
Abstract
Currently, the treatment and prevention of multiple sclerosis (MS) continue to encounter significant challenges. Mendelian randomization (MR) analysis has emerged as a crucial research method in the pursuit of new therapeutic strategies. Accordingly, we hypothesize that there exists a causal association between genetic variants of specific plasma proteins and MS through MR mechanisms, and that key therapeutic targets can be precisely identified by integrating multi-omics analytical approaches. In this study, we developed a comprehensive analytical framework aimed at identifying and validating potential therapeutic targets for MS. The framework commenced with a two-sample Mendelian randomization (MR) study utilizing two large plasma protein quantitative trait locus (pQTL) datasets. Building on this foundation, we performed Bayesian co-localization analysis of coding genes, followed by a full phenotype-wide association study (PheWAS) on the co-positive genes identified through both analytical methods. This approach allowed us to explore the functions of key genes and the mechanisms of co-morbidity associated with the disease. Subsequently, we integrated protein-protein interaction (PPI) network analysis, gene ontology (GO) analysis, and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis to facilitate drug prediction and molecular docking studies. This study conducted a systematic analysis between two large plasma pQTLs datasets and MS. In the MR analysis, the MR analysis of Icelandic plasma pQTLs and MS identified 88 positive plasma proteins, while the MR analysis of the UK Biobank database pQTLs and MS identified 122 positive plasma proteins. By comparison, uroporphyrinogen III synthase (UROS) and glutathione S-transferase theta 2B (GSTT2B) were found to be the positive proteins shared by the two datasets. After false discovery rate (FDR) correction, signal transducer and activator of transcription 3 (STAT3) was a significantly positive protein in the analysis of Icelandic plasma pQTLs. In the analysis of the UK Biobank database pQTLs, advanced glycosylation end product-specific receptor (AGER), allograft inflammatory factor 1 (AIF1), butyrophilin subfamily 1 member A1 (BTN1A1), cluster of differentiation 58 (CD58), desmoglein 4 (DSG4), ecotropic viral integration site 5 (EVI5), tumor necrosis factor (TNF), and tumor necrosis factor receptor superfamily member 14 (TNFRSF14) were significantly positive proteins. After Bonferroni correction, AGER, CD58, EVI5, and TNF remained significantly positive proteins in the analysis of the UK Biobank database pQTLs. In the Bayesian colocalization analysis, EVI5 (PPH4 = 0.9800), O-GlcNAcase (OGA) (PPH4 = 0.8569), and TNFRSF14 (PPH4 = 0.8904) were the common positive genes in the two analysis methods. In conclusion, EVI5, OGA, and TNFRSF14 may be potential therapeutic targets for MS. Through the comprehensive application of MR analysis and Bayesian colocalization analysis, we have successfully identified that EVI5, OGA, and TNFRSF14 may be key therapeutic targets for MS. These findings may provide a scientific basis for the development of novel immunotherapies, combination treatment regimens, or targeted intervention strategies.
Similar content being viewed by others


Background
Multiple sclerosis (MS) is one of the most prevalent idiopathic inflammatory demyelinating diseases of the central nervous system, closely associated with abnormalities in the immune system. It not only poses a significant threat to patients’ bone health but may also lead to a decline in joint function, thereby severely impacting their quality of daily life. As an inflammatory neurological disease that predominantly affects young adults, MS represents a major challenge to global health. A study on MS revealed that from 1990 to 2016, the number of global cases reached 2,221,188, with an age-standardized prevalence increase of 10.4% since 19901. Notably, the prevalence rate in high-income North America is as high as 164.6 cases per 100,000 people, while in eastern sub-Saharan Africa, it is only 3.3 cases per 100,000 people, highlighting significant geographical disparities. In China, the situation regarding MS is equally concerning2. Research indicates that from 1990 to 2019, the number of MS patients in China rose from approximately 21,286 to 42,571, while the age-standardized prevalence rate increased from 1.88 to 2.32 per 100,000 individuals. Notably, in 2019, the total number of disability-adjusted life years (DALYs) attributable to MS reached 71,439. The health threat posed by MS to women of childbearing age is particularly significant3. Data from 2019 reveal that the age-standardized prevalence rate of MS among women of childbearing age globally was 28.74 per 100,000, with an incidence rate of 1.63 per 100,000 and a mortality rate of 0.17 per 100,000. Although the prevalence and incidence rates have shown a downward trend from 1990 to 2019, the overall situation remains critical. For adolescents and young adults (AYA), the health impact of MS is also substantial4. Data from 2021 indicate that the global age-standardized incidence rate (ASIR), prevalence rate (ASPR), and mortality rate (ASMR) of MS among AYA are 1.40, 16.05, and 0.05 per 100,000 individuals, respectively. From 1990 to 2021, the global ASIR of MS exhibited an increasing trend, with an average annual percentage change (AAPC) of 0.22 (95% confidence interval: 0.19 to 0.26). The incidence of MS continues to rise, and studies predict that the global incidence of MS is severe and complex. It is anticipated that the global ASIR will continue to increase from 2020 to 2040, leading to a further rise in the number of cases5. Neglecting this issue will exacerbate the burden on the global public health system in managing this autoimmune disease. The pathogenesis of MS is highly complex and likely involves multiple factors, including genetics, diet, immune regulation, and environmental influences. Given its significant societal burden, conducting in-depth and comprehensive research is of paramount importance.
The treatment and prevention of MS represent some of the most complex medical challenges today. Despite the availability of new therapeutic options, including synthetic immunomodulatory agents, monoclonal antibodies, selective lymphomodulators, muscle relaxants, and corticosteroids, which can alleviate symptoms and enhance functionality, the field continues to encounter significant challenges.Nonetheless, treatment continues to encounter several obstacles. One major concern is the side effects associated with these drugs, which may include increased neurological complications6, heightened infection risk7, and potential damage to liver and kidney function8. Furthermore, the long-term use of immunosuppressants can compromise the immune system9. Another challenge lies in the variability of treatment response rates10, some patients experience limited symptom relief, while others may see their condition deteriorate or encounter a significant recurrence rate. This variability is influenced by factors such as the inherent characteristics of the drugs, individual patient differences, and the heterogeneity of the disease itself. To advance the exploration of MS treatments more effectively, it is essential to continue investigating new therapeutic targets. genome-wide association study (GWAS) can identify Single Nucleotide Polymorphism(SNPs) linked to the risk of MS; however, the findings from GWAS must be supplemented with comprehensive analyses to pinpoint causative genes and facilitate drug development. Without such analyses, it remains challenging to clearly and directly identify disease-causing genes or support drug development efforts. This difficulty arises because the associated loci identified by GWAS may reside in intergenic regions, and the mechanisms by which they influence gene expression and function remain unclear. Therefore, further in-depth research is necessary to elucidate the intrinsic relationships between these loci and the onset and progression of the disease.
MR is an approach that employs genetic variants as instrumental variables (IVs) to elucidate causal associations between exposure factors and disease. Recent advancements in platform aptamers and immunoassay technologies, such as SomaScan and Olink, enhance MR analysis, which integrates GWAS data with pooled results of pQTLs. This integration holds significant promise for identifying novel therapeutic targets. Notably, pQTLs situated within the vicinity of drug-acting genes serve as alternative biomarkers, reflecting gene expression levels that can indicate long-term exposure status. Building on this foundation, we conducted a comprehensive, drug-target-based MR study aimed at exploring potential therapeutic strategies for MS. In contrast to prior MR studies, we utilized two newly published datasets of large plasma protein pQTLs for our analysis, incorporating Bayesian co-localization methods to enhance the robustness and reliability of our findings. Furthermore, we employed bioinformatics strategies to analyze gene pathways and drug predictions with strong associations to the pathogenesis and treatment of MS. Ultimately, a phenome-wide association study (pheWAS) was conducted on the significant genes to systematically examine the relationship between exposures and a wide range of outcomes, comprehensively revealing the complete functions of the genes and the comorbidity mechanisms of the diseases. This series of comprehensive analysis steps provides new insights and guidance for the development of more efficient and targeted therapeutic drugs for MS, and is expected to drive the progress in the field of MS treatment.
Methods
Research design
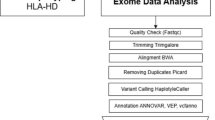
Figure 1 illustrates the analysis process of the full text. We established a systematic analysis framework comprising the following steps: First, positive proteins associated with MS were identified from two extensive plasma pQTL datasets using two-sample MR analysis. Second, we examined the functional roles of these candidate plasma protein targets in the context of MS. Subsequently, Bayesian co-localization analysis was employed to validate the coding gene checkpoints shared by the identified genes and MS. For the co-positive genes identified through these two analytical methods, PheWAS analysis was conducted with all GWAS projects in the 11th edition of the FinnGen database to comprehensively investigate the functional implications and comorbidities associated with these genes. We then explored the expression of significant positive genes across different tissues. Furthermore, we investigated the underlying biological mechanisms of the proposed protein targets through PPI analysis, GO enrichment analysis, and KEGG pathway analysis. Drug prediction and molecular docking studies were performed on the identified positive genes. Finally, we classified and assessed the evidence from this study based on the outcomes of the comprehensive analysis, in conjunction with existing research evidence.

