
Abstract
Bandavirus dabieense (DBV) causes severe fever with thrombocytopenia syndrome, which has a mortality rate of 6.18% of 27%. DBV has been classified into five to six genotypes using phylogenetic analyses. However, the absence of clear standards poses challenges in identifying new genotypes. We performed evolutionary and homology analyses using the sequences from GenBank and analysed nucleotide differences between the different genotypes. Nucleotide differences within the same genotype were mostly below 3%, whereas those between different genotypes ranged from 3 to 7%. Consequently, we established and validated a specific genotyping criterion for DBV using phylogenetic tree analysis with a 3% cut-off value and identified 8, 11, and 11 genotypes in the S, M, and L segments, respectively. Furthermore, we compared our method with the previous genotyping methods to elucidate the convenience and advantages of using a 3% cut-off value. Importantly, we also identified new genotypes and fragment reassortments in DBV using our new genotyping criterion. Additionally, we established two simplified genotyping methods for the rapid typing of DBV in clinical settings and demonstrated the existence of geographical and clinical variations among the different genotypes. Our findings provide a more reliable foundation for clinical typing of DBV.
Similar content being viewed by others


Introduction
Severe fever with thrombocytopenia syndrome (SFTS), characterised by clinical symptoms such as fever, thrombocytopenia, leukocytopenia, gastrointestinal issues, and various other manifestations such as muscular symptoms, neurological abnormalities, and coagulopathy is a newly identified haemorrhagic disease1. Disease severity in patients with SFTS is typically associated with factors such as age and complications such as haemorrhage, nerve damage, or respiratory failure2. Bandavirus dabieense (DBV; family Phenuiviridae) was initially identified as aetiological agent of SFTS by Li et al.3. DBV is also known as severe fever with thrombocytopenia syndrome virus (SFTSV) or Huaiyang Shan virus. Notably, thousands of SFTS cases have been reported in more than 10 provinces in China, along with reports from other countries, including Japan and South Korea4,5. Unfortunately, reliable vaccines or effective antiviral drugs against DBV remain unavailable, resulting in an average mortality rate of 6.18–27%4,6.
Variation in fatality rates may be associated with infections with DBV with different genotypes7,8,9. Therefore, sequence differences in viral genes could potentially influence pathogenicity based on the genotype or through immune responses triggered by the virus. Understanding these genetic differences is crucial for developing effective vaccines and neutralising antibodies for active or passive immunotherapy6.
Different methods for viral genotyping include phylogenetic tree analysis and other analyses based on genetic distance/nucleotide diversity and antigenicity or immunological differences. DBV genotyping has been previously performed using phylogenetic analyses10,11,12. Although this method is intuitive and highly reliable, it has several weaknesses, such as it lacks specific standards and potentially overlooks newly emerging genotypes. Moreover, current nomenclature often uses uppercase letters combined with numbers, leading to potential confusion, particularly in the context of the three viral segments, thus hindering readers’ comprehension. Additionally, using uppercase letters across multiple typing methods may create confusion among the different methodologies, and different segments of the same strain may exhibit varying genotypes, a complexity that is inadequately represented by existing naming conventions. Currently, typing methods rely heavily on whole-genome sequencing, without a faster and simpler approach for strain typing. Consequently, establishing a more prescriptive and concise genotyping method with specific standards is crucial.
In this study, we downloaded 1519 sequences from NCBI and categorised them into first and second stages based on chronological timeframes. These sets were then used to establish and validate genotyping criterion. We determined a pivotal cut-off threshold of 3% through evolutionary tree analysis and homology comparisons of the first-stage sequences. Subsequently, we established a more precise genetic typing standard by integrating an evolutionary tree with a 3% cut-off value. Furthermore, the reliability of the genotyping criterion was confirmed using second-stage sequences. Using this criterion, we then identified seven novel rare genotypes and established two simplified genotyping methods (nucleoprotein [NP] region-based typing and conventional PCR). These two methods not only aid in the rapid identification of DBV genotypes but also further validate the reliability of established genotyping standards.
Results
Screening process
We downloaded 1386 sequences from GenBank in the first stage to establish genotyping criterion. A final set of 522 sequences was used after excluding 864 irrelevant sequences and those with missing segments and other anomalies.
We then downloaded 2209 sequences in the second stage to validate the genotyping criterion established using the first-stage sequences. A final set of 997 sequences, including 471, 272, and 255 sequences of the S, M, and L segments, respectively, was used after excluding 1212 irrelevant sequences and those with recombination events (Supplementary Fig. 1; Supplementary Datas 1, 2, 3, 4).
