
Abstract
Heavy metals are commonly present in polluted soil in mining areas. In this study, we investigated 10 sites of farmland soil in the heavy metal tailing dam collapse area (TDCA) with the dominant phyla Acidobacteriota, Proteobacteria, Bacteroidetes, and Planctomycetes. The heavy metal dam collapse area is a composite contamination area of multiple heavy metals, with Cd, Pb and Zn being the most severely contaminated, and the levels of Hg and Cu exceeding the screening values at some of the sites. The Shannon, Chao1 and ACE indices revealed high microbial diversity but low relative abundance of microorganisms at the severely polluted TDCA1 and TDCA3 sites. The results of redundancy analysis (RDA) showed that Hg (Max = 4.31 mg/kg) and Cu (Max = 100 mg/kg) were important factors affecting soil microbial community in the TDCA compared to other heavy metals. Correlation analysis of heavy metals with microbial communities showed that RB41 (Acidobacteria) was more resistant to high concentrations of Cd, Pb, and Zn pollution. The genera of UTCFX1 (Chloroflxi) and norank_TRA3-20 had strong tolerance to the heavy metal Hg. Cu was significantly negatively correlated with norank_WD2101_soil_group (P < 0.05). Therefore these can be used as indicators for monitoring potential heavy metal contamination. The results can be used to predict the changes in the ecosystem of the mining area to maintain its ecological balance and health.
Similar content being viewed by others

Introduction
At present, with the rapid development of social industrialization, the speed of mining is accelerating, resulting in a gradual increase in the content of pollutants in the soil. Heavy metals are important pollutants in mining areas, and with the accumulation of a long time and rainfall erosion leading to mine damming make pollutants spread around the mine, increasing the concentration of heavy metals in the surrounding area. Accumulation of heavy metals in soil can lead to reduced soil enzyme activity1, decreased soil quality affecting crop growth, imbalance in microbial communities, and loss of biodiversity2,3,4,5. In addition, the accumulation of heavy metals in the biological chain can seriously jeopardize human health5. For example, exposure to high levels of Cr increases the risk of cancer6; Pb poisoning damages intellectual abnormalities in children7; Hg toxicity causes Minamata disease; and Cd poisoning causes itai-itai disease. In addition, heavy metals can cause toxicity to human organs, such as neurotoxicity, nephrotoxicity, and hepatotoxicity8. Therefore, it is essential to monitor heavy metals in soil.
Microorganisms are vital to the healthy operation of soil ecosystems9. The composition and diversity of microbial communities can quickly reflect changes in soil ecosystems, reflecting the sensitivity and resilience of microorganisms10. Numerous research endeavors have investigated the effects of heavy metals on the structure of soil microbial communities11,12,13. Therefore, it is crucial to understand the interrelationships between heavy metals and microbial communities. The response of microbial communities to heavy metal pollution can provide a scientific basis for soil pollution remediation14. In addition, the possibility of utilizing microorganisms for bioremediation can be explored by understanding the tolerance and degradation ability of microorganisms to heavy metals.
Bacterial communities are sensitive to changes in environmental conditions, especially in the presence of heavy metals15,16,17,18. Numerous studies have investigated the effects of heavy metals on soil microbial community structure19,20,21. The composition of microorganisms in different heavy metal contaminated soils is varied. Li et al. found that the dominant phyla in Pb/Zn smelters were Proteobacteria, Firmicutes, Actinobacteria, Bacteroidetes, and Acidobacteria22. Wang et al. found that the dominant phyla in Cd-contaminated soil were Firmicutes, Bacteroidetes, Actinobacteria, and Proteobacteria, respectively23. Liu et al. found that Proteobacteria, Actinobacteria, Acidobacteria, and Chloroflexi were the most abundant bacterial taxa24.
Under certain conditions, the presence of biodegradable microorganisms in soil can accelerate the degradation of pollutants such as heavy metals. The conversion of pollutants to less harmful states through adsorption, precipitation, or transformation processes is known as bioremediation25, which is more effective and environmentally friendly than current physicochemical remediation methods and does not cause problems such as secondary pollution. It has also been found that sulfate-reducing bacteria can convert Cr (VI) to Cr (III) in a short time with a conversion efficiency of up to 99%26. The study of the microbial community structure of polluted sites can help to understand the environmental adaptation mechanism of microorganisms and maintain a healthy land environment.