Flow chart of the analysis process in this article. MR: Mendelian randomization analysis; PheWAS: Phenome-wide association study; PPI: Protein-protein interaction; GO: Gene Ontology; KEGG: Kyoto Encyclopedia of Genes and Genomes; DSigDB: Drug Signatures Database.
The data source of the plasma proteome
This study investigates the GWAS data pertaining to plasma proteins, utilizing the SomaScan platform for analysis. Led by Egil Ferkingstad and colleagues, the research involved 35,559 Icelandic participants, resulting in the identification of 28,191 genetic associations corresponding to 4,907 aptamers11. The data primarily originate from two major projects: the Iceland Cancer Project (ICP), which comprises 52% of the participants, and various genetic initiatives conducted by deCODE Genetics in Reykjavik, Iceland, contributing the remaining 48%. Employing a recursive conditional analysis approach, the precomputed summary statistics highlight the most significant variant within each region (± 1 Mb) as a sentinel plasma pQTL ( n = 18,084), while other variants are categorized as minor plasma protein quantitative trait loci (n = 10,107). Notably, this GWAS successfully replicated 83% of the reported plasma pQTL based on SomaScan in the INTERVAL study and 64% of those reported in Olink’s SCALLOP consortium. The complete GWAS data is accessible for download at the following link: https://www.decode.com/summarydata/. All samples included in this study are of European ancestry.
Another GWAS dataset on plasma proteins derives from the pQTL research results released by the UK Biobank in October 2023. During the course of this study, the research team utilized the Olink platform to analyze the 2,923 proteins included in the UK Biobank Pharmaceutical Proteomics Project (UKB-PPP), and thus identified 23,588 preliminary genetic associations12. The P-values for these associations were significant, falling below the significance threshold, and within the ± 1 Mb interval, the linkage disequilibrium (LD) r² value was found to be less than 0.8. This GWAS study effectively verified 84% of the known pQTLs in the antibody study and 38% of the pQTLs in the aptamer study. The complete GWAS dataset is available for download at the following link (https://www.synapse.org/#!Synapse: syn51364943/files/). All samples included in this study are of European ancestry.
The data source of multiple sclerosis
The GWAS data of MS were sourced from the 11th version of the GWAS data released on the FinnGen database on June 24, 2024. This data involved 452,390 individuals, including 2,620 MS patients and 449,770 healthy control subjects. The complete GWAS dataset can be accessed and downloaded via the link (https://storage.googleapis.com/finngen-public-data-r11/summary_stats/finngen_R11_G6_MS.gz). All the samples included in this study were of European ancestry in the surveyed population.
Mendelian randomization analysis
In MR analysis, three fundamental assumptions must be met13: first, the genetic variation should be directly associated with the exposure factor; second, the genetic variation must not correlate with any confounding factors; and finally, if the genetic variation influences the outcome, this effect must be entirely mediated through the exposure factor. In this study, we conducted MR analysis utilizing the “TwoSampleMR” software package (version 0.6.8) within the R programming environment.We selected two large plasma pQTLs as exposure data and MS as outcome data. In constructing IVs14, we screened single nucleotides with a P value less than 5 × 10−8, as specified by the European 1000 Genomes Project, and located within ± 10,000 kb of the transcription start site (TSS) of each gene’s polymorphism (SNP). SNP analysis was conducted with the criterion that the linkage disequilibrium coefficient (r²) was less than 0.001, and the minor allele frequency (MAF) threshold was set to 0.01. To assess potential weak instrumental variable bias, we calculated the F statistic to quantify the strength of the instrumental variable using the formula: F = R²(NK-1)/[K(1-R²)], where R², N, and K represent the estimated exposure variance explained by the IV, the sample size, and the number of IVs, respectively. SNPs with an F statistic less than 10 were considered weak IVs and excluded from the analysis to mitigate bias caused by weak instruments15. We utilized LDtrait to remove confounding factors (https://ldlink.nih.gov/?tab=home), setting the threshold to 1 × 10−516. SNPs directly associated with MS and those traits directly linked to the disease were excluded. Following data reconciliation, MR analysis was performed on the filtered SNPs. When only one SNP was available for analysis, the Wald ratio method was employed17; for multiple SNPs, the IVW method with random effects was utilized18. Heterogeneity among individual causal effects of SNPs was assessed using Cochran’s Q test, and pleiotropy was evaluated with the MR Egger intercept, setting the MR Egger threshold at 0.05. Finally, we applied false discovery rate (FDR) correction to the resulting P values, establishing a significance threshold of P< 0.0519.
Bayesian colocalization analysis
We aimed to assess whether multiple genetic associations correspond to the same causal variant at identical genomic locations. To achieve this, we employed colocalization analysis to investigate whether the associations between positive proteins and positive gene loci, as identified through MR analysis, are derived from the same causal variant in the context of MS. This analysis utilizes a Bayesian model to calculate the posterior probabilities (PPH) for five hypotheses related to their association with traits20: (1) H0: MS is not associated with any trait; (2) H1: MS is solely associated with trait 1; (3) H2: MS is solely associated with trait 2; (4) H3: MS is associated with both traits, but through different causal variants; (5) H4: MS is associated with both traits and is caused by the same causal variant. During the analysis, we implemented the “coloc.abf” algorithm with default parameter settings. Specifically, these parameters include: the prior probability p1 of the SNP associated with trait 1 set at 1 × 10⁻⁴, the prior probability p2 of the SNP associated with trait 2 also set at 1 × 10⁻⁴, and the prior probability p12 for all correlations set at 1 × 10⁻⁵. Our criteria for analysis are as follows: if PPH4 (the posterior probability of hypothesis H4) exceeds 0.8, the association between plasma proteins and MS is determined to be a significant colocalization.
Whole-phenome group association analysis
We obtained SNP checkpoint information identified through MR analysis and Bayesian co-localization analysis, and conducted a PheWAS analysis based on the 11th edition of the FinnGen database (https://r11.finngen.fi/). The aim of this study is to systematically explore the relationship between significant positive results and a range of outcomes, as well as to enhance our understanding of gene function and comorbidity21.
Expression status of genes in different tissues
To analyze the expression status of significantly positive genes in human tissues, we utilized the Human Protein Atlas (https://www.proteinatlas.org), which provides information on the mRNA and protein levels of genes across various tissues in the human body22. The data encompasses protein expression profiles from 44 normal human tissues, representing 76% of human genes, totaling 15,323 genes. Investigating the expression of positively identified genes in specific tissues may offer insights into their potential mechanisms as therapeutic targets for MS.
Protein-protein interaction network construction
We utilized a protein-protein interaction (PPI) network to illustrate the interactions among gene-encoded proteins that hold significant potential for drug development. In constructing the network, we selected the STRING database (https://string-db.org/) as our data source and established a confidence score threshold of 0.4 to screen for valid interactions. All other default parameters of the STRING database were retained23. In addition, we used CytoNCA to conduct a centrality analysis and evaluation of the protein-protein interaction network in order to identify the significant core genes24.
GO enrichment analysis and KEGG pathway analysis
In our comprehensive analysis of the functional properties and biological connections of pre-screened potential druggable genes, we utilized the “clusterProfiler” package (version 4.12.6) in R software to conduct GO enrichment analyses25and KEGG enrichment analyses26,27,28. Our GO analysis encompasses three fundamental dimensions: biological process (BP), molecular function (MF), and cellular compartment (CC), with the objective of elucidating the activity patterns, functional roles, and cellular localization of these genes within biological systems. Furthermore, KEGG pathway analysis offers detailed insights into the metabolic pathways and signaling networks associated with these genes, which is essential for a comprehensive understanding of their biological functions and potential mechanisms of drug action. Through these analyses, we systematically delineated the functional network of these genes within cells, thereby providing a robust biological foundation for drug development.
Drug candidate prediction
The Drug Signatures Database (DSigDB, http://dsigdb.tanlab.org/DSigDBv1.0/) is a comprehensive repository of drug compounds and genes, comprising 22,527 gene sets and 17,389 compounds linked to 19,531 genes29. Through SMR analysis and Bayesian colocalization analysis of positively mapped protein genes associated with MS, significant druggable genes were identified and subsequently mapped to compounds within the DSigDB database. This approach aims to predict potential drug candidates and evaluate the pharmacological activity of the target genes. By leveraging the gene-compound information contained in the DSigDB database, we can explore potential drugs related to these druggable genes and gain a deeper understanding of the pharmacological properties of these target genes, thereby providing a robust data foundation for drug development and disease treatment research.
Molecular docking
Following an in-depth analysis of the DSigDB database, we proceeded with the molecular docking process. This approach aims to accurately evaluate the binding energy and interaction modes of candidate drugs with their corresponding targets. Through the screening and analysis of docking results, we identified ligands with higher binding affinities and favorable interaction profiles. Protein structure data were downloaded from the Protein Data Bank (PDB, http://www.rcsb.org/). In instances where certain protein structure data were missing, we utilized the online tool developed by Yang Jianyi’s research group at Shandong University (https://yanglab.qd.sdu.edu.cn/) to construct the PDB format of the missing data. Drug structure data were obtained from the U.S. National Library of Medicine Chemical Substances Database (https://pubchem.ncbi.nlm.nih.gov/), from which we downloaded the required data in SDF format30. Subsequently, we employed the OpenBabel 3.1.1 software tool (https://github.com) to convert the data to PDB format. During the molecular docking process, we selected key drugs with significant P values and the proteins encoded by their respective target genes, utilizing the computerized protein-ligand docking software AutoDock 4.2.6 (http://autodock.scripps.edu/) to perform the docking and analyze the binding energy. To visualize the docking results intuitively, we used PyMol 3.0.5 software (https://www.pymol.org/).
Results
Mendelian randomization analysis results
We conducted a two-sample MR analysis, with results presented in Appendix Table S1 and Fig. 2. Our analysis identified a total of 88 positive plasma proteins associated with plasma pQTLs and MS in the Icelandic cohort, and 122 positive plasma proteins in the UK Biobank cohort. Figure 3 illustrates a circular heat map depicting the positive results of the MR analysis of plasma proteins related to MS. A comparison of the positive MR results across the two datasets revealed that UROS and GSTT2B are common positive proteins. Following FDR correction, STAT3 emerged as a significantly positive protein in the MR analysis of plasma pQTLs and MS for the Icelandic cohort. In the UK Biobank analysis, the proteins AGER, AIF1, BTN1A1, CD58, DSG4, EVI5, TNF, and TNFRSF14 were found to be significantly positive. After applying Bonferroni correction, AGER, CD58, EVI5, and TNF were identified as significant positive proteins in the MR analysis of plasma pQTLs and MS within the UK Biobank.

