
Abstract
To examine the thalassemia genotypes and distribution among pregnant women in Shenzhen, as well as the diagnostic value of HbA2 in thalassemia screening, in order to provide scientific evidence for thalassemia prevention and control in this region. From January 2018 to June 2024, Shenzhen recorded 3243 cases of suspected thalassemia carriers. HbA2 was detected by hemoglobin electrophoresis analysis. The deletions of α-thalassemia mutation by gap-polymerase chain reaction (gap-PCR), and the non-deletion α‐thalassemia mutations and β‐thalassemia mutations were detected by reverse dot-blot hybridization (RDB-PCR). The receiver operating characteristic curve (ROC) was utilized to analyze the diagnostic value of HbA2 for pregnant women’s thalassemia. A total of 656 carriers were detected in 3243 pregnant women with suspected thalassemia carriers, with a positive detection rate of about 20.22%. 459 cases were defined to be α-thalassemia, with the main type of −−SEA/αα (47.71%). 170 cases were defined to be β‐thalassemia, with the main type of βCD41–42/βN (31.76%). 27 genotypes of αβ-thalassemia were noted in pregnant women. There was significant difference of HbA2 level between the pregnant women with different types of thalassemia and healthy controls (all P < 0.001). ROC curve analysis showed that the sensitivities of HbA2 for α‐thalassemia, β-thalassemia and αβ-thalassemia were 89.7%, 96.5% and 96.2%, with the optimal cut-off values of 2.71%, 3.71% and 3.86%, respectively, the specificities were 26.6%, 98.5% and 94.5%, and the area under the curve were 0.587, 0.979 and 0.972, respectively. The thalassemia genotypes of pregnant women in Shenzhen are diverse. It is necessary to strengthen the prevention and control measure of thalassemia in order to reduce birth defects and improve birth quality.
Similar content being viewed by others
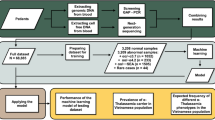

Introduction
Thalassemia is a common hereditary blood disease, which is an abnormal hemoglobin disorder caused by a mutation or deletion of the bead protein gene that results in a disruption of the production of bead protein, and is most common in α-degenerate and β-degenerate anemias1. Guangxi and Guangdong are areas with a high incidence of thalassemia in China, the incidence rates of α- and β-thalassemia in Guangxi population were 12.51–15.5% and 5.11–6.64% respectively2,3, and it is 9.46% in Guangdong2. Shenzhen is located in Guangdong Province, which has a relatively high rate of carrier and prevalence of thalassemia and thus increases the risk of births of children with moderate-to-severe anemia, as inferred from population exchanges and migrations. This study aims to analyze the results of genetic type testing and hematological characteristics of pregnant women in Shenzhen to explore the diagnostic value of HbA2 in screening pregnant women for thalassemia, and to provide reference for further improvement of prevention and control of thalassemia and clinical consultation in this region.
Method and materials
Study subjects
This study was approved by the ethic committee of Shenzhen Second People’s Hospital (Approval No:2024-339-01YJ) and all participants were informed and signed a written informed consent. During the period from January 1, 2018 to June 30, 2024, 3243 cases of pregnant women suspected to be thalassemia carriers were collected in Shenzhen Second People’s Hospital, the age distribution of these subjects ranged from 20 to.
47-year-old and all were with Shenzhen pregnant women. Inclusion criteria for pregnant women with suspected thalassemia: the pregnant woman has a mean cell volume (MCV) of < 82 fl. and/or a mean cell Hb (MCH) of < 27 pg on routine blood tests; Abnormal hemoglobin electrophoresis (In electrophoresis, the following patterns are considered abnormal:1.Presence of abnormal hemoglobin bands: If bands other than normal hemoglobin appear on the electrophoresis pattern, such as Hb S, Hb C or Hb H and others, these indicate the possibility of abnormalities; 2. Abnormal HbA₂ levels: In normal adults, the level of HbA₂ is 2.5–3.5%. If the HbA₂ level is HbA2 > 3.5% or HbA2 < 2.5%, it is considered abnormal; 3. Abnormal HbF levels: In normal adults, the level of HbF should be less than 2.3%. If the HbF level exceeds 2.3%, it may indicate abnormalities; 4. Abnormal HbA levels: Normally, HbA accounts for 95–98% of total hemoglobin, a significant decrease or absence of HbA may suggest abnormal), pregnant women with a parent or sibling who is a carrier of the thalassemia.