Based on the above research background this study was done as follows: (1) Determination of heavy metals concentrations; (2) microbial community composition in contaminated soils; (3) Relationship of soil microorganisms with heavy metals. This study aims to investigate the mechanism of heavy metal pollution on soil microorganisms through the investigation and analysis of the microbial community structure of heavy metals contaminated soil in the TDCA and to provide theoretical support for the ecological remediation of the mining area soil.
Results
Soil heavy metals concentration
The results of heavy metals concentration in soil are shown (Supplementary Table S1), compared with the screening values of farmland soil with heavy metals concentration, the heavy metals contamination was quite different in different sampling sites. According to the risk control standards for soil contamination of agricultural land (GB 15618–2018)27, the screening values for soil pollution in the study area (Supplementary Table S1) were Cd (0.6 mg/kg), As (25 mg/kg), Pb (170 mg/kg), Cr (250 mg/kg), Cu (100 mg/kg), Zn (300 mg/kg), Ni (190 mg/kg), and Hg (3.4 mg/kg). Overall, most of the sampling sites were contaminated with Cd, Pb, and Zn, with Pb and Zn being the most contaminated, followed by Cd. As, Hg, and Cu were the least contaminated. Specifically, except for point 2, the other 9 points were contaminated with Cd and Zn. Except for points 2, 7, and 8, all other points are contaminated with Pb. As, Hg and Cu contamination occurred at TDCA1, TDCA3 and TDCA6, respectively. In summary, the TDCA3 site was the most contaminated, with Pb concentrations 8.75 times the screening value, Zn concentrations 11.32 times the screening value, Cd concentrations 35.11 times the screening value, and Hg concentrations 1.27 times the screening value. Heavy metal concentrations were measured and analyzed using Surfer 23 software for kriging interpolation, aiming to predict the heavy metal pollution in the entire TDCA (Fig. 1).

Prediction of heavy metal concentrations (a: Cr; b: Ni; c: As; d: Cu; e: Zn; f: Cd; g: Pb; h: Hg. Red represents high concentrations of heavy metals. Maps were downloaded using BIGEMAP 30.0.0.0 (http://www.bigemap.com/). kriging interpolation analysis was performed by Sufer 23.2.176 (https://www.goldensoftware.com/products/surfer/) and microbial diversity indices (i).
The Spearman correlation analysis between heavy metals elements is shown in Supplementary Fig. S1. Firstly, overall, there is a positive correlation between most metal elements. Specifically, there was a significant positive correlation (P < 0.05) between the four elements Cd, Pb, Zn, and Hg. Cu was significantly positively correlated with Ni (P < 0.05), Pb (P < 0.05), and As (P < 0.01). As and Ni were also significantly positively correlated (P < 0.05), which suggests that the three elements, Cu, Ni, and As, are positively correlated with each other.
Diversity of bacterial communities
The alpha diversity indices showed that the richness and diversity of bacterial communities in the 10 samples were different (Fig. 1). Shannon indices are indicators of microbial diversity. As shown by the Shannon indices, the bacterial diversity varied from 6.64 to 7.23. ACE and Chao1 indices reflects microbial richness. The ordering of ACE and Chao1 indices is TDCA1 > TDCA3 > TDCA7 > TDCA10 > TDCA5 > TDCA2 > TDCA6 > TDCA4 > TDCA8 > TDCA9, indicating that the TDCA1 point has the highest number of Operational Taxonomic Unit (OTU). The Coverage index mainly embodied the depth of the sample detection, the coverage indexes are all over 0.98, indicating that the sequencing results can be used to respond to the composition of the bacterial community structure at sampling sites.
High-throughput sequencing results showed that the 10 soil samples consisted of bacteria from 56 phyla and 1369 genera, totaling 21,588 OTUs. The microbial composition varied greatly among different samples. TDCA1 had the highest number of species-specific OTUs at 1973 OTUs (9.14%), followed by TDCA3, TDCA10, TDCA2, and TDCA7 at 1532 (7.10%), 1524 (7.06%), 1310 (6.07%), and 1235 (5.72%), respectively (Fig. 2). The number of common microbial species at the 10 sites was 1426 OTUs (6.61%) (Fig. 2). The positive competition among soil microorganisms under multiple heavy metals contamination leads to a richer diversity of soil microbial communities.