Volcano plot of the results of the MR analysis between plasma pQTLs and MS.

Circular heat map of the positive results of the MR analysis between plasma pQTLs and MS.
Bayesian co-localization analysis results
In our analysis of positive genes identified in the previous MR study of Icelandic plasma pQTLs related to MS, we observed that ATF6B and TAPBP exhibited moderate co-localization. Similarly, in the UK Biobank’s MR analysis of plasma protein pQTLs associated with MS, we identified several significantly positive genes through Bayesian co-localization analysis: EVI5 (PPH4 = 0.9800), OGA (PPH4 = 0.8569), and TNFRSF14 (PPH4 = 0.8904). The results of the Bayesian co-localization analysis are presented in Appendix Table S2, while Figs. 4, 5 and 6 illustrates the scatter plots of these significant positive genes.

Bayesian co localization results of EVI5 and multiple sclerosis.

Bayesian co localization results of OGA and multiple sclerosis.

Bayesian co localization results of TNFRSF14 and multiple sclerosis.
Whole phenotype group association analysis results
We conducted a phenotype-wide association analysis between Bayesian co-localized significantly positive genes and the latest version 11 GWAS data released by the Finngen database. Our findings revealed that the gene EVI5 is associated with 163 diseases or phenotypes, with FDR correction indicating a strong association with MS. The gene OGA is linked to 216 diseases or phenotypes; after FDR correction, we found that OGA is associated with atrial fibrillation, atrial flutter, cervical disc disease, back disease, other disc diseases, spondylosis, and ovarian endometriosis. Additionally, TNFRSF14 is associated with 203 diseases or phenotypes. Post-FDR correction, we determined that TNFRSF14 is related to eosinophilic asthma, autoimmune diseases, body mass index, follicular lymphoma, primary lymphatic and hematopoietic malignancies, nasal polyps, dermatitis, eczema, and rheumatoid arthritis. Appendix Table S3 presents the MR-PheWas analysis results for significantly positive genes, while Fig. 7 illustrates the Manhattan plot of the MR-PheWas results for these genes.

Manhattan plot of significant positive gene PheWAS in multiple sclerosis.
Expression status of genes in different tissues
We utilized the Human Protein Atlas to identify differences in the expression of significantly positive genes across human tissues. Figures 8, 9 and 10 display their expression levels in various human tissues. Our study specifically found that EVI5 shows a primary expression in the placenta, along with significant expression in the liver, testis, parathyroid gland, adrenal gland, smooth muscle, and skeletal muscle. OGA is primarily expressed in the bone marrow, with notable expression in the lymph nodes, thymus, tonsils, and cerebral cortex. Lastly, TNFRSF14 is mainly expressed in the duodenum, with significant expression also detected in the small intestine, fallopian tube, lymph nodes, and liver.

Expression of EVI5 gene in human tissues.

Expression of OGA gene in human tissues.

Expression of TNFRSF14 gene in human tissues.
Protein-protein interaction network construction results
We imported the 208 positive drug target genes resulting from the MR analysis between plasma proteins and MS into the STRING database with the aim of constructing a PPI network. The formed protein interaction pathway contained 208 nodes and 372 edges. Attachment Table S4 shows the analysis results of the protein-protein interactions. Figure 11 displays the protein interaction network diagram constructed based on the STRING database, and Fig. 12 shows the results of the centrality analysis and evaluation of the protein interaction network conducted using CytoNCA.

STRING database protein interaction network diagram.

Results of the centrality analysis and evaluation of the protein-protein interaction network using CytoNCA, demonstrating the hierarchy of the core disease genes.
GO enrichment analysis and KEGG pathway analysis results
In our investigation of the properties of 208 potential drug targets identified through MR analysis of plasma proteins and MS, we employed GO analysis to determine that these genes are associated with a total of 231 pathways, primarily within biological processes (BP). Notably, these targets are significantly involved in several key processes, including carbohydrate derivative catabolism (GO: 1901136), leukocyte migration (GO: 0050900), leukocyte intercellular adhesion (GO: 0007159), chemotaxis (GO: 0006935), chemotaxis (GO: 0042330), positive regulation of the MAPK cascade (GO: 0043410), regulation of cell-cell adhesion (GO: 0022407), and positive regulation of cytokine production (GO: 0001819). Additionally, these targets exhibit specific molecular functions (MF), predominantly including glycosyltransferase activity (GO: 0016757), peptidase regulatory activity (GO: 0061134), and enzyme inhibitor activity (GO: 0004857). Regarding cellular sublocalization (CC), they are associated with the outer plasma membrane (GO: 0009897) and the collagen-containing extracellular matrix (GO: 0062023). The results of the GO enrichment analysis are presented in Annex Table S5 and Figs. 13 and 14.

