
Abstract
Sepsis-induced acute kidney injury (AKI) can lead to chronic renal dysfunction with accelerated renal aging. Activation of the mammalian target of rapamycin (mTOR) is implicated in the initiation and progression of renal injury. This study investigates the effectiveness of the mTOR inhibitor, rapamycin, in mitigating kidney injury and explores the underlying mechanisms. AKI was induced by intraperitoneal administration of a solution containing 10 mg/kg of lipopolysaccharide (LPS) in a mouse model. Two groups of endotoxemic mice received pre- and post- treatment with rapamycin. Whole-genome DNA methylation analysis was performed on renal proximal tubular epithelial cells (RPTEC). In the LPS-induced AKI mouse model, rapamycin treatment significantly reduced creatinine levels, preserved renal parenchyma, and counteracted the endothelial-to-mesenchymal transition (EndMT) by inhibiting the ERK pathway. Whole-genome DNA methylation analysis revealed that LPS induced aberrant methylation, particularly in genes associated with premature aging, including ectonucleoside triphosphate diphosphohydrolase 1 (ENTPD1/CD39) and wolframin ER transmembrane glycoprotein (WFS1). Accordingly, endotoxemic mice exhibited decreased CD39 expression and klotho down-regulation, both of which were reversed by rapamycin, suggesting an anti-aging effect in AKI. mTOR inhibition may represent a promising strategy to prevent accelerated renal aging in LPS-induced AKI and potentially slow the progression of chronic kidney disease.
Similar content being viewed by others
Introduction
Sepsis-induced acute kidney injury (SI-AKI) is a highly complex and multifactorial disease, characterized by hyperinflammation associated with systemic endothelial dysfunction and alterations in renal resident cells that may promote progression of renal disease and aging1. Lipopolysaccharide (LPS), the main structural component of the outer membrane of most Gram-negative bacteria, is well known for its role in sepsis pathophysiology. LPS is recognized by a specific innate immune receptor, Toll-like Receptor 4 (TLR4)2, which is expressed on immune and renal resident cells. LPS-induced signaling through the TLR4 pathway triggers a profound inflammatory response, leading to the release of several mediators, including DAMPs/PAMPs, and exerts several effects on renal parenchymal cells.
In this context, dysfunctional endothelial cells (EC) activate survival signaling pathways3,4 and acquire a fibroblast-like phenotype, through a process known as endothelial-to-mesenchymal transition (EndMT)5,6, contributing to vascular rarefaction and renal fibrosis. The pro-inflammatory microenvironment and local hypoxia resulting from vascular dysfunction further drive the metabolic adaptation of renal proximal tubular epithelial cells (RPTEC) through adaptive mechanisms of survival such as mitophagy, autophagy, and senescence, ultimately leading to a significant decline in kidney function7. Cellular senescence, a state of irreversible cell cycle arrest, has been identified as a key contributor to the detrimental effects of prolonged acute kidney injury (AKI). This phenomenon plays a crucial role in the acceleration of maladaptive repair mechanisms that ultimately link AKI to the development of chronic kidney disease (CKD)8,9. Epigenetic mechanisms, including DNA methylation and histone modifications, may play central roles in regulating RPTEC senescence in SI-AKI.
Identifying the potential sites of LPS/TLR4 signaling may represent a promising therapeutic strategy to reduce detrimental effects of endotoxin exposure10. Recent studies have highlighted the significance of the mammalian Target of Rapamycin (mTOR) in the pathogenesis of inflammatory diseases. mTOR, a serine/threonine kinase, plays a critical role in regulating protein synthesis, essential for cellular proliferation. Activation of mTORC1 in different types of immune cells11 has been observed upon exposure to LPS, likely mediated via the PI3K-Akt and ERK pathways12. The mechanistic target of mTORC1 has been shown to play a fundamental role in the pathogenesis and severity of acute lung injury (ALI) induced by LPS13. Recent studies have also emphasized the involvement of mTORC1 in modulating renal dysfunction by regulating various metabolic pathways implicated in cellular senescence, fibrotic processes, and klotho expression and function14.
However, the precise mechanism and significance of mTORC1 activation in SI-AKI are yet to be fully understood. Our findings suggest that mTORC1 is involved in the activation of LPS/TLR4 signaling in renal cells and contributes to the early development of renal fibrosis via the ERK pathway, as well as to cellular senescence through DNA methylation. Therefore, we hypothesize that inhibition of mTOR by rapamycin or its analogues could represent a promising therapeutic approach for SI-AKI.
Results
mTOR Inhibition by rapamycin prevents LPS-induced AKI
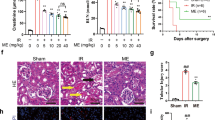
AKI was successfully induced in both endotoxemic cohorts (LPS24h and LPS48h group) (Fig. 1). After 24 h and 48 h from LPS infusion, significant morphological changes were observed in renal parenchyma, including tubular vacuolization, tubular dilation, tubular atrophy, epithelial flattening, necrosis, infiltration of inflammatory cells, and marked fibrin deposition with reduced number of capillaries in numerous glomeruli (Fig. 1A).

Recovery of renal damage in endotoxemic animals. (A) PAS staining showed significant proximal tubular epithelial cells damage (zoomed images), marked fibrin deposition, increased tubulointerstitial space, capillary rarefaction in numerous glomeruli, Bowman’s capsule expansion (zoomed images), and interstitial inflammatory infiltrates (zoomed images) at 24 h and 48 h after LPS infusion (LPS 24 h and LPS 48 h) compared to the control group (Sham group). LPS 48 h infusion induced more severe tubular and glomerular damage. Renal biopsies from endotoxemic animals receiving either rapamycin pre-treatment (RAPA/LPS) or post-treatment (LPS/RAPA) showed reduced inflammatory infiltrate and preservation of tubular and glomerular compartments (zoomed image). Rapamycin post-treatment also attenuated glomerular capillary loss, Bowman’s capsule expansion, and tubular injury, although the protective effect was less pronounced than that observed with pre-treatment (zoomed images). (B) Tubular and glomerular pathological scores were assessed as described in the Methods section and are expressed as mean ± SD from at least five mice for each group (Sham group n = 5; LPS 24 h group n = 8; LPS 48 h group n = 5; RAPA/LPS n = 8; LPS/RAPA n = 5). (C) Statistical significance was evaluated using one-way ANOVA, corrected for multiple comparisons of pairwise treatment group differences using Tukey’s method (****p < 0.0001 vs. Sham; ££££p < 0.0001 vs. LPS 24 h; ####p < 0.0001 vs. LPS 48 h).
The pathological scores of renal tubular and glomerular injury in the LPS groups were significantly higher than those observed in the sham groups (Fig. 1B). Rapamycin pre-treatment was more effective in limiting renal tissue damage compared to post-treatment group (Fig. 1B). Both rapamycin treatments significantly reduced tubular and glomerular damage and inhibited the recruitment of inflammatory infiltrates (Fig. 1A–C).
In addition, LPS injection led to a time-dependent increase of serum creatinine (Fig. 1C) compared to the sham group. Interestingly, rapamycin pre-treatment significantly improved serum creatinine level at 24 h compared to the LPS group. The rapamycin post-treatment was less effective in restoring baseline creatinine level.
Rapamycin reduces collagen deposition and renal fibrosis in LPS-induced AKI
Since glomerular thrombi and collagen deposits are considered hallmarks of SI-AKI15, we assessed the ability of LPS to induce early fibrosis and the potential mitigating effect of rapamycin by performing immunohistochemistry analysis for collagen III(Fig. 2A).

Anti-fibrotic role of rapamycin in LPS induced AKI. (A) Immunohistochemistry for collagen III showed increased collagen III deposition in endotoxemic mice (LPS 24 h and 48 h) at both interstitial and glomerular level (zoomed images). Notably, the duration of endotoxemia correlated with greater collagen III accumulation, indicative of interstitial fibrosis. Rapamycin pre-treatment (RAPA/LPS) significantly decreased collagen deposits (zoomed images). Similarly, rapamycin post-treatment (LPS/RAPA) also decreased collagen accumulation, although collagen III deposition remained detectable (zoomed images). (B) Quantitative analysis was performed as described in the Methods section. The mean intensity of strong positive (ISP) relative to the analyzed area was measured for collagen III immunohistochemistry. (Sham group n = 5; LPS 24 h group n = 8; LPS 48 h group n = 5; RAPA/LPS n = 8; LPS/RAPA n = 5). Error bars indicate standard deviation. ****p < 0.0001 vs. Sham; ££££p < 0.0001 vs. LPS 24 h; ####p < 0.0001 vs. LPS 48 h.