Distribution of species-specific OTUs in all soil samples in Venn diagram. The overlap in the Veen diagram indicates the bacterial OTUs shared between all soil samples.
The results of principal coordinate analysis (PCoA) of the bacterial community in the soil are shown in Fig. 3. The cumulative contributions of PCoA1 and PCoA2 were 31.43% and 20.32%, respectively. In addition, we found a stress value of 0.0537 (stress < 0.1) by NMDS analysis. Stress was used to check the NMDS results, and stress < 0.1 indicates a better sorting of the sample sites. Combined PCoA results indicate differences in microbial composition between samples. As shown in Fig. 4, the dominant phylum is not the same in different samples, for example, the dominant phylum of TDCA1 is Proteobacteria and the dominant phylum of TDCA6 is Acidobacteriota.

Bacterial community similarity analysis (PCoA). Different colored dots represent different groupings of samples. The closer the dots are to each other, the more similar the species composition of the two samples.

The relative abundance of bacterial community at the level of phylum (a) and genus (b) in samples. The horizontal axis is the number of each sample and the vertical axis is the proportion of relative abundance of species. The different widths of the color shapes indicate the relative abundance proportions of the different species.
Bacterial community composition
The results of bacterial community classification at the phylum level and genus level for the 10 soil samples are shown in Fig. 4. At the phylum level (Fig. 4a), the bacterial communities were dominated by Acidobacteriota, Proteobacteria, Bacteroidetes, and Planctomycetes, accounting for more than 60% of the total bacterial population in all samples. At the genus level (Fig. 4b), the dominant genus from TDCA1 to TDCA9 samples was RB41 (Actinobacteria), differing only in relative abundance (19 ~ 32%.) The dominant genus in the TDCA10 sample were Sphingomonas (Proteobacteria) (14%) and norank_Chloroplast (Cyanobacteria) (13%).
Correlation analysis between soil heavy metals concentration and bacterial community structure
Figure 5 shows the results of RDA analysis. The results of RDA heavy metals phylum level analysis showed that the first and second axes obtained from the RDA analysis explained 42.55% and 24.37% of the overall variation, suggesting that the result could explain the relationship between heavy metals concentration and bacterial community structure. Hg, Cu, and Ni were positively correlated with the first sorting axis, while the other heavy metals were negatively correlated with the first sorting axis (Fig. 5a). The results of RDA heavy metals genus level analysis showed that the first and second axes obtained from the RDA analysis explained 26.94% and 24.37% of the overall variation. All eight heavy metals were positively correlated with the first sorting axis (Fig. 5b), which indicated that the effects of heavy metals on microbial communities varied among different sorting levels. This indicates that the effects of heavy metals on microbial communities differed at different taxonomic levels.

RDA plots of heavy metals at phylum (a) and genus (b) level. The longer the arrow for an environmental factor, the greater the influence of that influencing factor.
Figure 6 is the Spearman correlation analysis of heavy metals with bacterial communities. At phlum level (Fig. 6a), Ni was significantly positively correlated with Nitrospirota and Patescibacteria (P < 0.05). The correlation between other heavy metals and phylum level microorganisms was not significant. Spearman correlation analysis between heavy metals and bacterial communities at the genus level found (Fig. 6b) that Ni was significantly positively correlated with Nitrospira (P < 0.05) and significantly negatively correlated with Sphingomonas (P < 0.05). As was significantly negatively correlated with norank_Vicinamibacterales (P < 0.05). Cu was significantly negatively correlated with norank_WD2101_soil_group (P < 0.05). Hg was significantly positively correlated with norank_TRA3-20 (P < 0.05) and UTCFX1 (P < 0.05).

The correlation analysis of heavy metals with bacterial communities at phylum (a) and genus (b) level. The red color represents a positive correlation and the blue color represents a negative correlation. The color shade is proportional to the correlation coefficient.