Histogram of the results of the GO enrichment analysis.

Bubble chart of the results of the GO enrichment analysis.
To further elucidate the relevance of these targets to MS treatment, we conducted an analysis using the KEGG. The results indicated that these target genes were significantly enriched in three specific pathways: the NF-kappa B signaling pathway (hsa04064), the interaction of viral proteins with cytokines and cytokine receptors (hsa04061), and the cytokine-cytokine receptor interaction pathway (hsa04060). The findings of the KEGG analysis are presented in Table S6 and Figs. 15, 16 attached hereto.

Histogram of the results of the KEGG enrichment analysis.

Bubble chart of the results of the KEGG enrichment analysis.
Candidate drug prediction results
We utilized the DSigDB database to predict potential intervention drugs and compiled a list of the top ten drug candidates based on the obtained P-values and corrected P-values. The analysis revealed that DL-mevulinic acid (CTD 00006329), simvastatin (CTD 00007319), and luteolin (CTD 00007430) were the three most significant drug candidates. Specifically, DL-mevulinic acid exhibited significant associations with the VCAM1, STAT3, HLA-DRA, TNF, and NFKB1 genes, while simvastatin demonstrated significant associations with VCAM1, STAT3, HLA-DRA, LTB, AGER, TNF, and NFKB1 genes. Luteolin was significantly associated with VCAM1, STAT3, AGER, TNF, and NFKB1 genes. Table S7 in the Annex presents the single-gene drug candidate prediction results, and Table S8 in the Annex provides the disease drug candidate prediction results for MS.
Molecular docking results
In this study, we identified significant positive target genes through MR analysis and Bayesian co-localization. We utilized AutoDock 4.2.6 software to analyze the binding checkpoints and interactions between the top six drug candidates associated with these significant positive genes and the proteins encoded by their respective genes. We calculated the binding energy for each interaction, successfully obtaining effective docking results between three genes and their corresponding drugs. Among all docking results, the interaction of TNFRSF14 with DMBA exhibited the lowest binding energy value of −8.8 kcal/mol, while the interaction of EVI5 with sanguinarine showed a binding energy value of −8.3 kcal/mol. Annex Table S9 presents the molecular docking results, while Fig. 10A-H illustrate the higher binding energies observed in the molecular docking analysis Figs. 17−23.

Molecular docking between EVI5 and Adehl.

Molecular docking between EVI5 and flunixin.

Molecular docking between EVI5 and imidurea.

Molecular docking between EVI5 and Sanguinarine.

Molecular docking between TNFRSF14 and DMBA.

Molecular docking between TNFRSF14 and EXEEMESTANE.