Tubulointerstitial and glomerular collagen deposits were absent in sham kidneys. On the contrary, LPS infusion determined extensive collagen deposition at both interstitial and glomerular levels. Notably, prolonged endotoxemia was associated with increased renal fibrosis, as observed in the LPS 48 h group compared to the LPS 24 h group. In endotoxemic animals, rapamycin post-treatment led to a slight reduction in collagen III expression at the tubulo-interstitial and glomerular levels. Interestingly, rapamycin pre-treatment significantly reduced collagen III deposition in both compartments (Fig. 2A,B).
mTOR Inhibition prevents EC dysfunction in endotoxemic AKI
Since EC play a pivotal role in pathogenesis of SI-AKI and EC dysfunction might be associated with the development of fibrosis5, we characterized endothelial phenotypic changes by double immunofluorescence staining for endothelial marker CD31 and α-SMA (Fig. 3A,B).

Rapamycin prevents EC dysfunction. (A) When activated by LPS, renal CD31+ (red) EC acquired myofibroblast marker α-SMA (green) within renal vessels, glomeruli and interstitium (LPS 24 h and LPS 48 h). Rapamycin pre-treatment (RAPA LPS) and post-treatment (LPS RAPA) restored the EC phenotype in all renal compartments. (B) Quantitative analysis was obtained as described in the Methods section. Magnification 630x. To-Pro 3 was used to counterstain nuclei (blue). (Sham group n = 5; LPS 24 h group n = 8; LPS 48 h group n = 5; RAPA/LPS n = 8; LPS/RAPA n = 5). ****p < 0.0001 vs. Sham; ££££p < 0.0001 vs. LPS 24 h; ####p < 0.0001 vs. LPS 48 h. (C,D) Primary endothelial cells (HUVEC) were exposed to LPS (4 µg/ml) for 48 h. For rapamycin pre-treatment, rapamycin (5nM) was added in medium 1 h before the LPS exposure and then maintained for 48 h. Post-Treatment was performed by incubating rapamycin 6 h after LPS stimulation. Upon LPS stimulation, EC showed a significant reduction in specific EC markers and an increased expression of dysfunctional-fibroblast markers, as determined by flow cytometry analysis. In the presence of rapamycin, EC preserved their phenotype. (C) Results are representative of three independent experiments. (****p ≤ 0.0001 vs. Basal; ££££p ≤ 0.0001 vs. LPS); (D) Representative plot.
Under basal condition, CD31+EC were localized in the intima of blood vessels as well as in peritubular and glomerular capillaries, while α-SMA+ cells were constitutively present within external tunica media of renal arteries (Fig. 3A). Following LPS infusion, EC underwent phenotypic changes, acquiring the mesenchymal marker α-SMA. Along large vessels, transitioning CD31+/α-SMA+ EC appeared detached from the underlying basement membrane and were detected within the media of the vascular wall (Fig. 3A). In renal interstitium, peritubular and glomerular EC co-expressed both CD31 and α-SMA markers (Fig. 3A,B).
Interestingly rapamycin pre- and post-treatment prevented endothelial dysfunction by reducing transitioning EC in both renal vessels and capillaries. The difference between the treated and untreated groups was statistically significant (Fig. 3B).
Next, we characterized the effects of rapamycin on LPS-induced EndMT through flow cytometry analysis (Fig. 3C,D). Our data revealed that rapamycin pre- and post-treatment inhibited endothelial dysfunction and restored constitutive endothelial and dysfunctional-fibroblastic markers to basal level (Fig. 3C,D).
Role of ERK pathway in endothelial dysfunction
Next, to identify the signaling involved in EndMT and directly activated by LPS/TLR4/mTORC-1, we analyzed the early phosphorylation of ERK and AKT, two key mediators shared by both the TLR4 and mTORC-1 pathways. Regarding mTORC-1 pathway, we assessed the activation levels of p-mTORC-1 and p-P70S6K. As shown in Fig. 4A,B, the expression of phosphorylated ERK, AKT, mTORC-1 and p70S6K were significantly increased in EC after LPS exposure. Rapamycin pre and post stimulation significantly decreased phosphorylation of mTORC-1 and p70S6K and, did not abrogate phosphorylation of the upstream modulators, AKT and ERK, as expected.

TLR4-mTORC1 axis induces EndMT through ERK signaling. (A,B) Cultured EC were stimulated with LPS (4µg/ml) for 30’. For rapamycin pre-treatment, rapamycin (5nM) was added in medium 30’ before the LPS exposure. Post-Treatment was performed incubating rapamycin after 30’ of LPS stimulation. EC were also pretreated with AKT-inh or ERK-inh for 30’ followed by a short stimulation with LPS (30’). LPS increased the expression of phosphorylated ERK, AKT, mTORC-1 and p70S6K. Rapamycin (RAPA) pre and post-stimulation significantly decreased p-mTORC1 and p-p70S6K but did not abrogate phosphorylation of AKT and ERK. Pre-treatment of EC with ERK-inh reduced LPS-induced phosphorylation of mTORC1 and p70S6K. (A) Representative plot of flow cytometry analysis; (B) Results are representative of three independent experiments. (C,D) EC were cultured with LPS or ERK-inh for 48 h; EC were pre-treated with ERK-inh for 1 h followed by LPS for 47 h. FACS analysis showed that EC did not modify their phenotype also upon LPS stimulation. (C) Representative plot of flow cytometry analysis; (D) Results are representative of three independent experiments; (****p ≤ 0.0001 vs Basal; ££££p ≤ 0.0001 vs LPS).
To selectively investigate the role of AKT and ERK signaling in LPS-induced mTORC-1 activation, we pre-treated EC with specific inhibitors targeting AKT and ERK (Fig. 4A,B). Interestingly, AKT-inhibition did not reduce the phosphorylation of mTORC1 and p70S6K triggered by LPS. However, ERK inhibition significantly reduced the phosphorylation of both mTORC-1 and p70S6K to basal level. Next, we cultured EC with LPS or ERK-inh for 48 h. For pre-treatment, EC were incubated with ERK inhibitor for 1 h, followed by LPS stimulation for 47 h, after which we analyzed their phenotype. We found that ERK-inhibited EC did not undergo phenotypic changes even after LPS stimulation, as shown by a significant decrease in α-SMA expression and an increase in endothelial markers (Fig. 4C,D). These data support the role of TLR4-ERK-mTORC1 axis in promoting EndMT and fibrosis, independently of the AKT pathway.
LPS induces DNA methylation changes in RPTEC
To evaluate whether LPS modulates DNA methylation in renal tubular cells, we stimulated human RPTEC with LPS (4 µg/ml) for 48 h and performed a whole-genome DNA methylation study.
Methylation analysis was performed for single CpG island for sets of predefined genomic regions such as CpG islands, promoters and tiling regions (5-kb spanning regions). In untreated cells (indicated as basal), the highest number of methylated tiling regions was found in chromosome 1, 8, 6, 2, 7 and 5. They cover the 50% of the total tiling regions, whereas chromosomes 9 and 14 contained the fewest methylated regions (Fig. 5A). LPS stimulation increased the DNA methylation in the overall genome when we consider the single CpG sites (Fig. 5B). However, considering the tiling regions, we found some DNA regions hypermethylated and some hypomethylated by the LPS treatment and located in specific chromosomes (Fig. 5C).

