K. W., Peiris, M. & Poon, L. L. M. Multivariate analyses of codon usage of SARS-CoV-2 and other betacoronaviruses. Virus Evol. 6, veaa032. https://doi.org/10.1093/ve/veaa032 (2020).
Suomalainen, M. & Greber, U. F. Virus infection variability by single-cell profiling. Viruses 13, 1568. https://doi.org/10.3390/v13081568 (2021).
Smatti, M. K. et al. Viruses and autoimmunity: A review on the potential interaction and molecular mechanisms. Viruses 11, 762. https://doi.org/10.3390/v11080762 (2019).
Novoa, E. M., Jungreis, I., Jaillon, O. & Kellis, M. Elucidation of codon usage signatures across the domains of life. Mol. Biol. Evol. 36, 2328–2339. https://doi.org/10.1093/molbev/msz124 (2019).
McInnes, L., Healy, J. & Melville, J. UMAP: Uniform manifold approximation and projection for dimension reduction. https://ui.adsabs.harvard.edu/abs/2018arXiv180203426M (2018).
Lemaître, G., Nogueira, F. & Aridas, C. K. Imbalanced-learn: A python toolbox to tackle the curse of imbalanced datasets in machine learning. J. Mach. Learn. Res. 18, 559–563 (2017).
Wu, F. et al. A new coronavirus associated with human respiratory disease in China. Nature 579, 265–269. https://doi.org/10.1038/s41586-020-2008-3 (2020).
Xu, C. et al. Conformational dynamics of SARS-CoV-2 trimeric Spike glycoprotein in complex with receptor ACE2 revealed by cryo-EM. Sci. Adv. 7, 5575. https://doi.org/10.1126/sciadv.abe5575 (2021).
Walls, A. C. et al. Structure, function, and antigenicity of the SARS-CoV-2 spike glycoprotein. Cell 181, 281–292. https://doi.org/10.1016/j.cell.2020.02.058 (2020).
Harrison, A. G., Lin, T. & Wang, P. Mechanisms of SARS-CoV-2 transmission and pathogenesis. Trends Immunol. 41, 1100–1115. https://doi.org/10.1016/j.it.2020.10.004 (2020).
He, Y., Shen, Z., Zhang, Q., Wang, S. & Huang, D. A survey on deep learning in DNA/RNA motif mining. Brief. Bioinform. 22, 229. https://doi.org/10.1093/bib/bbaa229 (2021).
Collaborators, C. E. M. Estimating excess mortality due to the COVID-19 pandemic: a systematic analysis of COVID-19-related mortality, 2020-21. Lancet 399, 1513–1536. https://doi.org/10.1016/S0140-6736(21)02796-3 (2022).
Crook, H., Raza, S., Nowell, J., Young, M. & Edison, P. Long covid-mechanisms, risk factors, and management. BMJ 374, n1648. https://doi.org/10.1136/bmj.n1648 (2021).
Brister, J. R., Ako-Adjei, D., Bao, Y. & Blinkova, O. NCBI viral genomes resource. Nucleic Acids Res. 43, D571–D577. https://doi.org/10.1093/nar/gku1207 (2015).
Hatcher, E. L. et al. Virus variation resource—improved response to emergent viral outbreaks. Nucleic Acids Res. 45, D482–D490. https://doi.org/10.1093/nar/gkw1065 (2017).
Rambaut, A. et al. A dynamic nomenclature proposal for SARS-CoV-2 lineages to assist genomic epidemiology. Nat. Microbiol. 5, 1403–1407. https://doi.org/10.1038/s41564-020-0770-5 (2020).
Pedregosa, F. et al. Scikit-learn: machine learning in Python. J. Mach. Learn. Res. 12, 2825–2830 (2011).
Akiba, T., Sano, S., Yanase, T., Ohta, T. & Koyama, M. Proceedings of the 25th ACM SIGKDD International Conference on Knowledge Discovery & Data Mining 2623–2631.
Alexaki, A. et al. Codon and codon-pair usage tables (CoCoPUTs): facilitating genetic variation analyses and Recombinant gene design. J. Mol. Biol. 431, 2434–2441. https://doi.org/10.1016/j.jmb.2019.04.021 (2019).
Acknowledgements
This research was fully supported by Data Science Institute (DSI) of the Faculty of Engineering and Information Technology (FEIT) of University of Technology Sydney (UTS) with high-performance computing resources and wonderful research environment. I also thank China Scholarship Council (CSC) Scholarship, Sydney Consulate General of China, and also Graduate Research School (GRS) of UTS to financially support my PhD study.I am grateful for advice and supports from Dr Xuan Zhang of Prof Jinyan Li’s research group, Ms Pattarasiri Rangsrikitphoti and Ms Sara Terer from Prof Gyorgy Hutvagner’s research group, and Dr Yanxiao Gao from Prof Jinling Tang’s research group.
Author information
Authors and Affiliations
Contributions
S.S. proposed the research idea and executed the research project, and he wrote the main manuscript text and prepared all the figures. Z.N., T.L. and P.P. provided important insights and suggestions in methodology. J.L. and G.H. are the supervisors of this project. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Su, S., Ni, Z., Lan, T. et al. Predicting viral host codon fitness and path shifting through tree-based learning on codon usage biases and genomic characteristics. Sci Rep 15, 12251 (2025). https://doi.org/10.1038/s41598-025-91469-z
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-91469-z
Keywords
This article is cited by
-
Deep learning decodes species-specific codon usage signatures in Brassica from coding sequences
Scientific Reports (2025)