Functional gene prediction
From the prediction results of FAPROTAX (Supplementary Fig. S2), it can be seen that chemoheterotrophs and aerobic_chemoheterotrophy are the dominant types of microorganisms in the TDCA, and are distributed at various locations. chemoheterotrophy and aerobic_chemoheterotrophy are functional groups of dominant bacteria (Acidobacteria, Actinobacteria, and Proteobacteria). Overall, TDCA10 has a wide range of functional microbial types. Among them, TDCA3, TDCA6, and TDCA10 have the highest levels of chemoheterotrophy and aerobic_chemoheterotrophy. In addition, there are also chloroplasts, phototrophy, photoautotrophy, cyanobacteria, and oxygenic photoautotrophy microorganisms present in the TDCA10 site. The photosynthetic (chloroplasts) and intercellular parasites microbial communities at the TDCA8 site are higher than those at other sites.
This study predicts the phenotypic function of soil microorganisms in the TDCA using BugBase (Supplementary Fig. S3). The results showed that the relative abundance of anaerobic and aerobic bacteria at each site showed an overall opposite trend. The content of mobile elements and the potential pathogenicity of microorganisms at each site showed an opposite trend overall. In contrast, the stress tolerance showed the same trend as the content of mobile elements. This can be explained by the lower content of mobile elements at this site, which may lead to a gradual increase in the relative abundance of pathogenic microorganisms and a smaller migration to other sites, resulting in an increased risk of pathogenicity and decreased stress tolerance.
Discussion
Soil is a complex ecosystem, which is an important habitat for microorganisms' survival and activities. Heavy metal pollution is mainly based on the compound pollution of multiple elements in the soil around the mining area, which leads to more complex environmental effects and affects public health28,29. In this study, spearman correlation analysis showed that there was a positive correlation between most of the heavy metals elements. This indicated that they had similar sources and transport and transformation behaviors, forming a certain degree of complex pollution. Microorganisms, as an important part of the soil ecosystem, are involved in the decomposition of organic matter, the release of nutrients, and the maintenance of soil structure30,31 The activities of soil microorganisms are intricate. Previous studies have demonstrated that microorganisms are highly sensitive to heavy metals in soil and that different microorganisms are not equally sensitive to various heavy metals32,33,34. For example, As pollution had a positive relationship with Chloroflexi, Betaproteobacteria, and Bacteroidetes relative abundance (p < 0.05). Firmicutes relative abundance decreased with an increase in As concentration24. Deng et al. 2022 found a significant positive correlation between Cd and Proteobacteria, Planctomycetes, Actinobacteria, and Acidobacteria35. Zhu et al. found a significant positive correlation between soil Hg and Chloroflexi abundance36. Liu et al. found a positive correlation between total soil Hg content and the relative abundance of Firmicutes and Bacteroidetes in the soil37.
At the phylum level (Fig. 4a), the bacterial communities were dominated by Acidobacteriota, Proteobacteria, and Bacteroidetes, which is consistent with the results of previous studies on the effects of heavy metals on the composition of microbial communities in industrially polluted soils19,22. Acidobacteria were the most abundant phylum, involved in the decomposition of organic matter and nutrient cycling in soil38,39,40, and were widely distributed in soil, water, and marine ecosystems41. The second most abundant phylum was the Proteobacteria, the largest of the bacterial phylum, which was closely related to the utilization of carbon sources and consists mainly of gram-negative bacteria responsible for fixing nitrogen42. The Proteobacteria phylum is the most abundant in varied environments due to its ability to rapidly change morphology to adapt to different environments43,44,45. Bacteroidetes have a strong capacity to adapt to the environment and degrade organic matter, and have been detected in extreme environments, such as high salinity46. At the genus level, the dominant genus in samples TDCA1-TDCA9 was RB41 (Acidobacteriota), differing only in relative abundance suggesting that RB41 was resistant to complex heavy metal contamination. The dominant genera in the TDCA10 samples were the Sphingomonas (Proteobacteria) (14%) and the norank_Chloroplast (Cyanobacteria) (13%). This may be because TDCA10 is located near agricultural soils where the influence of plant roots is stronger, resulting in different bacterial responses to heavy metals47.