LPS-associated changes in DNA methylation as indicated by whole-genome bisulfite assay in RPTEC. (A) Chart showing the number and the frequency of methylated regions (tiling regions) identified in RPTEC at basal level. The left vertical axis represents the number of methylated regions per each chromosome. The right vertical axis indicates the cumulative percentage of the total number of occurrences. The red concave curve is the cumulative function indicating that the 50% of the total methylated regions in RPTEC are covered by chromosomes 1, 8, 6, 2, 7 and 5. (B) DNA methylation levels at the single CpG sites for RPTEC at basal level (blue line) and for RPTEC stimulated by LPS (orange line). LPS increased the DNA methylation in the overall genome. (C) Graph showing the mean DNA methylation levels of tiling regions, shared for chromosomes, in RPTEC (blue line) and in LPS-stimulated RPTEC (red line). The central axis shows the mean β methylation value. (D) Scatterplot of the CpG site methylation comparison, colored according to the combined ranks of a given site. Values are represented as mean differences (mean.diff) between stimulated and unstimulated RPTEC for each CpG site. (Combined rank: difference in mean methylation levels of stimulated and non-stimulated RPTEC, the quotient in mean methylation and the t test are ranked for all regions. This value aggregates them using the maximum, i.e., worst rank of a site among the three measures.)
We then performed differential methylation analysis between LPS-stimulated and unstimulated RPTEC focusing on CpG sites and tiling regions based on the combined rank parameter (Fig. 5D). We then selected the tiling regions having the best 200 combined ranks and a threshold of Δβ > 0.05. The analysis identified 76 regions, including 18 hypomethylated and 58 hypermethylated regions (Supplementary Table 1). To investigate the biological processes involving genes included in these aberrantly methylated DNA regions, we conducted a pathway analysis. Two hypermethylated genes, CD39 and WFS1, were identified as central nodes in the most significant network (score 53) related to energy production and aging: (Supplementary Fig. 1).
Overall, these data demonstrate that LPS has a functional impact on re-programming the epigenetic state of tubular cells, promoting aberrant methylationin across different chromosomes.
LPS induces DNA methylation regulating CD39 and WFS1 gene expression
To confirm the critical role of DNA methylation in regulating both CD39 and WFS1, we assessed the gene expression profiles in LPS-stimulated RPTEC compared to untreated cells. Both genes were differentially expressed in LPS-stimulated RPTEC and their expression inversely correlated with DNA methylation levels (Fig. 6A,B). Specifically, CD39 and WFS1 genes were hypermethylated, leading to a significant down-regulation of their gene expression (Fig. 6B: **p < 0.001, *p < 0.01).

CD39 and WFS1 gene expression is regulated by the DNA methylation. (A) Methylation levels of CD39 and WFS1 in LPS-stimulated RPTEC or cells treated with 1 µM 5-aza-2’-deoxycytidine compared to basal condition. The percentage of PMR was calculated as described in the Materials and methods. (B) Gene expression levels of CD39 and WFS1 evaluated by qRT-PCR in the LPS-stimulated RPTEC or treated with 1 µM 5-aza, compared to untreated RPTEC. Expression levels were significantly different in LPS-stimulated RPTEC compared to untreated cells. Gene expression levels were normalized to GAPDH as a housekeeping gene. Results are presented as mean ± standard deviation (SD), n = 3. **p < 0.001, *p < 0.01.
These results suggest that the expression of CD39 and WFS1 is directly regulated by DNA methylation and highlight LPS as an epigenetic mediator that induces hypermethylation of genes involved, at least in part, in the anti-inflammatory and anti-fibrotic pathways.
Characterization of LPS-induced cellular senescence in RPTEC
In various conditions of AKI, tubular epithelial cells can become senescent, characterized by cell cycle arrest, mitochondrial dysfunction, and the release of pro-inflammatory and pro-fibrotic factors16. Therefore, we evaluated whether LPS could induce senescence in renal tubular cells in vitro by performing SA-β-gal staining, assessing klotho and p21 expression, and investigating CD39 and WFS1 markers, which are associated with anti-inflammatory, anti-fibrotic and anti-aging pathways. (Fig. 7A, B).

SA-β Gal staining in RPTEC and validation of target genes. (A) SA-β-Gal activity in early passage RPTEC exposed to LPS (4 µg/ml) for 48 h. For the pre-treatment group, rapamycin (5 nM) was added to the medium 1 h before LPS exposure and maintained for 48 h. Post-treatment was performed by incubating rapamycin 6 h after LPS stimulation. The inhibition of senescence was more pronounced in the pre-treatment group compared to post-treatment. SA-β-gal + cells were observed following LPS exposure with senescent RTEC appearing enlarged and morphologically distinct from the normal cells at the same passage, showing the formation of larger and polynucleated cells. Untreated cells are referred to as basal, whike cells exposed to H₂O₂ were used as a positive control for senescence. Representative images were acquired using phase contrast microscopy. Magnification 40X; scale bar: 400 μm (B) Quantification of SA-β-Gal + cells cultures. The ratio of cells positive for SA-β-gal activity was calculated by examining five non-overlapping fields per condition (6-well plate). Results are presented as mean ± SD from three independent experiments (**p < 0.001). (C–F) Real-time PCR analysis of four genes (KLOTHO, p21, CD39 and WFS1) differentially expressed in RPTECs under basal condition, after 48 h of stimulation, pre- and post-treatment with rapamycin, as well as rapamycin alone. Data are expressed as mean ± standard deviation (SD), n = 3. (KLOTHO *p < 0.05; p21, CD39 and WFS1 **p < 0.001and *p < 0.01).
We then stimulated RPTEC by LPS for 48 h (Fig. 7A) and observed that LPS-stimulated RPTEC acquired a senescent phenotype, as evidenced by a significant increase in SA-β-GAL positivity. LPS-exposed RPTEC appeared enlarged and polynucleated. Rapamycin treatment provided significant protection against LPS-induced senescence in tubular cells. RPTEC exposed to H2O2 were used as positive control of cellular senescence. Quantification of senescent cells in Fig. 7B showed that LPS- induced senescence in RPTEC was statistically significant (***p < 0.001).
To further investigate senescence, we performed real-time PCR for klotho and p21 gene expression in an independent set of RPTECs (Fig. 7C-F). Klotho expression was significantly decreased after 48 h of LPS stimulation (Fig. 7C). Rapamycin pre-treatment efficiently restored the klotho expression up to baseline. Whereas the addition of rapamycin after 6 h from LPS stimulation was less effective in restoring the basal expression of klotho and no significant difference was found between LPS/RAPA and LPS 48 h.
Additionally, LPS significantly increased p21 expression, indicative of senescence, but both pre- and post-treatment with rapamycin reduced p21 levels significantly (Fig. 7D).
Finally, we explored the expression of CD39 and WFS1 (Fig. 7E, F), which are potentially involved in the regulation of renal tubular senescence. After 48 h of LPS stimulation, both genes were significantly decreased. Interestingly, rapamycin pre-treatment significantly increased CD39 and WFS1 expression while post-treatment was not sufficiently effective in significantly enhancing their expression and no significant difference was found between LPS/RAPA and LPS 48 h.
Rapamycin treatment prevents LPS-induced CD39 downregulation in vivo
We further investigated whether LPS infusion might promote the downregulation of CD39 in vivo. The effect of LPS infusion and rapamycin treatment in the modulation of CD39 expression was investigated by CD39 immunohistochemistry on renal biopsies (Fig. 8).
Notably, CD39 expression was typically confined to the glomerular compartment and less evident in tubules. We demonstrated a significant CD39 expression in tubular compartment, even apical and intracellular, suggesting proximal tubular cells’ involvement (Fig. 8A).Sham mice presented high levels of CD39, whereas LPS 24 h and 48 h -animals showed decreased expression in renal tubules. Rapamycin pre-treatment protected the renal parenchyma in course of endotoxemic AKI, restoring tubular CD39 expression. However, the increase of CD39 levels was less evident in mice post-treated with rapamycin (Fig. 8B).