Genotyping criteria and validation
Phylogenetic analysis was performed using first-stage sequences. Sequences that clustered together in the evolutionary tree were considered to exhibit the same genotype. We classified the three segments of the same viral strain into the same genotype whenever possible. Subsequently, we performed pairwise comparisons among all first-stage sequences to calculate the nucleotide difference rate (%) between each genotype. Notably, nucleotide differences in the same segment among different viral strains within the same genotype were < 3%, whereas variations between viral strains of different genotypes ranged from 3 to 7%. Therefore, a 3% cut-off value was selected for genotypic classification. Ultimately, a new genotyping criterion for DBV was established by combining the phylogenetic tree analysis and the 3% cut-off value of nucleotide differences (Figs. 1, 2, 3).

Genetic evolutionary tree analysis of first-stage DBV sequences. (a) The genetic evolutionary tree analysis of S segments. (b) The genetic evolutionary tree analysis of M segments. (c) The genetic evolutionary tree analysis of L segments. (d) The genetic evolutionary tree analysis of nucleoprotein (NP). (e) All ORFs. Each genotype was labelled with a different colour. ▲ indicates that the S, M, and L segments belong to different genotypes. ORF: open reading frame.
The majority of first-stage sequences were classified into six genotypes (S1–6, M1–6, and L1–6) using a 3% cut-off value (Figs. 2, 3). We concatenated all ORFs and performed a phylogenetic analysis. Figures 1e and 3 present the results of the phylogenetic and homology analyses, respectively. The results of the concatenated analysis were generally consistent with those obtained using the analysis of the individual segments.

Nucleotide differences among first-stage sequences. (a–d) Intra-genotype nucleotide differences in the S segment (a), M segment (b), L segment (c), and NP region (d). (e–h) Inter-genotype nucleotide differences in the S segment (e), M segment (f), L segment (g), and NP region (h). (i–k) Intra- and inter-genotype nucleotide differences in S (i), M (j), and L (k) segments of DBV reassortant strains. N represents genotypes 7–11. B and W indicate intra-genotype and inter-genotype, respectively.

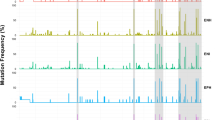
Nucleotide differences in the first-stage ORFs. (a) Intra-genotype nucleotide differences in ORFs (b) Inter-genotype nucleotide differences in ORFs. ORF: open reading frame.
We identified three viral strains with distinct sequences within the S segment. Consequently, two of these strains were named S7, and the other two were named S8. We determined nucleotide differences of 4.47% (3.27–5.22%) between S7 and viruses with other genotypes, whereas it was 4.82% (3.65–5.39%) between S8 and viruses with other genotypes (Fig. 4a). Similar to the S segment, the nucleotide differences between these newly designated genotypes (M7–11 and L7–11) and the other genotypes were > 3% (Fig. 4b,c). Ultimately, 8, 11, and 11 genotypes in the S, M, and L segments were identified, respectively, in the first stage based on the new genotyping criteria. Notably, most strains exhibited the same genotype across all three segments. However, nine strains exhibited the same genotype in the two segments and a different genotype in the third segment (Supplementary Tables 1, 2).

Inter-genotype nucleotide differences in S, M, and L segments in new genotypes among the first stage. (a) Inter-genotype nucleotide differences in the S segment (S7, S8). (b) Inter-genotype nucleotide differences in the M segment (M7–11). (c) Inter-genotype nucleotide differences in the L segment (L7–11) (d) Nucleotide difference between sequences belonging to different genotypes in the four genotyping methods.
To further validate the established genotyping criterion, we used the same methodology to identify the genotypes using the second-stage sequences (Supplementary Table 3). Notably, we obtained consistent results with < 3% of nucleotide differences within genotypes and > 3% differences between different genotypes (Fig. 5; Supplementary Fig. 2). Through validation in the second phase, we confirmed the reliability of the genotypic classification standards. Furthermore, the S segment in 19 strains exhibited significant dissimilarity (nucleotide differences of > 3%) compared with the other strains, indicating that they belong to a new genotype that is distinct from the genotypes S1 to S8. Consequently, these strains were denoted as N1 (17 sequences) and N2 (2 sequences), with inter-genotype nucleotide differences of 3.67% (2.69–4.70%) and 4.07% (3.10–4.70%), respectively (Fig. 5). Although a few viral strains displayed notable nucleotide diversity in the same genotypes (nucleotide differences > 3%), such as the S3, S8, and M5 genotypes, significant distinctions persisted between different genotypes.

Nucleotide differences in the second stage. (a–d) Intra-genotype nucleotide differences in S segment (a), M segment (b), L segment (c), and NP region (d). (e–h) Inter-genotype nucleotide differences in S segment (e), M segment (f), L segment (g), and NP region (h). “No” denotes that no more than two viral strains were included in the analysis.