Hematological analysis
Approximately 2 ml of peripheral blood was collected from each subject and anticoagulated with EDTA-K2 for Hematological Analysis. DNA was extracted with an automatic nucleic acid extractor at Kaishuo Biological Co., Ltd (Xiamen, China). we assessed the purity and concentration of the DNA by the Nanodrop 2000, with purity range is 1.8 –2.0 (A260/A280), with concentration range is 2 –50 ng/µl. All gene kits were purchased from Yilifang Biological Technology Company (Shenzhen, China). The hematological parameters were interpreted with a Sysmex XN-1000 automated blood cell counter (Sysmex Co. Ltd., Kobe, Japan), using the original matching reagents of the instrument. The hemoglobin components were detected by hemoglobin electrophoresis instrument (V8, Helena, Beaumout, USA), using the original matching reagents of the instrument.
Molecular diagnosis of thalassemia
DNA was extracted with an automatic nucleic acid extractor at Kaishuo Biological Co., Ltd (Xiamen, China). The deletions of α-thalassemia mutation (-α3.4, -α2.5, --SEA, --THAI deletions) using conventional Gap-PCR with the thalassemia gene detection kit (Shenzhen Yilifang Biological Products Co., Ltd.; Shenzhen, China). The non-deletion α‐thalassemia mutations (Hb CS, Hb QS and Hb Westmead), and 19 β‐thalassemia mutations, including - 28 (A > G) (HBB: c.-78 A > G), -29 (A > G) (HBB: c.-79 A > G), -30 (T > C) (HBB: c.-80T > C), -32 (C > A) (HBB: c.-82 C > A), -50 (A > G) (HBB: c.-100G > A), codons 14/15 (+ G) (HBB: c.45_46insG), codon 17 (A > T) (HBB: c.52 A > T), codon 26 (or Hb E) (G > A) (HBB: c.79G > A), codons 27/28 (+ C) (HBB: c.84_85insC), codon31 (-C) (HBB: c.94delC), codons 37 (G > A) (HBB: c.113G > A), codons 41/42 (-TTCT) (HBB: c.126_129delCTTT), codon 43 (G > T) (HBB: c.130G > T), codons 71/72 (+ A) (HBB: c.216_217insA), IVS-I-1 (G > T) (HBB: c.92 + 1G > T), IVS-I-5 (G > C) (HBB: c.92 + 5G > C), IVS-II-654 (C > T) (HBB: c.316–197 C > T), CAP + 1 (A > C) (HBB: c.-50 A > C) and initiation codon (T > G) (HBB: c.2T > G) using RDB-PCR with the thalassemia gene detection kit (Yilifang Biological Products Co., Ltd.; Shenzhen, China).
Statistical analysis
The ROC curve was plotted and the area under the curve (AUC) was calculated to evaluate the sensitivity, specificity, the positive predictive value (PPV), the negative predictive value (NPV) and Youden index of HbA2 screening for pregnant woman thalassemia. P < 0.001 indicates statistically significant difference. MedCalc® Statistical Software version 20.217 (MedCalc Software Ltd, Ostend, Belgium; https://www.medcalc.org;) was used for all statistical analyses.
Results
Overall prevalence of thalassemia
Of the 3243 suspected cases, 656 subjects were diagnosed with thalassemia, the detection rate was 20.22% (656/3243). Among them, 459 α-thalassemia carriers were detected, the detection rate was 14.15% (459/3243); 170 β‐thalassemia carriers were detected, the detection rate was 5.24% (170/3243); 27 αβ-thalassemia carriers were detected, the detection rate was 0.83% (27/3243).