Rapamycin treatment restores CD39 expression in LPS-induced AKI. (A) Immunohistochemistry for CD39 revealed a significant decrease of tubular CD39 expression in LPS mice (LPS 24 h), (zoomed image). At 48h, LPS exposure resulted in an even more pronounced reduction of tubular (zoomed image) and glomerular CD39 expression (zoomed image) compared to the 24h endotoxemic group. Rapamycin treatment restored CD39 expression, with pre-treatment (RAPA/LPS) being more effective than post-treatment (LPS/RAPA) in rescuing tubular CD39 levels (zoomed image). (B) Quantitative analysis was obtained as described in the Methods section. The mean intensity of strong positive (ISP) related to the area analyzed was measured for CD39 immunohistochemistry. Error bars indicate standard deviation. ****p < 0.0001 vs. Sham; ££££p < 0.0001 vs. LPS 24 h. (Sham group n = 5; LPS 24 h group n = 8; LPS 48 h group n = 5; RAPA/LPS n = 8; LPS/RAPA n = 5) (C,D) WB analysis revealed a significant decrease of CD39 expression after 24 h of LPS infusion, compared to sham level. A 48 h LPS infusion showed a marked reduction of CD39 protein. Rapamycin pre-treatment protected the kidney in course of endotoxemic AKI, restoring CD39 expression. However, post-treatment did not significantly reactivate CD39 expression. β-actin protein expression was used for normalization. (C) This gel is representative of three animals for group with similar results. (D) Data are expressed as mean ± standard deviation (SD). (Sham group n = 5; LPS 24 h group n = 8; LPS 48 h group n = 5; RAPA/LPS n = 8; LPS/RAPA n = 5). Statistically significant differences were assessed one-way ANOVA, corrected for multiple comparison of pairwise treatment group differences using Tukey’s method (****p < 0.0001 vs. Sham; ££p < 0.01 vs. LPS 24 h; ###p < 0.001 vs. LPS 48 h).
These results were confirmed by analyzing protein lysates from tissue in western blot experiments (Fig. 8C,D). CD39 protein analysis demonstrated a significant decrease in its expression in the renal cortex of both LPS 24 h and LPS 48 h-treated mice with a more pronounced reduction at 48 h. Rapamycin pre-treatment effectively restored CD39 expression. On the contrary, the expression levels of CD39 did not increase following rapamycin post-treatment and no significant difference was observed between LPS/RAPA and LPS 24 h.
mTOR Inhibition prevents LPS-induced klotho downregulation in vivo
Klotho, as previously described, was originally identified as an anti-aging protein and defects in gene expression have been associated with premature aging syndrome. Renal tubular epithelial cells are identified as the major producer of klotho and its decrease correlated with the severity of histological kidney damage.
The effect of LPS infusion and rapamycin treatment upon tubular klotho expression was investigated by klotho immunohistochemistry on renal biopsies (Fig. 9).

Characterization of tubular dysfunction and rapamycin effects. (A) Immunohistochemistry for klotho revealed a significant decrease of tubular klotho expression in LPS mice and its restoration by rapamycin treatment. After 48 h from LPS infusion, tubular klotho expression was dramatically decreased. Rapamycin pre-treatment (RAPA/LPS) more than post- (LPS/RAPA), restored tubular klotho expression. (B) The quantitative analysis was obtained as described in the Methods section. Mean number of strong positive (NSP) related to the area analyzed was measured for klotho immunohistochemistry. Error bars indicate standard deviation. (Sham group n = 5 LPS 24 h group n = 8 LPS 48 h group n = 5 RAPA/LPS n = 8 LPS/RAPA n = 5). ****p < 0.0001 vs. Sham; ££££p < 0.0001 vs. LPS 24 h; ####p < 0.0001 vs. LPS 48 h. (C,D) WB analysis revealed a significant decrease of klotho expression after 24 h of LPS infusion, compared to sham level. After 48 h from LPS infusion, there was a marked reduction of klotho expression. The rapamycin treatment, particularly in pre-treated group, preserved renal parenchyma, restoring klotho expression. β-actin protein expression was used for normalization. (C) This gel is representative of three animals for group with similar results. (D) Data are expressed as mean ± standard deviation (SD) and statistical differences were assessed one-way ANOVA, corrected for multiple comparison of pairwise treatment group differences using Tukey’s method. (Sham group n = 5 LPS 24 h group n = 8 LPS 48 h group n = 5 RAPA/LPS n = 8 LPS/RAPA n = 5) ****p < 0.0001 vs. Sham; ££p < 0.0001 vs. LPS 24 h; ###p < 0.0001 vs. LPS 48 h.
Notably, klotho expression was predominantly confined to the tubular compartment and was less evident in glomeruli, confirming its production by proximal tubular cells (Fig. 9A). Sham mice presented high levels of klotho, whereas LPS 24 h and 48 h infusions significantly reduced its expression in renal tubules. In our study, rapamycin pre-treatment protected the kidney from endotoxemic AKI by restoring tubular klotho expression and inhibiting the development of interstitial fibrosis. The increase of klotho level was less evident in mice post-treated with rapamycin as compared to those pre-treated (Fig. 9B). Although post-treatment with rapamycin also provided significant protection, increasing klotho levels, its effect was less pronounced compared to pre-treated groups.
This result has been confirmed by analyzing protein lysates from tissues in western blot experiments (Fig. 9C, D). The analysis of klotho protein demonstrated a significant decrease of this protein in renal cortex of both LPS 24 h and LPS 48 h mice and its restoration by both rapamycin treatments.
Discussion
This study provides novel insights on the short-terms effects of rapamycin in preventing endothelial dysfunction and cellular senescence, counteracting renal damage in the course of SI-AKI.
AKI, a common consequence of sepsis, is characterized by an exacerbated immune response associated with systemic endothelial impairment and alterations in renal resident cells1. A crucial mediator of SI-AKI is endotoxin, which exacerbates the host inflammatory response and has direct detrimental effects on renal parenchymal cells17.Activation of mTORC1 in response to endotoxin challenge is considered a major pathway involved in the LPS-TLR4 axis11, but the molecular mechanisms and significance in renal damage are not fully defined.
There are two distinct mTOR complexes in mammalian cells: mTORC1 and mTORC2. mTORC1 participates in the regulation of biosynthetic and metabolic events18,19 and is sensitive to rapamycin20. Evidence demonstrates that rapamycin reduces the risk of developing lung damage in severe sepsis, as it plays a central role in TLR2- and TLR4-induced pro-inflammatory responses in neutrophils and acute lung injury20. Additionally, mTOR is emerging as a crucial modulator of several chronic renal diseases, contribuiting to renal fibrosis by promoting fibroblast activation21,22. In ischemic AKI, mTORC1 has dual roles in renal repair and injury23. While its activation is necessary for renal tissue regeneration, prolonged activation contributes to persistent cellular dysfunct, leading to the progression of renal injury and fibrosis23.
Our study demonstrates that rapamycin treatment protects the kidneys during endotoxemia by preventing endothelial dysfunction, tubular senescence, and the development of interstitial fibrosis. We examined the molecular mechanisms involving mTORC1 in LPS-induced endothelial dysfunction and tubular senescence. Our findings provide novel insights into the activation of the mTORC1-S6K pathway in EC upon LPS exposure24. Contrary to previous reports, we show that this activation is independent of the PI3K-Akt pathway. Instead, our data suggest that the MEK-ERK pathway serves as an alternative route for mTORC1-S6K activation in EC upon LPS stimulation. Specifically, we propose that ERK phosphorylates TSC2, leading to the dissociation of the TSC1/TSC2 complex and consequent up-regulation of mTORC1, similar to the mechanism observed in tumor-like cells25.Furthermore, our findings suggest that ERK activation plays a pivotal role in the EndMT process in response to LPS. Taken together, our data point towards a potential mechanism of ERK-mediated endothelial dysfunction in response to LPS, involving mTORC1-S6K activation. These results have significant implications for understanding the molecular mechanisms underlying LPS-induced endothelial dysfunction, and may provide a basis for developing targeted therapeutic interventions.
Endothelial dysfunction, the direct interaction of RPTEC with bacterial components, the release of pro-inflammatory cytokines by pathogen-activated immune cells, and pro-coagulant mediators can activate pro-senescence pathways in RPTEC, accelerating the maladaptive repair linking AKI to CKD7. Epigenetic mechanisms have emerged as important regulators in AKI, leading to structural and functional changes26. Several endogenous mediators such as complement factors, inflammatory cytokines and bacterial components like LPS, can directly affect RPTEC chromatin status by histone acetylation and methylation7.