Virus segment reassortment
The genotypes of the S, M, and L segments of most virus strains were identical. However, these segments belonged to different genotypes in the nine viral strains, indicating segment reassortment. All the reassorted strains shared the same genotype for the two segments, with only one segment belonging to a different genotype. For instance, the S, M, and L segments of SD4 corresponded to genotypes S1, M9, and L9, respectively. The nucleotide differences between the SD4 and genotypes S1 or S2–8 were 2.81% (2.52–3.22%) or 4.30% (3.44–4.60%), respectively, with a statistically significant difference (P < 0.001). Similar results were obtained for strains JS4, AHL, SPL087A, LN2012-14, LN2012-34, LN2012-41, LN2012-42, and LN2012-58 (P < 0.001), suggesting that the three segments belonging to different genotypes may be associated with reassortment (Fig. 2; Supplementary Tables 1, 2).
Comparison of various genotyping methods for DBV
We analysed the same first-stage sequences using three genotyping methods from previous studies and the method from this study to further confirm the rationality of the established genotyping criterion7,11,12,13,14. Notably, the same dataset analysed using different genotyping methods yielded distinct outcomes for each genomic segment. In the S segment, notable differences were observed among S1, S4, S6, and S8. In the M segment, variations were most prominent in M4, M8, M10, and M11. In the L segment, the disparities were primarily noted in L4, L10, and L11 (Table 1). Therefore, we further calculated the nucleotide difference rates of the above sequences that were notably different between our method and previous methods (Fig. 4d; Supplementary Fig. 3). In Fig. 4d, S1–S1 exhibited nucleotide differences between two subsets (2.96%, 2.63–3.33%), which were categorised into one genotype using our method but were categorised as two separate genotypes using previous typing methods (one group comprised 2 sequences, whereas another comprised 56 sequences). Notably, a similar result was obtained for S4. Conversely, some sequences that were suggested to belong to two different genotypes using our method were categorised into the same genotype using other genotyping methods. For instance, the inter-type differences between S6–8 were 3.80% (3.50–4.10%). These sequences were classified into two genotypes based on a 3% cut-off value. Overall, the comparison of the results of various genotyping methods for DBV confirmed the specificity and rationality of our genotyping criteria.
Establishment of reference sequences and validation of genotype-specific primers
We established reference sequences and specific sites for genotypes 1–6 following the principles of reference sequence construction and specific site screening (Supplementary Data 5; Supplementary Table 4). These sites were used to design genotype-specific primers to distinguish types S1–6 (Supplementary Table 5). Plasmids containing the full-length S1–6 sequences were used as templates for amplification using genotype-specific primers. Hence, we established a conventional one-step RT-PCR method based on specific primers to detect the DBV genotype. Among the amplification outcomes using the six pairs of type-specific primers, only one distinct band was clearly visible using agarose gel electrophoresis, with the observed band size corresponding to the expected size, confirming the genotype of the serum samples (Fig. 6). The original agarose gel electrophoresis images are presented in Supplementary Data 6. Forty serum samples (S1: 21 cases, S3: 4 cases, S4: 7 cases, and S5: 8 cases) were included in the study. The samples were sequenced in parallel, and the results were consistent with the aforementioned experimental data. Overall, the one-step RT-PCR method could facilitate rapid identification of clinical genotypes and confirm the accuracy and rationality of our genotyping standards.

Verification of the specificity of the primer sequences. (a–f) The plasmids containing S1(a), S2(b), S3(c), S4(d), S5(e), S6(f) reference sequences were used as templates to verify six primer pairs, respectively. (g–j) The serum samples containing DBV types 1(g), 3(h), 4(i), and 5(j) were used as templates to verify six primer pairs.
Simple typing method
Considering the significance of the NP region within the DBV and its manageable length of approximately 700 base pairs, it is suitable for PCR amplification and sequencing. Therefore, we employed NP region sequence analysis to establish a straightforward genotyping method to conveniently identify clinical genotypes. Subsequently, a simple typing method based on the NP region, phylogenetic tree analysis, and a 3% cut-off value was employed separately for the NP region of all strains from both the first and second stages. The results revealed a high degree of consistency (99.2%) between the genotypic outcomes obtained using the analysis of the NP region and full-length S segment (Figs. 1, 4; Supplementary Table 6). Notably, in the second stage, the following discrepancies between S segment genotypes and NP-region simple genotypes were observed in four sequences: S3 → G4 (KU664009), N1 → G3 (JQ693002, MT114317), and N2 → G4 (KU664010, KR698328) (Supplementary Fig. 3).