Genotypes and mutation spectrum of α-thalassemia
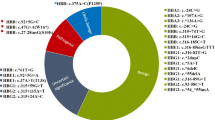
We identified 6 different variations with 10 distinct genotypes among 459 α-thalassemia carriers in this study (Table 1). The --SEA/αα was the most common α-thalassemia in Shenzhen, accounting for 47.71% of all α‐thalassemia genotypes. The next five most common genotypes were -α3.7/αα, -α4.2/αα, αWSα/αα, αCSα/αα, αQSα/αα, with frequencies of 32.90%, 9.59%, 3.48%, 3.05%, 1.09%, respectively (Table 1).
Genotypes and mutation spectrum of β-thalassemia
Among the 170 subjects detected with β-thalassemia, we found 11 β‐thalassemia mutations (Table 2). The most frequent genotypes were βCodons41/42(−TTCT)/βN, βIVS−II−654(A> G)/βN, with a remarkable proportion of 31.76% and 25.29%, respectively. The other common genotypes were βCodons17(A> T)/βN and βCodons−28(A> G)/βN with corresponding proportions of 17.65% and 12.94%, respectively (Table 2).
Genotypes and mutation spectrum of αβ-thalassemia
Twenty-seven subjects carried both α and β-globin variations. Among them, 88.88% of genotypes consisted of common deletions of α-globin gene (--SEA/αα, -α3.7/αα, -α4.2/αα) combined with a β-globin gene point mutation, and composite -α4.2/αα and βCodons41/42(−TTCT)/βN was the most common genotype (Table 3).
Comparison of HbA2 levels
Detecting HbA2 levels in pregnant women with α-, β-, and αβ-thalassemia and normal pregnant women, the results showed (2.42 ± 0.28) %, (5.13 ± 0.67) %, (5.27 ± 0.71) % and (2.50 ± 0.33) % respectively. The HbA2 values in the α-thalassemia group were lower than those in the normal group (P < 0.001), the HbA2 values in the β-, and αβ-thalassemia were significantly higher than those in the normal group (P < 0.001), also higher than the α‐thalassemia group (P < 0.001).
Differential diagnostic value of HbA2
Taking the normal group as a reference, the best cut-off value of HbA2 for α-thalassemia was 2.71, sensitivity 89.7%, specificity 26.6%, AUC 0.587, Youden 0.163, PPV 23.4%, NPV 91.1% (Fig. 1A; Table 4). The best cut-off value of HbA2 for β-thalassemia was 3.71, sensitivity 96.5%, specificity 98.5%, AUC 0.979, Youden 0.950, PPV 98.2%, NPV 97.0% (Fig. 1B; Table 4). The best cut-off value of HbA2 for αβ-thalassemia was 3.86, sensitivity 96.2%, specificity 94.5%, AUC 0.972, Youden 0.907, PPV 81.4%, NPV 99.0% (Fig. 1C and Table 4).
Discussion
Thalassemia is one of the most common monogenic one of the genetic diseases. Globally, more than 300 million people carry the thalassemia gene and about hundreds of thousands of children with thalassemia major are born each year6, bringing great burden to the family and society. Thalassemia is mainly distributed in the Mediterranean and other regions of Asia. In China, Guangdong, Guangxi and Hainan are the high incidence areas3,4,7. In 2020, it was reported that the incidence of thalassemia in Guangdong α- and β- was 15.07% and 3.8%, respectively, with a total incidence of 19.48%6. At present, there is no effective method to treat severe thalassemia, only symptomatic blood transfusion therapy or further hematopoietic stem cell transplantation and gene therapy, but it has certain technical and limitations, and the treatment cost is relatively high. Therefore, reducing the birth rate of children with moderate to severe thalassemia from the root is the most effective method, and the most fundamental measure is to conduct prenatal genetic screening and diagnosis.