Molecular docking of TNFRSF14 with folic acid.
Discussion
MS is a complex disease that affects the central nervous system, primarily targeting the brain and spinal cord. Although its exact pathogenesis remains incompletely understood, it is generally classified as an autoimmune disease. In MS, the patient’s immune system inappropriately attacks their own nerve tissue, specifically the myelin sheath, which is the protective covering of nerve fibers, resulting in damage. This damage disrupts the normal conduction of nerve signals, leading to a range of bodily dysfunctions. Currently, there is no complete cure for MS; however, timely diagnosis and appropriate treatment can significantly enhance patients’ quality of life and prognosis. Our study identified EVI5, OGA, and TNFRSF14 genes as potential targets for the treatment of MS. Additionally, metronidazole, sanguinarine, and 7,12-dimethylbenzo[a]anthracene (DMBA) may be significantly associated with the development of this disease.
The relationship between EVI5 and MS
Ecotropic Viral Integration Site 5 (EVI5) is a critical checkpoint in the retroviral integration process and is closely associated with various physiological processes, including the cell cycle. It plays a significant role in essential biological activities such as cell growth, division, and differentiation. In the context of MS, the proliferation and differentiation of neural stem cells and neural precursor cells may be disrupted. Mutations in the EVI5 gene can impair the normal cell cycle processes of these cells, potentially influencing the pathogenesis of MS. By regulating the expression of lymphocyte-specific factors, EVI5 has been shown to be crucial in T cell differentiation, particularly in the high expression of TH1 and TH2 cells31. The deletion of EVI5 adversely affects the differentiation of helper T cells, especially TH17 cells, leading to a significant decrease in IL-17 A production. Moreover, EVI5 also impacts genes such as HLA-DRB, CLEC16A, CD58, and IL7R, which may be linked to the immunomodulatory effects in MS32. Therefore, EVI5 may influence the pathogenesis of MS by modulating T cell differentiation and function. As a GTPase-activating protein (GAP) of Rab11, EVI5 activates the GTPase activity of Rab11, potentially participating in the regulation of the downstream RAB11 pathway33. This pathway is vital for the formation of immune synapses and T cell function. EVI5 interacts with GTP-Rab11 through its TBC domain, promoting the conversion of GTP to GDP, thereby inactivating Rab11. This regulatory mechanism may affect immune synapse formation and T cell functionality, subsequently influencing the immune response in MS. Additionally, EVI5 is involved in lipid metabolism-related pathways, which may further impact the pathological processes associated with MS34. As a member of the protein family containing the Tre-2/Bub2/Cdc16 (TBC) domain, EVI5 plays a regulatory role in the cell cycle, cell division, and cell membrane transport.The function of EVI5 may be linked to its regulatory role in cell membrane transport, which could indirectly influence lipid metabolism and inflammatory responses in MS. Additionally, EVI5 protein is closely associated with endocytosis and cell signaling. Research indicates that EVI5 may impact myelin repair by modulating the function of oligodendrocytes, the cells responsible for the formation and maintenance of myelin sheaths33. Variations in EVI5 activity can affect the survival, proliferation, and differentiation of these cells, thereby facilitating myelin regeneration. Furthermore, EVI5 may regulate immune responses by influencing cell signaling pathways. In MS, the immune system’s attack on the myelin sheath is a critical factor in initiating pathological changes. The regulatory effects of EVI5 help control immune cell activity, mitigate damage to the myelin sheath, and create favorable conditions for myelin repair35. Moreover, EVI5 may participate in the neuroinflammatory process by regulating the function of nerve cells. Neuroinflammation is a significant characteristic of MS, which can lead to myelin damage and neurodegeneration. EVI5 contributes to this process by affecting the recycling of endosomes and signaling pathways36. In summary, EVI5 is intricately linked to the onset and progression of MS and represents a crucial gene in the pathogenesis of the disease.
The potential therapeutic application of the EVI5 gene in MS shows considerable promise. Research has demonstrated that variations in the EVI5 gene are significantly associated with the risk of developing MS37. These variations may influence the function of the EVI5 protein and, consequently, play a role in the pathophysiological processes underlying MS38. Furthermore, variations in EVI5 may not only act as risk factors for MS but also serve as potential biomarkers for early diagnosis and prognostic assessment of the disease. By regulating the expression of EVI5, the activation of T cells and B cells, as well as their roles in MS, can be modulated, potentially improving patient symptoms38. These characteristics position EVI5 as a promising target for MS treatment, providing both a theoretical foundation and the possibility for the development of novel therapeutic strategies.
Sanguinarine is an alkaloid extracted from the bloodroot plant, which belongs to the poppy family. It exhibits a high binding energy with EVI5. Recent research has revealed its multifaceted effects on the immune system, demonstrating its ability to modulate the host’s immune response through various mechanisms. Sanguinarine shows potential in enhancing immune function and combating certain immune-related diseases. It can bolster the body’s defense against pathogens by adjusting the composition and diversity of intestinal flora, which may enhance the host’s immune response through the regulation of the intestinal microbiome39. Furthermore, sanguinarine plays a significant role in strengthening the body’s innate immune response by regulating reactive oxygen species (ROS) and activating the PMK-1/SKN-1 signaling pathway39. This mechanism has shown promising effects across different organisms, including improvements in the body’s resistance to oxidative stress. At the level of immune cells, sanguinarine influences the production of inflammatory cytokines by modulating multiple signaling pathways, such as MAPK, Wnt/β-catenin, NF-κB, JAK/STAT, TGF-β, and PI3K/Akt/mTOR pathways40. These interactions indicate that sanguinarine has dual effects on inflammatory and immune responses. Given its complex effects on the immune system and chronic inflammation, sanguinarine holds promise for applications in the treatment of immune-related diseases, chronic inflammation, and allergic reactions.
The relationship between TNFRSF14 and MS
TNFRSF14, a member of the tumor necrosis factor receptor superfamily 14 and also known as HVEM, is a protein that plays a crucial role in the immune response. It is significant in regulating lymphocyte activation and proliferation. Studies have demonstrated that HVEM can effectively modulate the immune response of T cells by activating the inflammatory response and transmitting inhibitory signals41,42. HVEM not only serves as a receptor for LIGHT and lymphotoxin-alpha but also interacts with immunoglobulin superfamily members BTLA and CD160. This versatility positions HVEM uniquely within the realm of immunomodulation. It is capable of binding to multiple ligands in various conformations, thereby forming complex and interconnected signaling networks that collectively regulate inflammatory and inhibitory responses. Abnormal expression of TNFRSF14 is observed in approximately 40% of patients with follicular lymphoma (FL). HVEM inhibits T cell activation through its interactions with receptors on both B cells and T cells, which is crucial for maintaining immune balance and preventing excessive immune reactions.
In the pathological process of MS, TNFRSF14 may influence disease progression by modulating the interactions between immune cells and central nervous system (CNS) cells. Abnormal function of TNFRSF14 can lead to the overactivation of immune cells, triggering a misdirected attack by the immune system on the myelin sheath of the CNS, which results in an inflammatory response—an established pathological feature of MS. Furthermore, TNFRSF14 may play a role in modulating the immune microenvironment within the CNS, where abnormalities could disrupt immune tolerance mechanisms and exacerbate the development of MS .Studies have shown that the polymorphism of the TNFRSF14 gene is associated with the susceptibility to MS43. In addition, TNFRSF14 can also affect the function of T cells, thereby regulating the intensity and duration of the inflammatory response44. Proper regulation of TNFRSF14 may mitigate neurological damage resulting from excessive inflammation in MS. Furthermore, studies indicate that TNFRSF14 can enhance the survival and activity of CD4 + memory T cells45. This enhancement facilitates effective immune surveillance, protecting against pathogens while minimizing attacks on the body’s own tissues, thereby alleviating symptoms of MS. Concurrently, TNFRSF14 plays a critical role in T cell costimulatory signaling46. On one hand, it can enhance the immune response by promoting the activation, proliferation, and function of T cells; on the other hand, it can suppress excessive immune responses through co-stimulation, thereby maintaining the balance of the immune system. This bidirectional regulatory function is crucial for maintaining immune tolerance and preventing the body from generating unnecessary immune responses to self-antigens. In the central nervous system (CNS), glial cells, such as microglia and astrocytes, play a central role in the inflammatory response. TNFRSF14 influences the activation state of these glial cells through its unique signal transduction mechanism, which in turn contributes to the development of neuroinflammation and the protective processes of nerve cells .Studies have found that the continuous activation of TNFRSF14 may trigger chronic neuroinflammation, which is closely related to the disease progression of MS47. In a chronic inflammatory state, glial cells persistently release cytokines, leading to ongoing damage and degeneration of nerve cells. Furthermore, the activation of TNFRSF14 exacerbates the inflammatory response of microglia, prompting them to release a range of pro-inflammatory cytokines48. This not only exacerbates the local inflammatory response but may also promote further activation of surrounding nerve cells and glial cells, thereby accelerating the development of MS. Consequently, the relationship between TNFRSF14 and MS primarily reflects its role in immunomodulation, particularly concerning the activation of T cells and B cells, as well as the overall immune response. Nevertheless, the specific mechanisms of action and therapeutic potential of TNFRSF14 in MS require further investigation for clarification.