Regional genotypic differences
The genotyping criterion established in this study was used to ascertain the distribution of DBV genotypes across diverse regions. The results revealed pronounced regional disparities in genotypes across China, Korea, and Japan. In China, genotype distribution has been statistically analysed by province. In Henan, the main genotype observed was S1, with S3 and S4 as supplementary genotypes. Genotypes S1 and S3 were predominantly observed in Hubei and Shandong, S1 and S4 were predominantly observed in Jiangsu, S5 was the dominant in Zhejiang, along with S1, S4, and S6, S2 in Japan, with other genotypes being relatively rare, and S2 and S5 in South Korea. Notably, similar patterns were observed for the M and L segments of the DBV. Therefore, regional differences in DBV genotypes could also be identified using our genotyping criterion (Fig. 7; Supplementary Fig. 4).

Genotypic distribution in different regions of all sequences in first and second stages. (a) Geographical distribution and names of the virus strains included in this study. (b–e) The genotypic distribution of S segment (b), M segment (c), L segment (d) and NP region (e). LN Liaoning, SD Shandong, HN Henan, HB Hubei, AH Anhui, JS Jiangsu, ZJ Zejiang, SK Korea, JP Japan.
Genotypes and clinical characteristics
We investigated the association between different DBV genotypes and clinical characteristics based on our genotyping criterion using 40 cases. The proportion of patients with neurological symptoms was as follows: genotype 1 (7/21), genotype 3 (0/4), genotype 4 (1/7), and genotype 5 (5/8). The proportion of patients experiencing vomiting symptoms was as follows: genotype 1 (1/21), genotype 3 (1/4), genotype 4 (0/7), and genotype 5 (4/8). No significant differences were observed in gastrointestinal bleeding or diarrhoea between the genotypes. Therefore, we hypothesise that clinical symptoms, particularly neurological and vomiting symptoms, may be associated with infection with different DBV genotypes, indicating that a specific and reasonable genotyping criterion is crucial for the clinical treatment and evaluation of disease progression following DBV infection.
Discussion
Viruses are conveniently classified based on various genetic criteria. For instance, Hepatitis B and C viruses are classified based on 8% and 15–30% nucleotide differences, respectively15,16. HIV is classified based on the infection sites and pathogenicity, whereas influenza virus is categorised by antigenic differences in haemagglutinin and neuraminidase17,18. Previous studies have classified DBV into five or six genotypes based on genetic evolutionary tree analyses. Currently, three genotyping nomenclature methods—using uppercase letters A–E, using uppercase letters A–F, and utilising geographic variations with C and J branches—exist for DBV7,11,12. The systematics-based classification method is highly scientific and intuitive but lacks standardised guidelines for genetic typing. Moreover, the multitude of genetic typing methods complicates the analysis of their interrelationships. In this study, we discovered < 3% nucleotide variation among viral strains within the same genotype using evolutionary tree analysis and sequence comparisons. Conversely, the nucleotide disparity between viral strains of different genotypes was > 3%. Therefore, we used 3% nucleotide variance as a threshold to distinguish different genotypes. After analysing and comparing 174 DBV strains with known S, M, and L segment sequences, we identified 8, 11, and 11 genotypes of the S, M, and L segments, respectively. Notably, genotypes 1–6 were the most prevalent in each segment. Consequently, S7–8, M7–11, and L7–11 may represent new genotypes that emerged during viral evolution. The complete viral strains of genotypes 1–6 were relatively abundant, and the differences between the genotypes were clear, thus confirming highly reliable results. In contrast, other genotypes had fewer or lacked complete viral strains (only one or two complete strains), thus exhibiting lower reliability. Notably, the usability of these genotypes may increase with the availability of more viral strains or sequences for validation in the future.
The S, M, and L segments of some DBV strains belonged to different genotypes in this study as well as the previous studies, indicating the possibility of segment reassortment19,20. Nonetheless, whether these reassortments occurred randomly or were triggered by immune-related stress remains unclear. Considering the reassortment in some DBV strains, we recommend using Arabic numerals rather than letters to classify markers to distinguish viral genotypes, thus minimising confusion and ambiguity. In the context of reassortment between fragments of different genotypes, any documented evidence regarding reassortment in B. dabieense under cell culture conditions remains lacking. However, similar genetic reassortment techniques have been used to prepare influenza vaccines21.
The genotyping approach used in this study differs from previous genotyping methods. For instance, we used a nucleotide difference of > 3% to distinguish S6 and S8 as separate genotypes, whereas earlier studies grouped these genotypes as the same. Similarly, previous typing methods have classified S1 sequences into two genotypes, although the nucleotide difference between these sequences (HM802204 and HQ141606) and the other S1 sequences was generally < 3%. Consequently, we conducted a supplementary analysis comparing the sequences of HM802204 and HQ141606 (S1 genotype) with those of the other genotypes. Notably, nucleotide differences were significantly higher than 3%. Therefore, we classified HM802204 and HQ141606 as the S1 genotype, thus highlighting the need to establish specific typing criteria. Based on the genetic distance data, our method indicated < 3% intra-genotype and > 3% inter-genotype differences. Thus, the nucleotide difference rate offers a strong theoretical foundation for genotyping and identification of new genotypes. If some data points within the same genotype exhibit > 3% differences, we suggest considering them subtypes in future research.