Heavy metals are the main factors affecting microbial communities48,49. Heavy metals have negative effects on microbial community structure. In the present study, the farmland soil in the TDCA was heavily contaminated with Cd, Pb, and Zn, and only a few points were contaminated with Cu and Hg, so the RDA analysis showed that Hg and Cu had the greatest effect on the microbial community. Both TDCA4 and TDCA10 samples were contaminated with varying degrees of Cd, Pb, and Zn (Supplementary Table S1), but the higher relative abundance of the genus RB41 in the TDCA4 samples suggested that the RB41 (Acidobacteria) was more resistant under the combined contamination of Cd, Pb, and Zn. Furthermore, correlation analysis revealed that Ni was positively correlated with Nitrospirota and Patescibacteria and was negatively correlated wth Sphingomonas (Fig. 6). This is mainly since nitrogen cycling is severely affected under heavy metals contamination in the soil, which coincides with the fact that Nitrospirota is an important bacterial phylum for nitrogen transformation50,51. Cu was significantly negatively correlated with norank_WD2101_soil_group (Fig. 6b). In addition, we also found that the Hg concentration was higher in TDCA3 and TDCA6 sites, and the relative abundance of norank_TRA3-20 was higher than in other sites. The results in Fig. 6b also showed a significant positive correlation (P < 0.05) between norank_TRA3-20 and Hg, which indicated that Norank_TRA3-20 was resistant to Hg. Furthermore, Hg was not correlated with Chloroflxi at the phylum level but was significantly positively correlated (P < 0.05) with the genus UTCFX1 (Chloroflxi) at the genus level, suggesting that UTCFX1 is potentially resistant to Hg.
Conclusion
Farmland soils in the TDCA were heavily contaminated with heavy metals, and it severely affected the community composition of microorganisms in the soil. At the phylum level, the bacterial communities were dominated by Acidobacteriota, Proteobacteria, and Bacteroidetes. Heavy metals analysis revealed that Hg, Cu, and Ni were the most important factors affecting microorganisms. In this work, Nitrospirota and Patescibacteria phylum had some tolerance to Ni. The genus UTCFX1 (Chloroflxi) and Norank_TRA3-20 were more tolerant to Hg. Ni was significantly negatively correlated with Sphingomonas (P < 0.05). As was significantly negatively correlated with norank_Vicinamibacterales (P < 0.05). Cu was significantly negatively correlated with norank_WD2101_soil_group (P < 0.05). By analyzing the structure of soil microbial communities in the TDCA, we can understand the interaction mechanism between heavy metals and soil microbial communities, and further provide theoretical guidelines for the restoration of heavy-metal-contaminated farmland soils in the mining area.
Materials and methods
Sites description
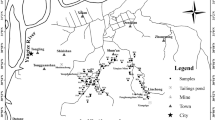
The tailing pond adopts decentralized ore discharge and belongs to the upstream method of dam construction, the height of the early dam is 13.5 m, the height of the late dam is 32.5 m, the total height of the dam is 46 m, and the total capacity of the bank is 228.40 × 104 m3, which belongs to IV class bank. The tailing pond is located in the longitudinal "U" shape, the width of the bottom of the ditch is 70–120 m, the geomorphology belongs to the hilly area, and the ground elevation is 228.36–280.83 m. The sudden heavy rainfall led to the occurrence of a tailing pond failure, forming a ditch 100 m long and 10 m wide. Figure 7 shows the distribution of sampling sites in the TDCA.

Distribution of sampling sites in the dam collapse area. The red area is the tailing dam collapse area and the blue area is the tailing pond. Maps were downloaded using BIGEMAP 30.0.0.0 (http://www.bigemap.com/). ArcGIS 10.7.0.10450 (https://www.esri.com/en-us/arcgis/products/arcgis-desktop/resources) mapped the distribution of sample sites.
Sample collection and preservation
The soil used in this study was extracted from the surface farmland soil (0–20 cm) of a lead–zinc mine tailing pond breaching area in Northeast China and was sampled using the grid method of laying out points and the plum sampling method. A surface 0–20 cm soil sample was collected using a stainless-steel sampler to remove plant residues and other biological residues. The samples were divided into two parts for processing: one for the determination of heavy metals content, and one for 16SrRNA analysis. The samples were immediately placed into sterile centrifuge tubes prepared in advance. The collected soil samples were put into sterile bags and placed into foam boxes with ice packs for low-temperature storage and transportation and an ultra-low-temperature refrigerator (− 80 °C) for long-term storage5.