This study conducted genetic testing for thalassemia in 3243 pregnant women, with a total detection rate of 20.23% for thalassemia. Among them, 27 cases of αβ-thalassemia were detected; α‐thalassemia was detected in 459 cases, of which the most common was --SEA/αα, followed by -α3.7/αα and -α4.2/αα, it is consistent with the common genotypes of a-thalassemia reported in Guangdong7, it is also consistent with common genotypes other reported in the country8. β-thalassemia was detected in 170 cases, of which the most common was βCD41–42/βN, followed by βIVS−II−654/βN and βCD17/βN, it is consistent with the common genotypes of β-thalassemia reported in Guangdong7. However, β-thalassemia is not consistent with the common genotypes reported in other provinces such as Hubei, Jiangxi and Zhejiang9,10,11; Moradi et al.12 reported that the most common α‐thalassemia was -α3.7/αα in Ilam Province, West Iram. Mustafa et al.13 reported that the most common β‐thalassemia was IVS1 + 5G > C in Maldives. The above studies have shown that there are regional differences in the distribution of thalassemia in China and even in Asia, and analyzing the genotypes and frequency of thalassemia pregnant women in this region is of great significance in guiding the clinical development of premarital examination, prenatal diagnosis, and postnatal childbearing.
Currently, genetic testing is widely used to diagnose thalassemia, and screening methods are critical to the clinical identification of patients with thalassemia. Hemoglobin electrophoresis analysis is an important method of screening for thalassemia, and the analysis of HbA2 cut-off values for different types of thalassemia can assist and guide clinicians in determining the direction of subsequent genetic diagnosis of thalassemia. The results of the present study showed that there was an overall statistically significant difference in HbA2 between the different pregnant women thalassemia groups compared to the normal group. However, the large sample size indicated that HbA2 levels were different in the pregnant and non-pregnant groups14, and differences in HbA2 levels are not fully understood and require further investigation. Given this pregnancy-specific variation, fixed universal reference intervals can lead to an excessive number of false-positive results, which can cause unnecessary financial and psychological stress for pregnant women. ROC curve analysis showed that HbA2 had the highest accuracy in diagnosing β-thalassemia in pregnant women, with an AUC of 0.979 and an optimal cut-off value of 3.71%; Followed by diagnosing αβ-thalassemia in pregnant women, with an AUC of 0.972 and an optimal cut-off value of 3.86%; In addition, the results of this study showed that the diagnostic value of HbA2 for the diagnosis of α-thalassemia in pregnant women was low (AUC = 0.587), with a cutoff value of 2.71%. This is slightly different from the findings of Kang et al.14 which showed that the critical value of HbA2 for diagnosis of β-thalassemia in pregnant women was 3.3% and that for α-thalassemia was 2.4%. Inconsistencies in hemoglobin electrophoresis results may be related to the size of the study data, geographic differences, and differences in the study populations. Therefore, different laboratories in different regions should develop their own databases to determine the optimal screening cutoff value for HbA2 diagnosis of thalassemia.
In summary, the thalassemia genotypes of pregnant women in Shenzhen are diverse. It is necessary to strengthen the prevention and control measure of thalassemia in order to reduce birth defects and improve birth quality. In this study, the optimal cut-off values of HbA2 for thalassemia in pregnant women of different genotypes in the laboratory were established by ROC curves, which provide a reference for the clinical implementation of thalassemia genetic testing and a scientific basis for thalassemia prevention, diagnosis, and genetic counseling in this region.

The ROC for HbA2 each group. (A) The α-thalassemia group. (B) The β-thalassemia group. (C) The αβ-thalassemia group.
Data availability
Data is provided within the supplementary information files.
Abbreviations
- GAP-PCR:
-
Gap-polymerase chain reaction
- RDB-PCR:
-
Reverse dot-blot hybridization
- ROC:
-
Receiver operating characteristic
References
Zheng, L. et al. Screening of some indicators for Alpha-Thalassemia in Fujian Province of Southern China. Int. J. Gen. Med. 14, 7329–7335. https://doi.org/10.2147/IJGM.S338419 (2021).
Lai, K., Huang, G., Su, L. & He, Y. The prevalence of thalassemia in Mainland China: Evidence from epidemiological surveys. Sci. Rep. 7, 920. https://doi.org/10.1038/s41598-017-00967-2 (2017).