We then identified sequences with > 3% differences between the S segments of the 19 strains and the known S1–8 genotypes. Evolutionary tree analysis revealed that these 19 strains exhibited significant differences from the established S1–8 genotypes and could not be confidently assigned to any of the S1–8 genotypes, suggesting the possibility of new genotypes. Among these strains, genotype N1 was reported from Hubei (KU664017) and Shandong (JQ693002). Genotype N2 was reported from Zhejiang (KR698328), with other DBV strains uploaded by the same author mainly belonging to genotype S622. Additionally, strains resembling genotype N2 were reported from Hubei (KU664010), whereas other strains uploaded by the same author were primarily genotypes S1 and S323. Notably, the N1 and N2 genotypes are unique. First, these genotypes have been reported only in the S segment, and complete viral genome sequences remain unavailable. In addition, they represent only a small proportion of the total sequences, with relatively low reliability. Therefore, the availability of more sequences is imperative for verification and more reliable classification. DBV continually adapts and evolves in response to shifting environmental factors, resulting in alterations to the reference sequence (for example, the C to T shift at position 373 in genotype S4). Therefore, monitoring viral mutations is essential for updating the reference sequence, and these variants may enhance our understanding of pathogenic mechanisms of DBV.
In this study, we established the cost-effective and convenient conventional PCR method, which also confirmed the accuracy and rationality of our genotyping criterion. However, this method has low sensitivity. Consequently, we are developing a TaqMan SNP genotyping method to obtain more precise and efficient results. Additionally, as all serum samples in this study were obtained from the same hospital, we could not access samples from patients with DBV genotypes 2 and 6, thus resulting in a lack of experimental data for these genotypes. Nevertheless, we confirmed the reliability of the specific primers using synthesised sequences for genotypes S1–6.
Furthermore, conducting full-length sequencing for genotype analysis is challenging and impractical because of the long lengths of the three DBV segments3. Our study revealed identical genotypes of the three segments in most viral strains in most cases (94.8%). The NP region of the S segment (738 base pairs) is well suited for PCR amplification and sequencing. This region is crucial for viral replication and immune responses, making it ideal for phylogenetic analyses and nucleotide difference comparisons. Notably, a 3% cut-off value as a typing criterion proved feasible in this study, further supporting the reliability and clinical utility of the NP region for genotypic classification.
Previous studies have confirmed regional variations in DBV genotype distribution11,24. Using our genotyping method, we identified that virus strains in Korea and Japan were predominantly of genotype 2, with over 85% of the strains in Japan belonging to this type. In contrast, genotype 2 is nearly absent in China, suggesting that it may be unique to Japan and Korea. In China, genotype 1 is prevalent in the Huaiyang Mountain region, genotype 3 in Shandong and Liaoning, and genotypes 5 and 6 in Zhejiang. These findings align with those of previous studies, supporting the extensive evolutionary history of DBV and validating our genotypic criteria10. Additionally, previous studies have indicated a slight variation in the DBV genotypes across different host species, with the genotypes being generally similar. Notably, epidemiological dynamics are closely related to regional locations25,26.
In the present study, we examined the association between clinical symptoms and DBV genotypes in patients with SFTS and found that individuals infected with genotype 5 were more likely to develop neurological symptoms than those infected with other genotypes. This suggests a potential association between the genotype and the mechanisms of DBV-induced damage and symptoms in the host. As the pathogenic mechanisms are still not fully understood, this insight could guide future research and underscore the importance of specific and rational genotyping criteria for investigating the pathogenic mechanisms and disease progression in DBV infections. The limited availability of clinical sample data is a limitation of this study, and we aim to collect and analyse more comprehensive data in future research.
We confirmed the feasibility of the proposed criterion by establishing new genotyping standards and validating them using clinical samples. However, variations in pathogenicity, replication capacity, and immunogenicity among the genotypes remain unproven and require further cellular and animal studies. In summary, we introduced a new genotyping criterion for DBV with 3% nucleotide differences and confirmed the existence of new DBV genotypes. We then established simplified genotyping methods for DBV based on the new genotyping criterion and demonstrated segment reassortment and geographical variations. Overall, our study provides a more reliable foundation for clinical typing and offers an expedited method for detecting viral genotypes in serum samples, thus offering significant implications for vaccine research and disease prognosis.
Materials and methods
Genomic sequence collection
DBV sequences were retrieved from the NCBI using search the term ‘Severe fever with thrombocytopenia syndrome virus’, with the complete genome of DBV strains including S, M, or L segments and each sequence being clearly annotated in terms of the sample source as the inclusion criteria. Non-DBV sequences and those with more than five “N” sequences in a segment or with internal recombinant segments were excluded.