Sample analysis
Grind the soil sample and pass it through a 100-mesh sieve. The sample is digested using a four-acid digestion method (nitric acid-perchloric acid-hydrofluoric acid-hydrochloric acid)52. The concentrations of Cd, Pb, Cr, Cu, Zn, As, and Ni in the soil were determined using inductively coupled plasma mass spectrometry (ICP-MS)53. Measure the concentration of Hg in soil using a catalytic pyrolysis-cold atomic absorption spectrophotometry54.
Soil microbiological determination
Total genomic DNA was extracted from soil samples using the Mag-Bind Soil DNA Kit. The quality of the extracted DNA was assayed using 1% agarose gel electrophoresis. The DNA concentrations were measured using a Quawell Q5000 microvolume UV spectrophotometer to meet testing requirements. The sequence of the V3-V4 region of the 16S rRNA gene was amplified using primers 341F and 805R on the Illumina high-throughput sequencing platform of Sangyo Bioengineering.
Statistical analyses
ArcGIS 10.2 was employed to create a distribution map of sampling points. Kriging interpolation analysis was performed using Surfer 23. Origin 2024 was used for data processing and data visualization.
Data availability
Data is provided within the manuscript or supplementary information files.
References
Angon, P.-B. et al. Sources, effects and present perspectives of heavy metals contamination: Soil, plants and human food chain. Heliyon 10, e28357 (2024).
Zhang, F., Xie, Y., Peng, R., Ji, X. & Bai, L. Heavy metals and nutrients mediate the distribution of soil microbial community in a typical contaminated farmland of South China. Sci. Total Environ. 947, 174322 (2024).
Chen, H., Ming, F., Hu, X., Ma, D. & Huo, Z. Biochar assists phosphate solubilizing bacteria to resist combined Pb and Cd stress by promoting acid secretion and extracellular electron transfer. J. Hazard. Mater. 452, 131176 (2023).
Yuan, X., Xue, N. & Han, Z. A meta-analysis of heavy metals pollution in farmland and urban soils in China over the past 20 years. J. Environ. Sci. 101, 217–226 (2021).
Pan, X. et al. Effects of soil chemical properties and fractions of Pb, Cd, and Zn on bacterial and fungal communities. Sci. Total Environ. 715, 136904 (2020).
Sun, H., Brocato, J. & Costa, M. Oral chromium exposure and toxicity. Curr. Environ. Heal. Rep. 2, 295–303 (2015).
Hou, S. et al. A clinical study of the effects of lead poisoning on the intelligence and neurobehavioral abilities of children. Theor. Biol. Med. Model. 10, 13 (2013).
Mitra, S. et al. Impact of heavy metals on the environment and human health: Novel therapeutic insights to counter the toxicity. J. King Saud Univ. Sci. 34, 101865 (2022).
Smith, M. B. et al. Natural bacterial communities serve as quantitative geochemical biosensors. mBio 6, e00326-e415 (2015).
Ho, J. & Chambers, L. G. Altered soil microbial community composition and function in two shrub-encroached marshes with different physicochemical gradients. Soil Biol. Biochem. 130, 122–131 (2019).
Lu, L. et al. Long-term metal pollution shifts microbial functional profiles of nitrification and denitrification in agricultural soils. Sci. Total Environ. 830, 154732 (2022).
Philippot, L., Chenu, C., Kappler, A., Rillig, M. C. & Fierer, N. The interplay between microbial communities and soil properties. Nat. Rev. Microbiol. 22, 226–239 (2024).
Mierzwa-Hersztek, M., Gondek, K., Klimkowicz-Pawlas, A., Baran, A. & Bajda, T. Sewage sludge biochars management—Ecotoxicity, mobility of heavy metals, and soil microbial biomass. Environ. Toxicol. Chem. 37, 1197–1207 (2018).
Gayan, A., Borah, P., Nath, D. & Kataki, R. Soil microbial diversity, soil health and agricultural sustainability. Sustain. Agricult. Environ. 4, 107–126 (2023).
Zhang, C. et al. Effects of heavy metals and soil physicochemical properties on wetland soil microbial biomass and bacterial community structure. Sci. Total Environ. 557–558, 785–790 (2016).
Jiang, R., Wang, M. & Chen, W. Heavy metal pollution triggers a shift from bacteria-based to fungi-based soil micro-food web: evidence from an abandoned mining-smelting area. J. Hazard. Mater. 459, 132164 (2023).