Our study provides evidence that LPS can trigger changes in DNA methylation, altering the expression of genes involved in inflammatory and senescence responses. We identified and validated two main genes as central nodes in the aging network: CD39 and WFS1. These hypermethylated genes are functionally linked to renal aging. The role of CD39 in regulating the response to septic-AKI is still under research27. We hypothesize an anti-inflammatory and reno-protective role for CD39, also known as ecto-apyrase ENTPD1, a vascular ectoenzyme expressed by EC and smooth muscle28, widely distributed in the kidneys (e.g., larger vessels, afferent arterioles, and glomeruli) and recently found in RPTEC27. CD39 metabolizes extracellular ATP and ADP to AMP, which is then converted to adenosine (ADO) by ecto-5’-nucleotidase (CD73). ADO has direct endothelial protection, anti-inflammatory, and antithrombotic effects29.
Extracellular ATP and ADP are released from damaged tissues, injured cells, and platelet degranulation, all leading to a proinflammatory environment30. By regulating extracellular nucleotide levels, CD39 controls ligand availability to a large family of type 2 purinergic receptors (P2) involved in acute rejection and chronic allograft dysfunction31,32. CD39 also serves as a vascular protective factor in various pathophysiological conditions such as cancer33, autoimmune diseases30, I/R injury29 and diabetic nephropathy32. Recent studies show that CD39 ameliorates myocardial infarction and improves survival in mouse models of SI-AKI by decreasing systemic inflammation and renal damage34. Notably, one recent study demonstrated that CD39 effectively enhances survival in a mouse model of SI-AKI by reducing systemic inflammation and renal parenchyma damage27. The authors revealed that CD39 overexpression in RPTEC counteracts the detrimental effects of LPS by suppressing the activation of the NLRP3-related inflammatory pathway, thereby modulating cell apoptosis and cell cycle arrest27.
In aging-related diseases, there is a complex crosslink between endoplasmic reticulum (ER) stress and p21 signaling35. In diabetic RPTECs, ER stress markers correlate with enhanced SA-β-gal-positive cells and premature senescence. WFS1 encodes a transmembrane protein localized to the ER, regulating the unfolded protein response36. Mutations or altered expression of WFS1 are associated with rapid progression to renal failure37,38. Our analyses demonstrated LPS-induced hypermethylation of WFS1, reducing its expression and increasing cell cycle regulator p21. Considering ER stress’s role in AKI progression, WFS1 signaling could be crucial in premature senescence of RTECs in sepsis-induced AKI.
Several studies confirm rapamycin’s efficacy in prolonging lifespan and attenuating age-related diseases39. Since mTOR activation promotes p21 expression, mediating cell cycle arrest and senescence in renal cells, rapamycin could prevent renal aging40. Our study assessed whether rapamycin could restore CD39 and WFS1 expression and prevent RPTEC senescence. Rapamycin pre-treatment counteracted LPS effects by upregulating CD39 and WFS1, restoring klotho expression, and reducing SA-βgal and p21 positivity. In vivo results confirmed that LPS accelerates renal aging, while rapamycin abrogated these detrimental effects.
We acknowledge that our study has some limitations. First, methylation data were obtained from only three different lots of human RPTEC and we lack data on the methylation status of our mouse model or on rapamycin-induced methylation. Additionally, the 48-hour LPS stimulation may not fully capture the dynamic effects of LPS on DNA methylation in RPTEC cells.
Despite this, our study focused on evaluating the short-term effects of rapamycin in mitigating AKI under septic conditions, emphasizing the immediate molecular and cellular mechanisms involved. These findings provide valuable insights into the acute phase, supporting rapamycin’s role in reducing inflammation, endothelial dysfunction, and premature RPTEC senescence, thereby attenuating renal damage. The long-term renal recovery and functional impact of rapamycin warrant further investigation in future studies.
Materials and methods
All methods were performed in accordance with relevant guidelines and regulations, as outlined in the editorial policies of Scientific Reports.
Animal model
All experiments were conducted using 12- to 16-week-old male and female C57BL/6 N mice (Charles River Laboratories, Inc.) hosted in five animals per cage. Mice were maintained and used at the Molecular Biotechnology Center “Guido Tarone” (University of Turin, Italy) and treated following institutional animal welfare guidelines and legislation, approved by the Animal Ethical Commission of the University of Turin and the Italian Ministry of Health (742/2018-PR prot. N. CC652.92). All the reported methods are in accordance with the ARRIVE guidelines.
Briefly, mice were weighted (22–25 g/each) and endotoxemia was induced by intraperitoneal administration of a saline solution containing 10 mg/kg of LPS (lipopolysaccharide membrane of Escherichia coli, L2630-25MG) (Sigma-Aldrich Saint Louis, MO, USA). Specifically, under anesthesia using 2% isoflurane, the LPS was injected intra-peritoneally (ip) into the right iliac fossa, with either 10 mg/kg of LPS or an equivalent volume of n-saline solution as a control. Subsequently, 1 ml n-saline solution was administered intra-peritoneally into the left iliac fossa as fluid replacement.
The mice (N = 36) were randomized using block randomization to ensure equal distribution of male and female mice across the following groups:
-
Sham group:5 mice treated with 100 µl PBS ip injection.
-
LPS group 24 h: 8 mice received 10 mg/Kg of LPS ip injection and were sacrificed after 24 h from LPS challenge.
-
LPS group 48 h:5 mice received 10 mg/Kg of LPS ip injection and were sacrificed after for 48 h from LPS challenge.
-
Rapamycin (RAPA):5 mice received 5 mg/Kg of rapamycin (Sigma R0395) ip injection.
-
Rapamycin/LPS (RAPA/LPS, pre-treatment): 8 mice received two ip injections of 5 mg/kg of rapamycin separated by a period of 24 h, followed by ip injection of 10 mg/Kg of LPS.
-
LPS/Rapamycin (LPS/RAPA, post-treatment): 5 mice received one ip injection of LPS 10 mg/Kg and after 6 h they were treated with 5 mg/Kg rapamycin. Mice received the second injection of Rapamycin after 24 h from LPS infusion.
Only one of the two researchers in charge for animal handling was aware of the group allocation at the different stage of experiment. No specific confounders were considered. Endotoxemic animals were sacrificed 24 h (LPS 24 h) and 48 h (LPS 48 h) after LPS administration, respectively. Rapamycin-pretreated animals (RAPA/LPS) were sacrificed 24 h after LPS injection (48 h after the first rapamycin injection). LPS/RAPA animals were sacrificed 48 h after LPS injection. Sham mice and RAPA mice were sacrificed 24 h after PBS and Rapamycin injections, respectively.
No exclusion criteria were set. A sample size of five mice per group was calculated to detect a difference of 20% (SD 0.2) in both tubular and glomerular pathological scores with a power of 80% and alpha error of 0.05.
Collection of samples
Mice were sacrificed under anesthesia with 2% isoflurane with a cardiac puncture to collect blood followed by cervical dislocation to confirm animal death. At sacrifice, kidneys were collected from all animals to assess tubular and glomerular damage. A portion of each biopsy specimen was immediately snap-frozen in Optimal Cutting Temperature (OCT, Tissue-Tek) medium and stored in liquid nitrogen. Another portion was fixed in buffered formalin (4%) for 12 h and embedded in paraffin by using standard procedures. Mice sera and plasma samples were collected and were stored at − 80 °C until their use. Renal function was assessed by serum creatinine.