We filtered and classified the obtained sequences into two stages: the first stage and the second stage. The first-stage sequences included only those with complete S, M, and L segments obtained from the initial period until July 2017. The second stage concluded in February 2021 and includes all remaining sequences not part of the first stage (Supplementary Fig. S1).
Integrated sequence analysis
All gene sequences were grouped according to segments and input into a database using ClustalW software (version 2.0). Default parameters were used for multi-alignment. The alignment results were then input into a new database using RDP 4.0 Beta software for virus sequence integration analysis. Recombination events were detected using seven methods: RDP, GENECONV, Maxchi, BootScan, SiScan, Chimaerea, and 3Sep. The window size was set to 100, and the bootstrap value was set to 70. Recombinant events detected by five of the seven methods were considered a positive result, reflecting an integrated sequence, and thus were not included in subsequent analyses.
Evolutionary tree analysis and homology comparisons
Evolutionary tree analysis was divided into the first and second stages. The three segments were analysed by generating a genetic evolution tree using MEGA 7.0 software27. Homology analysis was performed for each of the three segments, and sequence differences were obtained via pairwise comparisons of all sequences. The alignment results were input into MEGA 7.0, and the “Compute Pairwise in Distance” algorithm was selected to determine the differences between sequences. Different segments of the same strain were labelled with the same genotype when possible, and the differences between the internal and external sequences of each genotype were compared. Data were analysed using GraphPad Prism 8. The median is marked with a thick solid line, the box delimits the first and third quartiles, and the whiskers extend to the 2.5% and 97.5% percentiles.
Genotyping criteria and validation
Based on the classification in the initial stage, the cut-off value was further refined, ultimately establishing the classification criteria. The genotyping criterion adhered to the following specifications: the cut-off value was preferably an integer to facilitate practical implementation. Homologous analysis alone demonstrated accurate discrimination of over 95% of viral strains, and the accuracy approached 100% when coupled with evolutionary tree analysis. We aimed to minimise the number of genotypes, typically limiting them to approximately 10, which is conducive for both clinical applications and research. Additionally, we concatenated the open reading frames of all complete strains from the first stage in the order NP–NSS–GN–GC–RDRP and performed phylogenetic and homology analyses.
Following the establishment of the genotyping criterion and methodology, the second-stage strains were genotyped. Thereafter, we conducted evolutionary tree analysis and homology comparisons to assess the reliability and accuracy of the criterion and classification methods.
Establishment of reference sequence and screening of specific sites
Reference sequences were determined using the following established principles:
(1) Consensus sequence selection: Reference sequences were constructed by selecting the most common nucleotides at each position. (2) Initial construction: In the first stage, reference sequences were constructed using a dataset including 174 strains. (3) Classification and naming: Reference sequences were assigned to different branches according to a unified classification system and labelled with Arabic numerals, such as S1-ref. (4) Sequence refinement: In the second stage, additional sequences were used for refinement. If different nucleotides were found at the same position, sequences from both stages were compared, and a nucleotide with a frequency > 50% was selected as the reference sequence.
Specific sites were determined according to the following guidelines: (1) High consistency within genotypes: Specific sites for each genotype should have a presence of > 95% within that genotype. (2) Low consistency across other genotypes: These specific sites should have a presence of < 5% across other genotypes. (3) Validation across multiple stages: The selected sites should meet the above criteria in both the first and second stages of data analysis.
Comparison of different DBV genotyping methods
We used first-stage sequences for comparison. Reference sequences from each genotype were selected for each genotyping method. Subsequently, MEGA7 was used to conduct phylogenetic tree analyses and enumerate the sequences contained within each genotype to elucidate the similarities and differences among these methods. Furthermore, we scrutinised the non-matching sequences from the four methods using homology analysis as previously described.
Genotype-specific primer design and validation
Based on the specificity and sensitivity of the specific sites, we designed genotype-specific primers for S1–6 with each genotype-specific mutation site as the 3ʹ and then performed PCR to distinguish different genotypes. Subsequently, we used these primers to detect genotypes in both plasmid and serum samples. Full-length sequences of S1–6 were synthesised and inserted into the PUC-SP vector. Blood serum samples were collected in 2019 from 40 patients with SFTS during the acute phase of the disease by the Department of Infectious Disease at the Second Affiliated Hospital of Anhui Medical University. RNA was extracted from the patient serum using a TaKaRa MiniBEST Viral RNA/DNA Extraction Kit (Ver. 5.0; TaKaRa, Dalian, China) and reverse transcribed into cDNA. PCR was performed using a PrimeScriptTM RT-PCR Kit (TaKaRa).