Liu, Y. et al. Effects of heavy metals on bacterial community surrounding Bijiashan mining area located in Northwest China. Open Life Sci. 17, 40–54 (2022).
Song, J. et al. Effects of Cd, Cu, Zn and their combined action on microbial biomass and bacterial community structure. Environ. Pollut. 243, 510–518 (2018).
Abbaszade, G. et al. Exploring the relationship between metal(loid) contamination rate, physicochemical conditions, and microbial community dynamics in industrially contaminated urban soils. Sci. Total Environ. 897, 166094 (2023).
Chu, D. Effects of heavy metals on soil microbial community. In: IOP Conf. Ser.: Earth Environ. Sci. 113, 012009 (2018).
Wang, X. et al. Heavy metal contamination collapses trophic interactions in the soil microbial food web via bottom-up regulation. Soil Biol. Biochem. 184, 109058 (2023).
Li, S. et al. A comprehensive survey on the horizontal and vertical distribution of heavy metals and microorganisms in soils of a Pb/Zn smelter. J. Hazard. Mater. 400, 123255 (2020).
Wang, X. et al. Improvement of alfalfa resistance against Cd stress through rhizobia and arbuscular mycorrhiza fungi co-inoculation in Cd-contaminated Soil. Environ. Pollut. 277, 116758 (2021).
Liu, Y. et al. Arsenic pollution from human activities drives changes in soil microbial community characteristics. Environ. Microbiol. 25, 1–12 (2023).
Abbas, S. Z. et al. A review on mechanism and future perspectives of cadmium-resistant bacteria. Int. J. Environ. Sci. Technol. 15, 243–262 (2018).
Han, X. et al. Removal of Cr (VI) and phenol coupled with the reduction of sulfate by sulfate-reducing bacteria sludge. Int. J. Environ. Sci. Technol. 14, 2173–2180 (2017).
Ministry of Ecology and Environment of the People’s Republic of China. National Standards of the People’s Republic of China-Soil environment quality GB 15618–2018-Risk control standard for soil contamination of agricultural land. (2018).
Briffa, J., Sinagra, E. & Blundell, R. Heavy metal pollution in the environment and their toxicological effects on humans. Heliyon 6, e04691 (2020).
Qin, G. et al. Soil heavy metal pollution and food safety in China: Effects, sources and removing technology. Chemosphere 267, 129205 (2021).
Souza, L. F. & Billings, S. A. Temperature and pH mediate stoichiometric constraints of organically derived soil nutrients. Global Change Biol. 28, 1630–1642 (2022).
Havlin, J. L. Soil: Fertility and nutrient management landscape and land capacity 251–265 (CRC Press, 2020).
Chun, S.-J., Kim, Y.-J., Cui, Y. & Nam, K.-H. Ecological network analysis reveals distinctive microbial modules associated with heavy metal contamination of abandoned mine soils in Korea. Environ. Pollut. 289, 117851 (2021).
Wang, Y. Heavy metal induced shifts in microbial community composition and interactions with dissolved organic matter in coastal sediments. Sci. Total Environ. 927, 172003 (2024).
Qi, Q. et al. Contamination with multiple heavy metals decreases microbial diversity and favors generalists as the keystones in microbial occurrence networks. Environ. Pollut. 306, 119406 (2022).
Deng, Y. et al. Ecological responses of bacterial assembly and functions to steep Cd gradient in a typical Cd-contaminated farmland ecosystem. Ecotox. Environ. Safety 229, 113067 (2022).
Zhu, D., Wang, Z. & Zhang, Z. Effects of heavy metal pollution and soil physicochemical properties on the Sphagnum farmland soil microbial community structure in Southern Guizhou. China. Int. J. Phytoremediat. 25, 1762–1773 (2023).
Liu, Y.-R., Delgadi-Baquerizo, M., Bi, L., Zhu, J. & He, J.-Z. Consistent responses of soil microbial taxonomic and functional attributes to mercury pollution across China. Microbiome 6, 183 (2018).
Chen, L.-F., He, Z.-B., Zhao, W.-Z., Kong, J.-Q. & Gao, Y. Empirical evidence for microbial regulation of soil respiration in alpine forests. Ecol. Indic. 126, 107710 (2021).