Histological analysis
Two-µm-thick sections of formalin-fixed paraffin-embedded C57BL/6 kidney sections were deparaffinized and rehydrated with alcohol. Slides were then washed with distilled water and processed for histologic staining [Hematoxylin and Eosin (H&E), Periodic Acid-Schiff (PAS). Then, digital slides were analyzed and acquired by the Aperio ScanScope CS2 device (Aperio Technologies, Vista, CA, USA) and the entire digital slides analyzed with ImageScope V12.1.0.5029 (Aperio Technologies). H&E and PAS staining were performed to evaluate histological injury. Tubular and glomerular injury was scored semi quantitatively by two blinded observers who examined at least 20 visual field of each kidney section. The tubular damage was defined by evident signs of tubular dilation, tubular atrophy, tubular epithelial cell necrosis, and cast formation. The score of tubular lesions was assessed by the following score: 0 = normal kidney without tubular injury; 1 = < 10% of tubules injured; Score 2: 10–25% of tubules injured; Score 3: 25–50% of tubules injured; Score 4: 50–74% of tubules injured; Score 5: >75% of tubules injured. The glomerular lesion score: 0 = normal glomeruli; 1 = mild deposition of fibrin and reduced number of capillaries in a few glomeruli; 2 = moderate fibrin deposition and reduced number of capillaries in numerous glomeruli; 3 = marked fibrin deposition and reduced number of capillaries in numerous glomeruli; 4 = marked fibrin deposition and reduced number of capillaries in numerous glomeruli and glomerulosclerosis; 5 = glomerulosclerosis. The score index in each animal was expressed as a mean value of all scores obtained. Both tubular and glomerular pathological score of each group was expressed as median ± interquartile range (IQR) (Sham group n = 5; LPS 24 h group n = 8; LPS 48 h group n = 5; RAPA/LPS n = 8; LPS/RAPA n = 5).
Immunohistochemistry
For the immunohistochemical evaluation of collagen III (Abcam, ab7778), Klotho (LS Bio, b6625) and CD39 (ectonucleoside triphosphate diphosphohydrolase 1, ENTPD1) (Abcam, ab223842), two-µm-thick sections of formalin-fixed paraffin-embedded renal sections underwent deparaffinization and heat-mediated antigen retrieval (citrate buffer, pH = 6.00 for collagen III and klotho sections; Tris/EDTA buffer pH 9.0 for CD39 sections). After epitope unmasking, the slides were incubated with H2O2 (3%), klotho sections were permeabilized with Tween-20 (0.1%) and collagen III sections were permeabilized with Triton 0.25%. Then, blocked by Protein Block Solution (Dako Cytomation, USA) and primary antibody incubation. The positive staining was detected by the Peroxidase/DAB Dako Real EnVision Detection System (Dako, Glostrup, Denmark). The peroxidase reaction was shown by a brown precipitate, counterstained with Mayer hematoxylin (blue). Negative controls were prepared by incubation with a control irrelevant antibody. Images were acquired by Aperio ScanScope CS2 device (Aperio Technologies) and signals were analyzed using the ImageScope V12.1.0.5029 (Aperio Technologies, Vista, CA). Collagen III and CD39 staining were quantified as the ratio of total intensity of strong positive (ISP) / the total analyzed area (ISP/Area) for each animal. For klotho staining, results were expressed as the ratio of number of strong positive (NSP) over the total analyzed area (NSP/Area).
Confocal laser scanning microscopy
Renal sections were double stained for α-SMA (Santa Cruz Biotechnologies, Santa Cruz, CA) and CD31 (Abcam, Cambridge, USA). All the antibodies cross-react with mouse tissue. Tissue sections were deparaffinized through xylene and alcohol and underwent epitope retrieval through three microwave (750 W) cycles of 5 min in citrate buffer (pH = 6). After that they were incubated with specific blocking solution, primary antibodies (anti-α-SMA 1:100, anti-CD31 1:30) and the corresponding secondary antibodies (Alexa Fluor 488 anti-mouse; Alexa Fluor 555 goat anti-rabbit, Molecular Probes, Eugene, OR). All sections were counterstained with TO-PRO-3 (Molecular Probes) and mounted with fluoromount. Negative controls were prepared by omitting the primary antibody.
Image acquisition was performed by confocal microscope Leica TCS SP2 (Leica, Wetzlar, Germany). The number of CD31+/α-SMA+ was quantified in at least 10 high power (×630) fields (HPF)/sections by two independent observers blinded to the origin of the slides. The final counts were the mean of the two measures. In no case inter-observer variability was higher than 20%.
Western blot
Protein lysates were homogenized by RIPA buffer with phosphatase and protease inhibitors. Proteins (20 µg) were separated in 4–15% polyacrylamide gel (Bio-Rad, Hercules, CA) and then transferred to PVDF membrane (Trans-Blot Turbo Midi PVDF, 0.2 mM; Bio-Rad) by Trans-Blot Turbo (Bio-Rad). After blocking in 2% of BSA, the membranes were incubated overnight with the following primary antibodies: Klotho (LS Bio, b6625) [1:500] and CD39 (Abcam, ab223842) [1:1000] and then with secondary antibody Goat Anti-Rabbit IgG HRP-conjugated (Abcam, ab6721) [1:2000]. The same membrane was probed with mouse monoclonal anti-β-actin antibody (1:20.000; Sigma). The Electrochemiluminescence (ECL) system was used to detect the binding antibody, (Amersham, UK). The chemiluminescent signal was acquired by Chemidoc and quantified using Image J software.
EC culture and immunophenotypic analysis
Human umbilical vein endothelial cells lines (HUVEC) (ATCC-LGC Standards, Italy) were grown in EndGro (Merck Millipore) at 37°C in an atmosphere of 5% CO2. Cells were stimulated with LPS (4 µg/ml) for 30’. For pre-treatment, rapamycin (5 nM) was added to the medium 30 ‘before LPS exposure. For post-treatment, rapamycin was added after 30’ of LPS stimulation.
For pERK inhibition, cells were pretreated with SC1 (Pluripotin, Abcam) at 1 µM for 30’ and 1 h. For pAKT inhibition assay, cells were preincubated with Akt inhibitor IV (Akt-inh IV, Calbiochem) for 30’ before the LPS exposure. EC phenotype was analyzed by FACS analysis as already described41.
For Intracellular staining of phosphorylated proteins cells were fixed and permeabilized with IntraPrep™ kit (Instrumentation Laboratory) and incubated for unconjugated primary antibody, anti- Phospho-Akt (T308) (Cell Signaling Technology, USA), anti-Phospho-p44/42 MAPK (Erk1/2) (Thr202/Tyr204) (Cell Signaling Technology, USA), anti-Phospho-mTOR (Ser2448) (Cell Signaling Technology, USA), anti-Phospho-p70 S6 Kinase (Thr389) (Cell Signaling Technology, USA), 25 min at 4 °C. Cells were then washed and labeled with secondary Antibody AlexaFluor 488 (Molecular Probes) and secondary Antibody AlexaFluor 555 (Molecular Probes) for 25 min at 4 °C. Finally, cells were washed twice and resuspended in FACS buffer for acquisition as previously described41.
To evaluate phenotypic changes related to the EndMT process, ECs were cultured with LPS or ERK-inh for 48 h. Pre-treatment with ERK-inh was conducted for 1 h, followed by LPS exposure for the remaining 47 h.
The EC phenotype was analyzed by surface staining, as previously described41, using allophycocyanin-conjugated anti-CD31 (Miltenyi Biotec, Bergisch Gladbach, Germany), and phycoerythrin-conjugated anti-vascular endothelial (VE) cadherin (Miltenyi Biotec). Following surface incubation, cells were washed with FACS buffer and FACs analysis was performed.
Intracellular staining for unconjugated α-SMA (Abcam) was performed following cell fixation and permeabilization using the IntraPrep™ kit, with incubation for 25’ at 4 °C. Cells were then washed and labeled with the AlexaFluor 488 secondary antibody (Molecular Probes) for an additional 25 min at 4 °C. Finally, cells were washed twice, and data acquisition was conducted.
RPTEC culture and DNA/RNA extraction
RPTEC (human Renal Proximal Tubular Epithelial Cells) between 3 and 5 passages were grown to confluence in RGEM medium (Renal epithelial cell growth medium, Lonza) in which a basal medium RBEM was supported with rhEGF (recombinant human EGF), transferrin, insulin, hydrocortisone, epinephrine, triidothyronine, and Fetal Bovine Serum (FBS), preserved at + 4 °C and changed every two days in the culture. Cells with medium were incubated at 37 °C with CO2 at 5%. Once they became confluent, cells were exposed to LPS (4 µg/ml) or rapamycin (5nM) for 48 h. For the pre-treatment group, rapamycin (5 nM) was added to the medium 1 h before LPS exposure and maintained for 48 h. Post-treatment was performed by incubating rapamycin 6 h after LPS stimulation.