Simple genotyping method
In this study, we selected an NP region that may significantly influence DBV characteristics for streamlined genotyping and clinical applicability. We then established a simplified typing method using evolutionary tree analysis and differential comparison. Subsequently, we compared the results obtained using this simplified method with those obtained using a conventional genotyping approach to assess the level of concordance between the two methods for clinical application.
Statistical analysis
Skew distributional data were expressed as medians (inter-quartile range [2.5–97.5 percentile]). Continuous variables were compared using the Students’ t-test or Mann–Whitney U test as appropriate. All the statistical analyses were performed using SPSS, version 17.0 (SPSS Inc., Chicago, US), and P < 0.05 in a two-sided test was considered statistically significant. Graphs were prepared using GraphPad Prism 9.
Data availability
Viral genome sequences were downloaded from the National Center for Biotechnology Information (NCBI, http://www.ncbi.nlm.nih.gov/). Due to the extensive number of sequences involved, it is not feasible to display all accession numbers (such as HM745930–HM745932, etc.) in this section. Therefore, the accession numbers and other sequence information will be provided in the Supplementary Data 1, Data 2, Data 3, Data 4.
References
Liu, Q., He, B., Huang, S. Y., Wei, F. & Zhu, X. Q. Severe fever with thrombocytopenia syndrome, an emerging tick-borne zoonosis. Lancet Infect. Dis. 14, 763–772. https://doi.org/10.1016/S1473-3099(14)70718-2 (2014).
Dong, Y., Lin, S. H., Jiang, L. & Liu, H. Clinical characteristics and risk factors of 267 patients having severe fever with thrombocytopenia syndrome-new epidemiological characteristics of fever with thrombocytopenia syndrome: Epidemiological characteristics of SFTS. Medicine (Baltimore). 101, e31947. https://doi.org/10.1097/MD.0000000000031947 (2022).
Yu, X. J. et al. Fever with thrombocytopenia associated with a novel bunyavirus in China. N. Engl. J. Med. 364, 1523–1532. https://doi.org/10.1056/NEJMoa1010095 (2011).
Casel, M. A., Park, S. J. & Choi, Y. K. Severe fever with thrombocytopenia syndrome virus: Emerging novel phlebovirus and their control strategy. Exp. Mol. Med. 53, 713–722. https://doi.org/10.1038/s12276-021-00610-1 (2021).
Tran, X. C. et al. Endemic severe fever with thrombocytopenia syndrome, Vietnam. Emerg. Infect. Dis. 25, 1029–1031. https://doi.org/10.3201/eid2505.181463 (2019).
Bopp, N. E. et al. Baseline mapping of severe fever with thrombocytopenia syndrome virology, epidemiology and vaccine research and development. NPJ Vaccines 5, 111. https://doi.org/10.1038/s41541-020-00257-5 (2020).
Yun, S. M. et al. Genetic and pathogenic diversity of severe fever with thrombocytopenia syndrome virus (SFTSV) in South Korea. JCI Insight. https://doi.org/10.1172/jci.insight.129531 (2020).
Yokomizo, K., Tomozane, M., Sano, C. & Ohta, R. Clinical presentation and mortality of severe fever with thrombocytopenia syndrome in Japan: A systematic review of case reports. Int. J. Environ. Res. Public Health. https://doi.org/10.3390/ijerph19042271 (2022).
Li, Z. et al. Susceptibility of spotted doves (Streptopelia chinensis) to experimental infection with the severe fever with thrombocytopenia syndrome phlebovirus. PLoS Negl. Trop. Dis. 13, e0006982. https://doi.org/10.1371/journal.pntd.0006982 (2019).
Liu, L., Chen, W., Yang, Y. & Jiang, Y. Molecular evolution of fever, thrombocytopenia and leukocytopenia virus (FTLSV) based on whole-genome sequences. Infect. Genet. Evol. 39, 55–63. https://doi.org/10.1016/j.meegid.2015.12.022 (2016).
Liu, J. W. et al. Molecular evolution and spatial transmission of severe fever with thrombocytopenia syndrome virus based on complete genome sequences. PLoS One 11, e0151677. https://doi.org/10.1371/journal.pone.0151677 (2016).
Lv, Q. et al. Novel sub-lineages, recombinants and reassortants of severe fever with thrombocytopenia syndrome virus. Ticks Tick Borne Dis. 8, 385–390. https://doi.org/10.1016/j.ttbdis.2016.12.015 (2017).
Li, Z. et al. Increased prevalence of severe fever with thrombocytopenia syndrome in Eastern China clustered with multiple genotypes and reasserted virus during 2010–2015. Sci. Rep. 7, 6503. https://doi.org/10.1038/s41598-017-06853-1 (2017).
Liu, B., Zhu, J., He, T. & Zhang, Z. Genetic variants of Dabie bandavirus: Classification and biological/clinical implications. Virol. J. 20, 68. https://doi.org/10.1186/s12985-023-02033-y (2023).