Crits-Christoph, A., Diamond, S., Butterfield, C. N., Thomas, B. C. & Banfield, J. F. Novel soil bacteria possess diverse genes for secondary metabolite biosynthesis. Nature 558, 440–444 (2018).
Woodcroft, B. J. et al. Genome-centric view of carbon processing in thawing permafrost. Nature 560, 49–54 (2018).
Gonçalves, O. S. et al. Insights into plant interactions and the biogeochemical role of the globally widespread Acidobacteriota phylum. Soil Biol. Biochem. 192, 109369 (2024).
Chaudhry, V., Rehman, A., Mishra, A., Chauhan, P. S. & Nautiyal, C. S. Changes in bacterial community structure of agricultural land due to long-term organic and chemical amendments. Microb. Ecol. 64, 450–460 (2012).
Huang, W. et al. Comparison among the microbial communities in the lake, lake wetland, and estuary sediments of a plain river network. MicrobiologyOpen 8, e00644 (2019).
Lee, H. Environmental drivers affecting the bacterial community of intertidal sediments in the Yellow Sea. Sci. Total Environ. 755, 142726 (2021).
Lu, M. et al. Nutrients and heavy metals mediate the distribution of microbial community in the marine sediments of the Bohai Sea. China. Environ. Pollut. 255, 113069 (2019).
Wolińska, A. et al. Bacteroidetes as a sensitive biological indicator of agricultural soil usage revealed by a culture-independent approach. Appl. Soil Ecol. 119, 128–137 (2017).
Qian, F., Huang, X., Su, X. & Bao, Y. Responses of microbial communities and metabolic profiles to the rhizosphere of Tamarix ramosissima in soils contaminated by multiple heavy metals. J. Hazard. Mater. 438, 129469 (2022).
Wang, L. et al. Shift in the microbial community composition of surface water and sediment along an urban river. Sci. Total Environ. 627, 600–612 (2018).
Yang, X., Huang, S., Wu, Q., Zhang, R. & Liu, G. Diversity and vertical distributions of sediment bacteria in an urban river contaminated by nutrients and heavy metals. Front. Environ. Sci. Eng. 7, 851–859 (2013).
Hou, D. Metal contamination and bioremediation of agricultural soils for food safety and sustainability. Nat. Rev. Earth Environ. 1, 366–381 (2020).
Rijk, I. J. C. & Ekblad, A. Carbon and nitrogen cycling in a lead polluted grassland evaluated using stable isotopes (δ13C and δ15N) and microbial, plant and soil parameters. Plant Soil 449, 249–266 (2020).
Ministry of Ecology and Environment of the People’s Republic of China. The People’s Republic of China National Environmental Protection Standards HJ 832–2017-Soil and sediment-Digestion of total metal elements-Microwave assisted acid digestion method. (2017a).
Ministry of Ecology and Environment of the People’s Republic of China. The People’s Republic of China National Environmental Protection Standards HJ 803–2016-Soil and sediment-Determination of aqua regia extracts of 12 metal elements-Inductively coupled plasma mass spectrometry. (2016).
Ministry of Ecology and Environment of the People’s Republic of China. The People’s Republic of China National Environmental Protection Standards HJ 923–2017-Soil and sediment-Determination of total mercury-Catalytic pyrolysis-cold atomic absorption spectrophotometry. (2017b).
Acknowledgements
This work was supported by the National Key R&D Program of China (NO. 2019YFD1100504) and the National Water Pollution Control and Treatment Science and Technology Major Project of China (NO. 2018ZX07601). The assistance of the coordinating authors is gratefully acknowledged. Thanks to the proof-readers and editors for their help.
Author information
Authors and Affiliations
Contributions
J.D. proposed the ideas, performed all the experiments, wrote the manuscript text, prepared the tables and analyzed the data. Y.W., X.L. found funding and reviewed the paper. D.Y., J.Y. prepared the figures and reviewed the paper.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Deng, J., Wang, Y., Yu, D. et al. Effects of heavy metals on variation in bacterial communities in farmland soil of tailing dam collapse area. Sci Rep 15, 8100 (2025). https://doi.org/10.1038/s41598-025-93244-6
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-93244-6