After LPS stimulation, cells were washed 3 times in PBS1X, and fresh medium was replaced. In addition, cells were exposed to H2O2 (100–300 μm) as positive control of senescence. For the in vitro experiments, including the whole-genome methylation analysis, we used three independent batches of RPTEC cells DNA and RNA were simultaneously extracted from RPTEC using the All Prep DNA/RNA Mini Kit (Qiagen) according to the manufacturer’s protocol. Nucleic acid concentration and quality were assessed using a NanoDrop Spectrophotometer (NanoDrop Technologies).
Real-time PCR analysis
Total RNA (500 ng) was used in a reverse transcription reaction by using the iScript cDNA Synthesis Kit (Bio-Rad, Hercules, CA, USA) according to the manufacturer’s instructions. Real-time quantitative PCR was performed on an iCycler Thermal Cycler (Bio-Rad) by using Klotho (Kl), CDKN1A p21/Waf1, CD39 and WFS1 primers (Integrated DNA Technologies, Coralville, IA, USA) (Supplementary Table 2)in combination with SYBR Green Master Mix (Bio-Rad) and the Light Cycler@96 (Roche). The relative amounts of mRNA were normalized to β-actin mRNA as the housekeeping gene. Data was analyzed using the ΔΔCt method.
DNA methylation analysis
DNA methylation analysis was performed utilizing the Illumina methylation profiling platform consisting of HiScanSQ system and Infinium HumanMethylation450 BeadChips as previously described42. Bisulfite conversion of DNA extracted from RTEC was performed with the EZ-96 DNA Methylation Kit (ZymoResearch), and subsequent hybridization of this DNA was carried out on the InfiniumHumanMethylation450BeadChips. Microarray data of the LPS stimulated RTEC and normal cultured RTEC are available in BioStudies database, under accession number E-MTAB-13,396 at https://www.ebi.ac.uk/biostudies/arrayexpress.
All statistical analysis was performed using R and the RnBeads R package. Differential methylation analysis on site and region level was computed considering the difference in mean methylation levels LPS stimulated RTEC and normal cultured RTEC, the quotient in mean methylation and the t test assessing whether the methylation values in the two groups originate from distinct distributions. Additionally, each site was assigned a rank based on each of these three criteria. A combined rank was computed as the maximum (i.e., worst) rank among the three ranks. Coverage of differential methylation analysis on region level was targeted across gene regions with sites in the promoter region, 5’-UTR, first exon, gene body and 3’-UTR in order to provide the broadest most comprehensive view of methylation state possible.
Quantitative methylation-specific PCR assay (qMSP)
Bisulfite converted DNA was used to perform the qMSP by Methylamp MS-qPCR Fast Kit (Epigentek Group) according to the manufacturer instructions. For each reaction 20 ng of bisulfite-treated DNA was used as template. Primers for the genes of interest were designed using MethPrimer (http://www.urogene.org/methprimer/index.html) (Supplementary Table 2). ACTB (β-actin) gene was used as a reference gene. No- template controls were included in each run as negative controls. An EpiTect Control DNA, a 100% methylated DNA (Qiagen), was used as a positive control for all genes studied. The PMR (percentage of methylated reference) (i.e., degree of methylation) was used to define the percentage of fully methylated molecules at a specific locus and was calculated as reported previously. Briefly, the PMR value was calculated by dividing the gene/ACTB ratio in a sample by the gene/ACTB ratio in SssI-treated leucocyte DNA (Qiagen) and multiplied by 100. Parallel PCRs were carried out for the genes of interest and reference. PMR values were detected using the comparative CT method. The relationship between the percentages of methylated DNA molecules and CT is described as PMR = 2 − DDCT×100%.
Ingenuity pathway analysis (IPA) for gene network construction in differentially methylated regions
The analysis of gene networks within differentially methylated regions was conducted using the Ingenuity Pathway Analysis (IPA) software (IPA®, IPA Analysis Match 50 dataset LNUL, QIAGEN Redwood City, http://www.qiagen.com/ingenuity).
SA-β-gal test
Senescence-associated SA-β-gal staining was performed as described in manufacturer’s protocol (Cell Signaling Technology). After washing with PBS (pH 6.0), the cells were fixed in 4% paraformaldehyde for 15 min and stained with freshly prepared SA-β-gal solution (1 mg/mL X-gal, 40 mM citric acid/sodium phosphate (pH = 6.00), 5 mM potassium ferrocyanide, 5 mM potassium ferricyanide, 150 mM NaCl, and 2 mM MgCl). Next, cells are incubated at 37 °C overnight in a dry incubator. Then, we removed the staining solution, immersed the samples in 70% glycerol, and assessed the development of blue color. The number of SA-β-gal- positive cells was determined by counting at least 500 cells per dish (60 mm) in five not overlapping fields. The % of the number of SA-β-gal strong positive cells that appeared with increased dimension is calculated as ratio considering the total numbers of cells counted in each dish. described as PMR = 2 − DDCT×100%.
Statistical analysis
Graphs were displayed using GraphPad Prism Software 8.0 (GraphPad software, Inc., San Diego, CA, USA). Data were expressed as mean ± standard deviation (SD) and were analyzed using one-way ANOVA, corrected for multiple comparison of pairwise treatment group differences using Tukey’s method (Supplementary Table 3). A p-value of < 0.05 was considered significant.
Data availability
Microarray data that support the findings of this study have been deposited in the BioStudies database, with the primary accession code E-MTAB-13396.
Abbreviations
- AKI:
-
Acute kidney injury
- SI-AKI:
-
Sepsis-induced AKI
- mTOR:
-
Mammalian Target of Rapamycin (mTOR)
- LPS:
-
Lipopolysaccharide
- ENTPD1:
-
Ectonucleoside triphosphate diphosphohydrolase 1
- WFS1:
-
Wolframin ER transmembrane glycoprotein
- TLR4:
-
Toll-like receptor 4
- EC:
-
Endothelial cells
- RPTEC:
-
Renal proximal tubular epithelial cells
- ALI:
-
Acute lung injury
- SD:
-
Standard deviation
References
Gómez, H. & Kellum, J. A. Sepsis-induced acute kidney injury. Curr. Opin. Crit. Care. 22, 546–553 (2016).
Dauphinee, S. M. & Karsan, A. Lipopolysaccharide signaling in endothelial cells. Lab. Invest. 86, 9–22 (2006).
LeBleu, V. S. et al. Origin and function of myofibroblasts in kidney fibrosis. Nat. Med. 19, 1047–1053 (2013).
Singla, S. et al. Pro-inflammatory endothelial cell dysfunction is associated with intersectin-1s down-regulation. Respir. Res. 12, 46 (2011).
Castellano, G. et al. Endothelial dysfunction and renal fibrosis in endotoxemia-induced oliguric kidney injury: possible role of LPS-binding protein. Crit. Care. 18, 520 (2014).
Echeverría, C. et al. Lipopolysaccharide induces a fibrotic-like phenotype in endothelial cells. J. Cell. Mol. Med. 17, 800–814 (2013).
Franzin, R. et al. Inflammaging and complement system: A link between acute kidney injury and chronic graft damage. Front. Immunol. https://doi.org/10.3389/fimmu.2020.00734 (2020).
Lin, X., Jin, H., Chai, Y. & Shou, S. Cellular senescence and acute kidney injury. Pediatr. Nephrol. 37, 3009–3018 (2022).
Li, Y. & O Lerman, L. Cellular senescence: A new player in kidney injury. Hypertens. (Dallas Tex. : 1979). 76, 1069–1075 (2020).
Nijland, R., Hofland, T. & van Strijp, J. A. G. Recognition of LPS by TLR4: potential for anti-inflammatory therapies. Mar. Drugs. 12, 4260–4273 (2014).