Okamoto, H. et al. Typing hepatitis B virus by homology in nucleotide sequence: Comparison of surface antigen subtypes. J. Gen. Virol. 69(Pt 10), 2575–2583. https://doi.org/10.1099/0022-1317-69-10-2575 (1988).
Smith, D. B. et al. Expanded classification of hepatitis C virus into 7 genotypes and 67 subtypes: Updated criteria and genotype assignment web resource. Hepatology 59, 318–327. https://doi.org/10.1002/hep.26744 (2014).
Nahmias, A. J. & Roizman, B. Infection with herpes-simplex viruses 1 and 2. II. N. Engl. J. Med. 289, 719–725. https://doi.org/10.1056/NEJM197310042891404 (1973).
Yang, R. et al. Human infection of avian influenza A H3N8 virus and the viral origins: A descriptive study. Lancet Microbe 3, e824–e834. https://doi.org/10.1016/S2666-5247(22)00192-6 (2022).
Fu, Y. et al. Phylogeographic analysis of severe fever with thrombocytopenia syndrome virus from Zhoushan Islands, China: Implication for transmission across the ocean. Sci. Rep. 6, 19563. https://doi.org/10.1038/srep19563 (2016).
Shi, J. et al. Migration, recombination, and reassortment are involved in the evolution of severe fever with thrombocytopenia syndrome bunyavirus. Infect. Genet. Evol. 47, 109–117. https://doi.org/10.1016/j.meegid.2016.11.015 (2017).
Long, Y. et al. Preparation and evaluation of a novel, live, attenuated influenza H1N1 vaccine strain produced by a modified classical reassortment method. Hum. Vaccin. Immunother. 14, 615–622. https://doi.org/10.1080/21645515.2017.1380761 (2018).
Ni, H. et al. Apodemus agrarius is a potential natural host of severe fever with thrombocytopenia syndrome (SFTS)-causing novel bunyavirus. J. Clin. Virol. 71, 82–88. https://doi.org/10.1016/j.jcv.2015.08.006 (2015).
Cheng, J. et al. Prevalence and molecular phylogenetic analysis of severe fever with thrombocytopenia syndrome virus in domestic animals and rodents in Hubei Province, China. Virol. Sin. 34, 596–600. https://doi.org/10.1007/s12250-019-00119-y (2019).
Yun, S. M. et al. Molecular genomic characterization of tick- and human-derived severe fever with thrombocytopenia syndrome virus isolates from South Korea. PLoS Negl. Trop. Dis. 11, e0005893. https://doi.org/10.1371/journal.pntd.0005893 (2017).
Wang, Y., Yang, M., Zhou, H., Quan, C. & Kang, H. Outbreak of natural severe fever with thrombocytopenia syndrome virus infection in Farmed Minks, China. Emerg. Infect. Dis. 30, 1299–1301. https://doi.org/10.3201/eid3006.240283 (2024).
Kang, J. G. et al. Severe fever with thrombocytopenia syndrome virus in ticks in the Republic of Korea. Korean J. Parasitol. 60, 65–71. https://doi.org/10.3347/kjp.2022.60.1.65 (2022).
Kumar, S., Stecher, G. & Tamura, K. MEGA7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 33, 1870–1874. https://doi.org/10.1093/molbev/msw054 (2016).
Acknowledgements
We thank Dr. Dexin Li for his carefully reading this article and presenting valuable amendments.
Funding
The study was supported by Anhui Provincial Natural Science Foundation (2108085MH298), The Second Hospital of Anhui Medical University Natural Science Foundation (2019GMFY02), and the Clinical Medicine project of Anhui Medical University (2021lCXK027). The funders had no role in the study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Author information
Authors and Affiliations
Contributions
Bingyan Liu was responsible for the experimental design and writing the articles. Tengfei He and Shanshan Sun was responsible for bioinformatic analysis and geographic distribution statistics. Changtai Wang takes responsibility for proofreading the manuscript. Guomei Xia, Shijun Zhou and Xinlei Chen was responsible for samples and clinical data collections. Xiaodan Hong and Jie Zhu were responsible for guiding the experiment and mapping. Zhenhua Zhang was responsible for the research idea and study design.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethical approval and consent to participate
The protocols and sample collection for present study was reviewed and approved by Anhui Medical University Ethics Committee Review Board (approval number: S20210059). Signed informed consent was provided by patients or surrogates. All methods were performed in accordance with the relevant guidelines and regulations (Declaration of Helsinki).
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Liu, B., He, T., Wang, C. et al. Establishment of a genotyping criteria for Bandavirus dabieense and confirmation of new genotypes. Sci Rep 15, 11269 (2025). https://doi.org/10.1038/s41598-025-94203-x
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-94203-x