In this study, we observed that 7,12-dimethylbenzo[a]anthracene (DMBA) binds significantly to TNFRSF14. As a carcinogen, DMBA’s role extends beyond direct DNA damage; it also promotes tumor development by influencing cell proliferation. This process is accompanied by complex immune regulation, including an increase in regulatory T cells and modulation of both humoral and cellular immune responses. Studies have demonstrated that DMBA can elevate the number of regulatory T cells49. As the dosage of DMBA increases, the immune response diminishes, leading to Treg cell proliferation and an immunosuppressive state. Additional research has indicated that DMBA exerts inhibitory effects on both cellular and humoral immunity in mice50,51. Regarding humoral immunity, DMBA significantly reduces antibody production. In our experiments, the quantity of antibodies produced by DMBA-treated mice post-vaccination was markedly lower than that of control mice. In terms of cellular immunity, DMBA treatment resulted in diminished cytotoxic T cell function, abnormal immune cell activation, and reduced cytokine secretion, suggesting that DMBA has a direct toxic effect on immune cells. The immunosuppressive effects of DMBA intensify with increasing doses, and the observed increases in T cells, antibody production, and cytotoxic T cell function are all dose-dependent. As research deepens and the immune mechanisms affected by DMBA are further explored, novel strategies may emerge for the prevention and treatment of related diseases.
The relationship between OGA and MS
O-GlcNAcase (OGA) plays a critical role in regulating protein glycosylation modifications, primarily by catalyzing the hydrolysis of O-GlcNAc on proteins52. In the dynamic process of O-GlcNAcylation, O-GlcNAc transferase (OGT) and OGA function in concert. OGT is responsible for adding monosaccharides to proteins, while OGA removes these monosaccharides, thereby maintaining a dynamic balance of protein O-GlcNAcylation levels. This precise regulation is essential for the structural integrity, functional performance, and stability of proteins, and it plays a central role in key biological processes, including cell signal transduction, metabolic regulation, immune response, and tumor initiation and progression. Consequently, the balance of OGA and OGT activities is vital for sustaining the stability of the intracellular environment.
MS is a chronic inflammatory disease of the central nervous system that is mediated by the immune system. It is characterized by the destruction of the myelin sheath, which exposes nerve fibers and adversely affects the transmission of nerve signals, leading to a range of neurological dysfunctions. Previous studies have indicated that OGA plays a crucial role in various signaling pathways, primarily by removing glycosylation modifications that regulate numerous cellular processes. In particular, the MAPK/ERK signaling pathway is significant for cell proliferation, differentiation, and survival. OGA influences cell proliferation and differentiation through the regulation of key genes. Some research has demonstrated that differentially expressed genes targeted by OGA are associated with the MAPK/ERK signaling pathway, where gene upregulation can enhance cell proliferation and alter differentiation53. In the G-protein coupled receptor (GPCR) signaling pathway, GPCRs play a crucial role in various physiological processes as a significant mechanism for signal transduction. The activity of OGA influences the function of GPCRs by modulating their glycosylation state, which can alter the affinity and signaling capacity of these receptors. Consequently, this modulation may impact cellular responses to external stimuli54. OGA is associated with various growth factor signaling pathways, including insulin and epidermal growth factor. These pathways are crucial for cell growth and metabolism. OGA modulates the efficiency of growth factor signaling by removing O-GlcNAc modifications, thereby influencing cellular responses to growth factors. In pathways related to the extracellular matrix (ECM), the ECM serves a vital role in providing cell support and facilitating signaling. OGA regulates the interaction between cells and their microenvironment by modulating the expression of ECM-related genes. Notably, multiple ECM-related genes are identified among the differentially expressed genes targeted by OGA, which may enhance cell migration and tissue reconstruction53. OGA activity is closely linked to mitochondrial function, as mitochondria serve as the energy production factories of the cell and play a crucial role in cell signaling. Research has demonstrated that the copy number of mitochondrial DNA and the activity of mitochondrial enzymes are associated with OGA activity and the levels of OGT protein. This suggests that OGA may influence the cellular metabolic state by modulating mitochondrial function55. OGA may play a crucial role in the regulation of the cell cycle. Glycosylation modifications impact the stability and activity of cyclins. OGA functions by removing these modifications, thereby regulating cell cycle progression and influencing cell proliferation and growth53. In addition, OGA is also associated with the immune response. It may influence the secretion of cytokines and the transmission of immune signals by regulating the glycosylation status of immune cells, and thus play a role in inflammatory responses and autoimmune diseases .Studies have indicated that OGA may influence the function and signaling of nerve cells by regulating the glycosylation state of specific neural proteins56. Abnormal glycosylation can lead to structural instability of myelin-related proteins, thereby increasing the susceptibility of myelin to immune attack or dysfunction, and contributing to the pathological processes associated with MS57. OGA may influence nerve cell function and disease progression in MS by regulating the glycosylation state of neuroproteins. The potential application of OGA inhibitors in the treatment of MS has garnered significant attention. OGA is crucial for the regulation of O-GlcNAcylation; by inhibiting OGA, the levels of O-GlcNAc can be elevated, thereby modulating signaling pathways associated with inflammation and neuroprotection58,59. The inhibition of OGA can also improve the metabolic state of cells, such as increasing glucose metabolism, which is crucial for the neuroprotection and functional recovery of patients with MS60. Clinical studies have shown that OGA inhibitors perform well in terms of tolerability and safety, demonstrating their potential in the treatment of MS61. In conclusion, OGA plays a regulatory role in various signaling pathways and has significant implications for the immune system, inflammatory response, myelin regeneration, and receptor alterations. These factors not only influence the physiological state of cells but also contribute to the onset and progression of diseases. Future inhibitors targeting OGA have demonstrated considerable potential in the treatment of MS. By modulating O-GlcNAcylation, enhancing metabolic function, and exhibiting anti-inflammatory effects, these compounds may pave the way for new treatment options for MS patients.
Despite implementing a rigorous data analysis process and employing a series of the latest GWAS data, this study acknowledges several limitations. Firstly, the focus on populations of European ancestry raises concerns regarding the generalizability of the conclusions to other ethnic groups, necessitating further in-depth exploration. Variations among ethnic groups in genetic makeup and living environments may influence the broader applicability of the research findings. Secondly, during the integration of original data for the meta-analysis of differentially expressed genes in plasma proteins, the use of diverse data sources, such as microarrays and bulk RNA sequencing with varying sample sizes, may introduce biases. The differences in data collection and analysis methods can complicate the accurate identification of differentially expressed genes. Thirdly, the expression quantitative trait loci (eQTLs) vary with disease progression due to differences in cell types. The eQTLs derived from bulk RNA sequencing face significant constraints when elucidating the key molecular mechanisms associated with diseases. Specifically, the mechanisms governing plasma protein expression in vivo differ from those observed in vitro, making it inappropriate to directly extrapolate in vitro data to represent all plasma protein functions. Additionally, alterations in various cell types throughout the disease development process may further hinder the identification of critical molecular mechanisms. Lastly, a small sample size and unbalanced grouping can lead to errors in the analysis. Although high thresholds and multiple corrections have enhanced analytical accuracy, there remains a risk of overlooking genuine associations that may lack statistical significance in smaller samples.Fifthly, the subtle effects of genetic variations may diminish statistical power and elevate the likelihood of false positives. Sixthly, the pathogenic mechanisms of diseases are exceedingly complex, involving genetic factors, environmental influences, and numerous unknown variables. To address these gaps, large-scale, multi-center, and rigorously designed studies are essential. Only through such comprehensive investigations can the associations between various factors and diseases be accurately elucidated, thereby establishing a scientific foundation for effective prevention and treatment strategies.