He, S. et al. Molecular characterization of alpha- and beta-thalassemia in the Yulin region of Southern China. Gene 655, 61–64. https://doi.org/10.1016/j.gene.2018.02.058 (2018).
Wang, M., Zhang, X., Zhang, Y. & Xiao, M. Prevalence and genetic analysis of thalassemia and hemoglobinopathy in different ethnic groups and regions in Hainan Island, Southeast China. Front. Genet. 13, 874624. https://doi.org/10.3389/fgene.2022.874624 (2022).
He, S. et al. Prevalence and genetic analysis of alpha- and beta-thalassemia in Baise region, a multi-ethnic region in Southern China. Gene 619, 71–75. https://doi.org/10.1016/j.gene.2016.02.014 (2017).
Ngim, C. F., Lai, N. M. & Ibrahim, H. Counseling for prenatal diagnosis and termination of pregnancy due to thalassemia major: A survey of health care workers’ practices in Malaysia. Prenat Diagn. 33, 1226–1232. https://doi.org/10.1002/pd.4233 (2013).
Xian, J. et al. Molecular epidemiology and hematologic characterization of thalassemia in Guangdong Province, Southern China. Clin. Appl. Thromb. Hemost. 28, 10760296221119807. https://doi.org/10.1177/10760296221119807 (2022).
Huang, H. et al. Molecular characterization of thalassemia and hemoglobinopathy in southeastern China. Sci. Rep. 9, 3493. https://doi.org/10.1038/s41598-019-40089-5 (2019).
Zhu, Y., Shen, N., Wang, X., Xiao, J. & Lu, Y. Alpha and beta-Thalassemia mutations in Hubei area of China. BMC Med. Genet. 21, 6. https://doi.org/10.1186/s12881-019-0925-5 (2020).
Wang, K. et al. Investigation of the distribution of thalassemia in children in Jiangxi Province, the People’s Republic of China. Hemoglobin 46, 272–276. https://doi.org/10.1080/03630269.2022.2138429 (2022).
Ding, Z. Y., Shen, G. S., Zhang, S. & He, P. Y. Epidemiology of hemoglobinopathies in the Huzhou region, Zhejiang Province, Southeast China. Hemoglobin 40, 304–309. https://doi.org/10.1080/03630269.2016.1200988 (2016).
Moradi, K. et al. α-Thalassemia mutations in Ilam Province, West Iran. Hemoglobin 46, 147–152. https://doi.org/10.1080/03630269.2019.1694033 (2022).
Mustafa, I. et al. Genetic epidemiology of beta-thalassemia in the Maldives: 23 years of a beta-thalassemia screening program. Gene 741, 144544. https://doi.org/10.1016/j.gene.2020.144544 (2020).
Kang, L., Yi, S., Tan, S., Li, Q. & Li, C. Establishment of pregnant-specific intervals for hemoglobin (Hb) A2, HbF and cut-off points for HbA2 for thalassemia in Chongqing, China. Saudi Med. J. 43, 353–359. https://doi.org/10.15537/smj.2022.43.4.20210729 (2022).
Funding
The work was supported by the National Key Research and Development Program of China (No. 2022YFC2302700), the Guangdong Science and Technology Foundation (No. 2023B0101200003), the Shenzhen Science and Technology Foundation (No. KJZD20230923115359001).
Author information
Authors and Affiliations
Contributions
H.Qian., D.Gu. and H.Zhang. designed the present study. W.L., W.Y., W.Z., X.L. and J.H. performed the present study and analyzed the data. H.Qian. wrote the main manuscript. H.Qian. and D.Gu. confirm the authenticity of all the raw data. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval and consent to participate
The study protocol was in line with the declaration of Helsinki (as revised in Brazil 2013). This study was approved by the Ethics Committee of Shenzhen Second People’s Hospital (Approval No: 2024-339-01YJ) and all participants were informed and signed a written informed consent.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Qian, H., Zhao, W., Li, W. et al. Analysis of thalassemia genotypes and HbA2 test results in pregnant women in Shenzhen, China. Sci Rep 15, 7483 (2025). https://doi.org/10.1038/s41598-025-91908-x
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-91908-x