Weichhart, T. et al. The TSC-mTOR signaling pathway regulates the innate inflammatory response. Immunity 29, 565–577 (2008).
Lee, P. S. et al. mTORC1-S6K activation by endotoxin contributes to cytokine up-regulation and early lethality in animals. PloS One. 5, e14399 (2010).
Hu, Y. et al. Activation of MTOR in pulmonary epithelium promotes LPS-induced acute lung injury. Autophagy 12, 2286–2299 (2016).
Xu, J., Zhou, L. & Liu, Y. Cellular senescence in kidney fibrosis: pathologic significance and therapeutic strategies. Front. Pharmacol. 11, 1–17 (2020).
Stasi, A. et al. PMMA-Based continuous hemofiltration modulated complement activation and renal dysfunction in LPS-Induced acute kidney injury. Front. Immunol. https://doi.org/10.3389/fimmu.2021.605212 (2021).
Li, Z. L. et al. Renal tubular epithelial cells response to injury in acute kidney injury. EBioMedicine 107, 105294 (2024).
Opal, S. M. Endotoxins and other sepsis triggers. Contrib. Nephrol. 167, 14–24 (2010).
Hay, N. & Sonenberg, N. Upstream and downstream of mTOR. Genes Dev. 18, 1926–1945 (2004).
Wullschleger, S., Loewith, R. & Hall, M. N. TOR signaling in growth and metabolism. Cell 124, 471–484 (2006).
Lorne, E. et al. Participation of mammalian target of Rapamycin complex 1 in Toll-like receptor 2- and 4-induced neutrophil activation and acute lung injury. Am. J. Respir. Cell Mol. Biol. 41, 237–245 (2009).
Lieberthal, W. & Levine, J. S. The role of the mammalian target of Rapamycin (mTOR) in renal disease. J. Am. Soc. Nephrology: JASN. 20, 2493–2502 (2009).
Gui, Y. & Dai, C. mTOR signaling in kidney diseases. Kidney360 1, 1319–1327 (2020).
Lieberthal, W. & Levine, J. S. Mammalian target of Rapamycin and the kidney. II. Pathophysiology and therapeutic implications. Am. J. Physiol. Renal. Physiol. 303, F180–F191 (2012).
Thomson, A. W., Turnquist, H. R. & Raimondi, G. Immunoregulatory functions of mTOR Inhibition. Nat. Rev. Immunol. 9, 324–337 (2009).
Ma, L., Chen, Z., Erdjument-Bromage, H., Tempst, P. & Pandolfi, P. P. Phosphorylation and functional inactivation of TSC2 by Erk implications for tuberous sclerosis and cancer pathogenesis. Cell 121, 179–193 (2005).
Shiels, P. G., McGuinness, D., Eriksson, M., Kooman, J. P. & Stenvinkel, P. The role of epigenetics in renal ageing. Nat. Rev. Nephrol. 13, 471–482 (2017).
Yang, M., Lu, L., Kang, Z., Ma, T. & Wang, Y. Overexpressed CD39 mitigates sepsis–induced kidney epithelial cell injury via suppressing the activation of NLR family Pyrin domain containing 3. Int. J. Mol. Med. 44, 1707–1718 (2019).
de Bertoncheli, C. et al. M. Increased NTPDase activity in lymphocytes during experimental sepsis. TheScientificWorldJournal 941906 (2012). (2012).
Crikis, S. et al. Transgenic overexpression of CD39 protects against renal ischemia-reperfusion and transplant vascular injury. Am. J. Transplantation: Official J. Am. Soc. Transplantation Am. Soc. Transpl. Surg. 10, 2586–2595 (2010).
Antonioli, L., Pacher, P., Vizi, E. S. & Haskó, G. CD39 and CD73 in immunity and inflammation. Trends Mol. Med. 19, 355–367 (2013).
Roberts, V., Stagg, J. & Dwyer, K. M. The role of ectonucleotidases CD39 and CD73 and adenosine signaling in solid organ transplantation. Front. Immunol. 5, 64 (2014).
Friedman, D. J., Rennke, H. G., Csizmadia, E., Enjyoji, K. & Robson, S. C. The vascular ectonucleotidase ENTPD1 is a novel renoprotective factor in diabetic nephropathy. Diabetes 56, 2371–2379 (2007).
Bastid, J. et al. ENTPD1/CD39 is a promising therapeutic target in oncology. Oncogene 32, 1743–1751 (2013).
Ziegler, M. et al. A single-chain antibody-CD39 fusion protein targeting activated platelets protects from cardiac ischaemia/reperfusion injury. Eur. Heart J. 39, 111–116 (2018).
Yamada, T. et al. WFS1-deficiency increases Endoplasmic reticulum stress, impairs cell cycle progression and triggers the apoptotic pathway specifically in pancreatic beta-cells. Hum. Mol. Genet. 15, 1600–1609 (2006).
Liu, J. et al. Receptor for advanced glycation end-products promotes premature senescence of proximal tubular epithelial cells via activation of Endoplasmic reticulum stress-dependent p21 signaling. Cell. Signal. https://doi.org/10.1016/j.cellsig.2013.10.002 (2014).
Yuca, S. A. et al. Rapidly progressive renal disease as part of Wolfram syndrome in a large inbred Turkish family due to a novel WFS1 mutation (p.Leu511Pro). Eur. J. Med. Genet. 55, 37–42 (2012).
Franceschini, N. et al. The association of genetic variants of type 2 diabetes with kidney function. Kidney Int. 82, 220–225 (2012).
Weichhart, T. mTOR as regulator of lifespan, aging, and cellular senescence: A Mini-Review. Gerontology 64, 127–134 (2018).
Zhuo, L. et al. Expression and mechanism of mammalian target of Rapamycin in age-related renal cell senescence and organ aging. Mech. Ageing Dev. 130, 700–708 (2009).
Sallustio, F. et al. Renal progenitor cells revert LPS-induced endothelial-to-mesenchymal transition by secreting CXCL6, SAA4, and BPIFA2 antiseptic peptides. FASEB J. https://doi.org/10.1096/fj.201900351r (2019).
Castellano, G. et al. Complement component C5a induces aberrant epigenetic modifications in renal tubular epithelial cells accelerating senescence by Wnt4/betacatenin signaling after ischemia/reperfusion injury. Aging 11, 4382–4406 (2019).
Acknowledgements
The authors thank Dr. Angela Picerno and Dr Chiara Divella for excellent technical assistance.
Funding
This study was supported by University of Bari “Aldo Moro” and the Italian Ministry of Health (Giovani Ricercatori 2011-2012, GR‐2011‐02351027, granted to GC; Fondo Sociale Europeo, Azione I.2 “Attrazione e Mobilità Internazionale dei Ricercatori”‐AIM‐1810057‐activity 2 granted to AS).
Author information
Authors and Affiliations
Contributions
A.S. (Alessandra Stasi) and G.C. (Giuseppe Castellano) conceived and coordinated the study; A.S. (Alessandra Stasi) performed all the experiments, statistical analyses and wrote the manuscript. R.F. and F.S. performed experiments with A.S. (Alessandra Stasi), critically interpreted data, statistical analysis, and revised the manuscript. E.S. performed histological staining with A.S. (Alessandra Stasi) and contributed to revisions. P.C., A.S. (Alessandro Scagliotti) and A.C. carried out animal study and helped to revise the manuscript. G.C. (Gianvito Caggiano), R.L. and M.P. contributed to ex vivo and in vitro experiments. V.C., C.C., P.P., G: S., L: G. and V.F. contributed to design the study and critically revised the manuscript. G: C. (Giuseppe Castellano)supervised the research and critically revised the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethical approval
All animal procedures (see Methods for summary) were approved by the local Animal Welfare Body and by the Ethics Committee (EC) of the Italian National Institute of Health (ISS), the Centre for animal research and welfare prior to study initiation.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Stasi, A., Franzin, R., Sallustio, F. et al. mTOR Inhibition limits LPS induced acute kidney injury and ameliorates hallmarks of cellular senescence. Sci Rep 15, 9635 (2025). https://doi.org/10.1038/s41598-025-93815-7
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-93815-7