Future studies should prioritize multi-ethnic correlation research to enhance the applicability of findings. Currently, correlation studies focused on Asian populations are progressing rapidly, and we anticipate the opportunity to conduct multi-ethnic correlation studies by gathering datasets from various ethnicities and regions to increase sample diversity. Simultaneously, we will employ meta-analysis techniques to synthesize results from different datasets, thereby enhancing the generalizability of our findings. To our knowledge, published pQTL data is available for Asian populations, and we plan to perform MR analysis of drug targets specific to these groups in the future. Additionally, in the realm of molecular docking, evaluating binding energy is critical for predicting ligand-receptor interactions; however, its application faces several limitations that hinder its accuracy and practicality in drug design and screening.In response to this limitation, future experimental studies will play a crucial role that cannot be overlooked. First, to investigate expression differences, we will utilize RT-qPCR and Western blot (WB) techniques to analyze the expression levels of the EVI5, OGA, and TNFRSF14 genes and proteins in Jurkat, Raji, C6, OLN-93, and PC12 cells. Secondly, for gene function exploration, we will employ cell transfection technology to knock down and overexpress the corresponding genes, monitoring various indicators at the gene and protein levels (qPCR, Western blot), as well as assessing cell function (CCK-8 proliferation, Annexin V-FITC/PI double staining for apoptosis), and evaluating cytokines and signaling pathways (ELISA for cytokines, Western blot for signal protein phosphorylation). Finally, regarding animal gene research, we will select adeno-associated virus (AAV) or lentiviral vectors to deliver the CRISPR-Cas12a system and target crRNA for gene editing, assessing the effects of gene knockout from multiple levels. At the molecular level, tissue sample DNA will be extracted, and qPCR and gene sequencing will be employed to verify the knockout of the target gene, while mRNA expression changes will be assessed via qPCR. At the protein level, Western blot or immunohistochemistry will be utilized to detect the expression of the target protein and evaluate its impact on protein synthesis. At the cellular level, we will analyze the phenotype, activation status, and quantity changes of oligodendrocytes through flow cytometry by isolating target tissue cells.
Conclusion
In this study, the proteins EVI5, OGA, and TNFRSF14 were identified as key targets for the treatment of MS through MR and Bayesian co-localization analysis. These proteins, along with their corresponding coding genes, demonstrated significant therapeutic relevance in the context of MS.
Data availability
All types of data involved in this study have clear sources for easy access. The complete GWAS information for UK Biobank plasma pQTLs can be downloaded directly from s3://ukbiobank.opendata.sagebase.org/. The GWAS data for Icelandic plasma proteins is available in the literature titled “Large-scale integration of the plasma proteome with genetics and disease,” which can be accessed at https://www.nature.com/articles/s41588-021-00978-w#Sec36. Complete GWAS data for RA, including seropositive and seronegative RA, as well as data for conditions such as ulcerative colitis, AS, MS, and PsA, can be downloaded from https://www.finngen.fi/en. Additionally, the complete GWAS data for juvenile RA can be obtained from https://www.nature.com/articles/s41588-021-00931-x. Other relevant data can be sourced from original literature and respective websites.
References
Wallin, M. T. et al. Global, regional, and national burden of multiple sclerosis 1990–2016: a systematic analysis for the Global Burden of Disease Study 2016. The Lancet Neurology 18(3), 269–285 (2019).
Zhang, C. et al. Prevalence and burden of multiple sclerosis in China, 1990–2019: findings from the global burden of disease study 2019. Neurology 102 (11), e209351. https://doi.org/10.1212/wnl.0000000000209351 (2024).
Cao, F. et al. Age-standardized incidence, prevalence, and mortality rates of autoimmune diseases in women of childbearing age from 1990 to 2019. Autoimmun. Rev. 22 (11), 103450. https://doi.org/10.1016/j.autrev.2023.103450 (2023).
Zhao, M. et al. Age-standardized incidence, prevalence, and mortality rates of autoimmune diseases in adolescents and young adults (15–39 years): an analysis based on the global burden of disease study 2021. BMC Public. Health. 24 (1), 1800. https://doi.org/10.1186/s12889-024-19290-3 (2024).
Li, D. P. et al. A global assessment of incidence trends of autoimmune diseases from 1990 to 2019 and predicted changes to 2040. Autoimmun. Rev. 22 (10), 103407. https://doi.org/10.1016/j.autrev.2023.103407 (2023).
Gogulescu, A. et al. Neurological side effects of TNF-α inhibitors revisited: A review of case reports. Med. (Kaunas). 60 (9). https://doi.org/10.3390/medicina60091409 (2024).
So, M. W., Kim, A. R. & Lee, S. G. Drug persistence and incidence of active tuberculosis of tumor necrosis factor alpha inhibitors versus Tocilizumab as the First-Line biological treatment in patients with rheumatoid arthritis: A nationwide Population-Based retrospective cohort analysis. Rheumatol. Ther. 11 (4), 881–895. https://doi.org/10.1007/s40744-024-00674-1 (2024).
Varisco, P. A. & So, A. [A new therapeutical option for chronic inflammation in rheumatology: Janus kinases inhibitors (JAK)]. Rev. Med. Suisse. 10 (414), 187–191 (2014).
Hennessee, I., Benedict, K., Bahr, N. C., Lipner, S. R. & Gold, J. A. W. Incidence and risk factors for invasive fungal infections in patients initiating TNF-alpha inhibitors for inflammatory bowel disease and rheumatoid arthritis. Clin. Infect. Dis. https://doi.org/10.1093/cid/ciae444 (2024).
Gonzalez-Lorenzo, M. et al. Immunomodulators and immunosuppressants for relapsing-remitting multiple sclerosis: a network meta-analysis. Cochrane Database Syst. Rev. 1 (1), Cd011381. https://doi.org/10.1002/14651858.CD011381.pub3 (2024).
Ferkingstad, E. et al. Large-scale integration of the plasma proteome with genetics and disease. Nat. Genet. 53 (12), 1712–1721. https://doi.org/10.1038/s41588-021-00978-w (2021).
Sun, B. B. et al. Plasma proteomic associations with genetics and health in the UK biobank. Nature 622 (7982), 329–338. https://doi.org/10.1038/s41586-023-06592-6 (2023).
Davies, N. M., Holmes, M. V. & Davey Smith, G. Reading Mendelian randomisation studies: a guide, glossary, and checklist for clinicians. Bmj 362, k601. https://doi.org/10.1136/bmj.k601 (2018).
Lin, Z., Deng, Y. & Pan, W. Combining the strengths of inverse-variance weighting and Egger regression in Mendelian randomization using a mixture of regressions model. PLoS Genet. 17 (11), e1009922. https://doi.org/10.1371/journal.pgen.1009922 (2021).
Burgess, S. & Thompson, S. G. Avoiding bias from weak instruments in Mendelian randomization studies. Int. J. Epidemiol. 40 (3), 755–764. https://doi.org/10.1093/ije/dyr036 (2011).
Lin, S. H., Thakur, R. & Machiela, M. J. LDexpress: an online tool for integrating population-specific linkage disequilibrium patterns with tissue-specific expression data. BMC Bioinform. 22 (1), 608. https://doi.org/10.1186/s12859-021-04531-8 (2021).
Li, X., Xie, Z., Qiu, H., Xie, X. & Liu, L. Exploring the causal associations between diet-derived Circulating antioxidants and the risk of endometriosis: a Mendelian randomization study. Front. Nutr. 11, 1453147. https://doi.org/10.3389/fnut.2024.1453147 (2024).
Burgess, S., Butterworth, A. & Thompson, S. G. Mendelian randomization analysis with multiple genetic variants using summarized data. Genet. Epidemiol. 37 (7), 658–665. https://doi.org/10.1002/gepi.21758 (2013).
Zhou, L. et al. Metabolic syndrome and cancer risk: A two-sample Mendelian randomization study of European ancestry. Int. J. Surg. 111 (1), 311–321. https://doi.org/10.1097/js9.0000000000001926 (2024).
Bulik-Sullivan, B. et al. An atlas of genetic correlations across human diseases and traits. Nat. Genet. 47 (11), 1236–1241. https://doi.org/10.1038/ng.3406 (2015).
Francis, C. M. et al. Genome-wide associations of aortic distensibility suggest causality for aortic aneurysms and brain white matter hyperintensities. Nat. Commun. 13 (1), 4505. https://doi.org/10.1038/s41467-022-32219-x (2022).
Uhlén, M. et al. Proteomics. Tissue-based map of the human proteome. Science 347 (6220), 1260419. https://doi.org/10.1126/science.1260419 (2015).
Szklarczyk, D. et al. The STRING database in 2023: protein-protein association networks and functional enrichment analyses for any sequenced genome of interest. Nucleic Acids Res. 51 (D1), D638–d46. https://doi.org/10.1093/nar/gkac1000 (2023).
Tang, Y., Li, M., Wang, J., Pan, Y. & Wu, F. X. CytoNCA: a cytoscape plugin for centrality analysis and evaluation of protein interaction networks. Biosystems 127, 67–72. https://doi.org/10.1016/j.biosystems.2014.11.005 (2015).
Expansion of the Gene Ontology knowledgebase and resources. Nucleic Acids Res. ;45(D1):D331–d8. https://doi.org/10.1093/nar/gkw1108 (2017).
Kanehisa, M. & Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 28 (1), 27–30. https://doi.org/10.1093/nar/28.1.27 (2000).
Kanehisa, M., Furumichi, M., Sato, Y., Matsuura, Y. & Ishiguro-Watanabe, M. KEGG: biological systems database as a model of the real world. Nucleic Acids Res. 53 (D1), D672–d7. https://doi.org/10.1093/nar/gkae909 (2025).
Kanehisa, M. Toward Understanding the origin and evolution of cellular organisms. Protein Sci. 28 (11), 1947–1951. https://doi.org/10.1002/pro.3715 (2019).
Yoo, M. et al. DSigDB: drug signatures database for gene set analysis. Bioinformatics 31 (18), 3069–3071. https://doi.org/10.1093/bioinformatics/btv313 (2015).
Kim, S. et al. PubChem 2023 update. Nucleic Acids Res. 51 (D1), D1373–d80. https://doi.org/10.1093/nar/gkac956 (2023).
Hoppenbrouwers, I. A. et al. EVI5 is a risk gene for multiple sclerosis. Genes Immun. 9 (4), 334–337. https://doi.org/10.1038/gene.2008.22 (2008).
Jafari, N., Broer, L., van Duijn, C. M., Janssens, A. C. & Hintzen, R. Q. Perspectives on the use of multiple sclerosis risk genes for prediction. PLoS One. 6 (12), e26493. https://doi.org/10.1371/journal.pone.0026493 (2011).
Soltani, S., Webb, S. M., Kroll, T. & King-Jones, K. Drosophila Evi5 is a critical regulator of intracellular iron transport via transferrin and ferritin interactions. Nat. Commun. 15 (1), 4045. https://doi.org/10.1038/s41467-024-48165-9 (2024).
Cai, T. et al. EVI5 is an oncogene that regulates the proliferation and metastasis of NSCLC cells. J. Exp. Clin. Cancer Res. 39 (1), 84. https://doi.org/10.1186/s13046-020-01585-z (2020).
Dashti, M., Ateyah, K., Alroughani, R. & Al-Temaimi, R. Replication analysis of variants associated with multiple sclerosis risk. Sci. Rep. 10 (1), 7327. https://doi.org/10.1038/s41598-020-64432-3 (2020).
Rida Zainab, S., Zeb Khan, J., Khalid Tipu, M., Jahan, F. & Irshad, N. A review on multiple sclerosis: unravelling the complexities of pathogenesis, progression, mechanisms and therapeutic innovations. Neuroscience 567, 133–149. https://doi.org/10.1016/j.neuroscience.2024.12.029 (2025).
Mazdeh, M. et al. Ecotropic viral integration site 5 (EVI5) expression analysis in multiple sclerosis patients. Hum. Antibodies. 26 (3), 113–119. https://doi.org/10.3233/hab-170328 (2017).
D’Anca, M. et al. Extracellular vesicles in multiple sclerosis: role in the pathogenesis and potential usefulness as biomarkers and therapeutic tools. Cells 10 (7). https://doi.org/10.3390/cells10071733 (2021).
Liu, F. et al. Sanguinarine promotes healthspan and innate immunity through a conserved mechanism of ROS-mediated PMK-1/SKN-1 activation. iScience 25 (3), 103874. https://doi.org/10.1016/j.isci.2022.103874 (2022).
Huang, L. J. et al. Bioactivity and mechanism of action of sanguinarine and its derivatives in the past 10 years. Biomed. Pharmacother. 173, 116406. https://doi.org/10.1016/j.biopha.2024.116406 (2024).
Kotsiou, E. et al. TNFRSF14 aberrations in follicular lymphoma increase clinically significant allogeneic T-cell responses. Blood 128 (1), 72–81. https://doi.org/10.1182/blood-2015-10-679191 (2016).
Steinberg, M. W., Cheung, T. C. & Ware, C. F. The signaling networks of the herpesvirus entry mediator (TNFRSF14) in immune regulation. Immunol. Rev. 244 (1), 169–187. https://doi.org/10.1111/j.1600-065X.2011.01064.x (2011).
Blanco-Kelly, F. et al. Members 6B and 14 of the TNF receptor superfamily in multiple sclerosis predisposition. Genes Immun. 12 (2), 145–148. https://doi.org/10.1038/gene.2010.42 (2011).
Croft, M. The role of TNF superfamily members in T-cell function and diseases. Nat. Rev. Immunol. 9 (4), 271–285. https://doi.org/10.1038/nri2526 (2009).
Jernås, M. et al. MicroRNA regulate immune pathways in T-cells in multiple sclerosis (MS). BMC Immunol. 14, 32. https://doi.org/10.1186/1471-2172-14-32 (2013).
Ward-Kavanagh, L. K., Lin, W. W., Šedý, J. R. & Ware, C. F. The TNF receptor superfamily in Co-stimulating and Co-inhibitory responses. Immunity 44 (5), 1005–1019. https://doi.org/10.1016/j.immuni.2016.04.019 (2016).
Mazziotti, V. et al. The contribution of tumor necrosis factor to multiple sclerosis: a possible role in progression independent of relapse? J. Neuroinflammation. 21 (1), 209. https://doi.org/10.1186/s12974-024-03193-6 (2024).
Conti, P. et al. Impact of TNF and IL-33 cytokines on mast cells in neuroinflammation. Int. J. Mol. Sci. 25 (6). https://doi.org/10.3390/ijms25063248 (2024).
Nasti, T. H. et al. Regulatory T cells play an important role in the prevention of murine melanocytic nevi and melanomas. Cancer Prev. Res. (Phila). 14 (2), 165–174. https://doi.org/10.1158/1940-6207.Capr-20-0360 (2021).
Gao, J., Lauer, F. T., Dunaway, S. & Burchiel, S. W. Cytochrome P450 1B1 is required for 7,12-dimethylbenz(a)-anthracene (DMBA) induced spleen cell immunotoxicity. Toxicol. Sci. 86 (1), 68–74. https://doi.org/10.1093/toxsci/kfi176 (2005).
Ladics, G. S., Kawabata, T. T. & White, K. L. Jr Suppression of the in vitro humoral immune response of mouse splenocytes by 7,12-dimethylbenz[a]anthracene metabolites and Inhibition of immunosuppression by alpha-naphthoflavone. Toxicol. Appl. Pharmacol. 110 (1), 31–44. https://doi.org/10.1016/0041-008x(91)90287-o (1991).
Cai, H. et al. Protein O-GlcNAcylation in multiple immune cells and its therapeutic potential. Front. Immunol. 14, 1209970. https://doi.org/10.3389/fimmu.2023.1209970 (2023).
Liu, Q. et al. O-GlcNAcase regulates pluripotency States of human embryonic stem cells. Stem Cell. Rep. 19 (7), 993–1009. https://doi.org/10.1016/j.stemcr.2024.05.009 (2024).
Nelson, Z. M., Leonard, G. D. & Fehl, C. Tools for investigating O-GlcNAc in signaling and other fundamental biological pathways. J. Biol. Chem. 300 (2), 105615. https://doi.org/10.1016/j.jbc.2023.105615 (2024).
Huynh, V. N. et al. Acute Inhibition of OGA sex-dependently alters the networks associated with bioenergetics, autophagy, and neurodegeneration. Mol. Brain. 15 (1), 22. https://doi.org/10.1186/s13041-022-00906-x (2022).
Lee, B. E., Suh, P. G. & Kim, J. I. O-GlcNAcylation in health and neurodegenerative diseases. Exp. Mol. Med. 53 (11), 1674–1682. https://doi.org/10.1038/s12276-021-00709-5 (2021).
Carotenuto, A., D’Ursi, A. M., Nardi, E., Papini, A. M. & Rovero, P. Conformational analysis of a glycosylated human Myelin oligodendrocyte glycoprotein peptide epitope able to detect antibody response in multiple sclerosis. J. Med. Chem. 44 (14), 2378–2381. https://doi.org/10.1021/jm010811t (2001).
Sharma, A., Singh, A., Debnath, R., Gupta, G. D. & Sharma, K. Role of O-GlcNAcylation in Alzheimer’s disease: insights and perspectives. Eur. J. Med. Chem. Rep. 12, 100195. https://doi.org/10.1016/j.ejmcr.2024.100195 (2024).
Ostojic, S. M. Guanidinoacetic acid as a nutritional adjuvant to multiple sclerosis therapy. Front. Hum. Neurosci. 16, 871535. https://doi.org/10.3389/fnhum.2022.871535 (2022).
Abdel-Magid, A. F. Inhibition of O-GlcNAcase (OGA): A potential therapeutic target to treat Alzheimer’s disease. ACS Med. Chem. Lett. 5 (12), 1270–1271. https://doi.org/10.1021/ml500450c (2014).
Pratt, M. R. & Vocadlo, D. J. Understanding and exploiting the roles of O-GlcNAc in neurodegenerative diseases. J. Biol. Chem. 299 (12), 105411. https://doi.org/10.1016/j.jbc.2023.105411 (2023).
Acknowledgements
We would like to express our gratitude to the European Bioinformatics Center, the GWAS Catalog database, the FinnGen database, the British Biobank database, the IEU OpenGWAS database, the eqtlgen database, the GTEx database, the YANG LAB database, the Human Protein Atlas database, the STRING database, and the DSigDB database for providing shared data. We also extend our thanks to all data providers mentioned in this article and to the anonymous reviewers for their constructive comments.
Funding
Not applicable.
Author information
Authors and Affiliations
Contributions
W.Y. contributed to the conception and design of this study. Z.H.L. and M.C. provided the conceptualization ideas and analysis and supervised the whole research process. W.Y. conducted the data analysis. W.Y. authored the first draft, then W.Y., C.L.L. and M.C. revised and refined the manuscript. W.Y. and M.C. carried out the visualization analysis of the research results. All authors reviewed and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
For this study, all our GWAS data were derived from published statistical sources, thus negating the need for additional ethical approval.
Consent for publication
All authors unanimously agreed to submit this research achievement for publication.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Yang, W., Liu, c., Li, Z. et al. Multi-omic biomarkers associated with multiple sclerosis: from Mendelian randomization to drug prediction. Sci Rep 15, 9421 (2025). https://doi.org/10.1038/s41598-025-94303-8
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-94303-8
Keywords
This article is cited by
-
EVI5 unveils a role of the oncogene in modulating the Rab11/PD-L1 pathway in lung adenocarcinoma
Scientific Reports (2025)
-
Multiple sclerosis pathophysiology: a comprehensive review of genetic, environmental, and immunological drivers
Inflammopharmacology (2025)
