
Abstract
This study aims to predict and diagnose pediatric septic shock through the screening of immune infiltration-related biomarkers. Three gene expression datasets were accessible from the Gene Expression Omnibus repository. The differentially expressed genes were identified using the R 4.3.2 (https://www.r-project.org/), followed by gene set enrichment analysis. Thereafter, the genes were identified utilizing machine-learning algorithms. The receiver operating characteristic curve was employed to assess the discrimination and effectiveness of the hub genes. The inflammatory and immune status of pediatric septic shock was evaluated through cell-type identification by estimating relative subsets of RNA transcripts (CIBERSORT). The correlation between diagnostic markers and infiltrating immune cells was further examined. Overall, we detected 12 differentially expressed genes. CD177, MCEMP1, MMP8, and OLAH were examined as diagnostic indicators for pediatric septic shock, revealing statistically significant differences (P < 0.01) and diagnostic efficacy in the validation cohort. The immune cell infiltration analysis suggests that various immune cells may contribute to the onset of pediatric septic shock. Furthermore, all diagnostic characteristics may exhibit varying degrees of correlation with immune cells. This study identifies four potential biomarkers—CD177, MCEMP1, MMP8, and OLAH—that provide diagnostic value and novel insights into immune dysregulation in pediatric septic shock. Through the integration of bioinformatics and machine learning methodologies, we offer a novel perspective on the immune mechanisms involved in pediatric septic shock, potentially facilitating more targeted and personalized therapies for individual patients.
Similar content being viewed by others
Introduction
The management of sepsis continues to be one of the most enduring and significant challenges in human healthcare. Pediatric septic shock is a category of sepsis, defined by cardiovascular dysfunction, evidenced by at least one cardiovascular parameter from The Phoenix Sepsis Score. These encompass significant hypotension, blood lactate concentrations surpassing 5 mmol/L, or the necessity for vasopressor administration1. Pediatric septic shock is linked to elevated mortality rates and imposes a considerable burden on global healthcare systems. In high-resource environments, the in-hospital mortality rate for pediatric septic shock persists at 10.8%1. Therefore, identifying biomarkers with predictive and diagnostic significance is essential for the early detection and intervention of pediatric septic shock.
The comprehension of sepsis has progressed, transitioning from the “bacterial theory” to the “host theory”2. Infection is increasingly acknowledged to elicit a complex and extended host response, encompassing both pro-inflammatory and anti-inflammatory mechanisms. Pro-inflammatory responses facilitate the eradication of invading pathogens but also lead to tissue damage in severe sepsis. Conversely, anti-inflammatory responses reduce local and systemic tissue damage but may also heighten vulnerability to secondary infections, resulting in immune suppression in numerous sepsis patients3. Additionally, pharmacological interventions and environmental influences can precipitate immunosuppression in septic patients4,5. Meanwhile, several prospective observational studies have shown that children with severe sepsis or septic shock display early congenital and adaptive immune suppression, which correlates with extended organ dysfunction6,7. Therefore, the assessment of immune infiltration-related biomarkers may facilitate the monitoring of early immune function in pediatric septic shock and identify potential targets for future immune-modulating therapies.
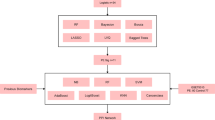
Sepsis, septic shock, and their interactions with the immune system, as well as the influences of genetics and epigenetics, constitute significant research domains within the scientific community8. The amalgamation of high-throughput sequencing technologies and machine learning has facilitated a more comprehensive analysis of genetic-level biological inquiries, encompassing diagnosis and disease monitoring—subjects that were formerly challenging to tackle9. This research, in contrast to prior studies, emphasizes the immune infiltration characteristics of diagnostic genes, integrating identification and validation efforts to furnish evidence for early detection and targeted intervention of pediatric septic shock. Figure 1 delineates the comprehensive research workflow.

The research workflow.
Methods
Gene expression profiles
We utilized the publicly accessible Gene Expression Omnibus (GEO) database (https://www.ncbi.nlm.nih.gov/geo/)10, a thorough repository for high-throughput gene expression data, encompassing microarrays and chips. By employing the search terms “Pediatric septic shock” [MeSH Terms] AND “Homo sapiens” [porgn: txid9606] AND “Expression profiling by array” [All Fields], we identified pertinent datasets. The chosen datasets were derived from specific criteria, concentrating on genome-wide gene expression profiles from blood samples of pediatric septic shock patients and healthy controls, without any association to other diseases. Datasets were included solely if they possessed sample sizes exceeding 10 for both groups. This study incorporated three distinct gene expression datasets: GSE812111 and GSE1390412 for analysis, and GSE2637813,14,15 for validation.
Identification of the differentially expressed genes
We merged the GSE8121 and GSE13904 datasets, normalizing the data and batch-adjusting expression values utilizing the “sva” package16. The LIMMA package17 was utilized to identify differentially expressed genes (DEGs) between the pediatric septic shock and control cohorts. A volcano plot was created to illustrate the differential expression of DEGs. Adjusted p-values were utilized to mitigate potential false positives. DEGs that met the criteria of an adjusted p-value < 0.05 and |log2FC| > 2 were deemed statistically significant. A heatmap of the screened DEGs was generated utilizing the heatmap package in R 4.3.2 (https://www.r-project.org/).
Gene set enrichment analysis (GSEA)
To achieve a more intuitive understanding of gene expression levels within highly enriched functional pathways, we conducted GSEA18 analysis utilizing R. Pathways with a corrected p-value less than 0.05 were deemed statistically significant for differential expression.
Functional enrichment analysis
To clarify the potential roles of candidate target genes, we conducted functional enrichment analysis. Gene Ontology (GO) was utilized to categorize molecular functions (MF), biological processes (BP), and cellular components (CC)19. KEGG is an extensive database encompassing genomics, bioinformatics, and systems biology, offering information on gene function, metabolic pathways, diseases, and drugs20. The Human Disease Ontology (DO) database, accessible at www.diseaseontology.org, enhances research and understanding of diverse disease conditions by providing standardized and structured representations of human diseases. To enhance our understanding of the pathogenic relevance of target genes, we employed R’s “ggplot2” package21 and “cluster profiler” to examine the GO functions, KEGG pathways, and Disease Ontology of the candidate target genes.
Screening and validation of diagnostic markers
Two machine-learning algorithms, specifically least absolute shrinkage and selection operator (LASSO) logistic regression22,23 and support vector machine-recursive feature elimination (SVM-RFE)24, were utilized to identify novel and significant biomarkers for pediatric septic shock. This study employed LASSO logistic regression utilizing the R package “glmnet”25, with the minimal lambda value deemed optimal. We utilize 10-fold cross-validation and comply with the minimum criterion for partial likelihood. This method allows us to ascertain the ideal parameter configuration and improve the dependability of our results. The feature genes derived from the aforementioned two models were intersected and illustrated using a VENN diagram, which will be utilized for subsequent research. The validation set for the thorough analysis of biomarker efficacy will employ the dataset from GSE26378. Assessment will rely on receiver operating characteristic (ROC) curves, with the algorithms’ predictive efficacy quantified by determining the area under the curve (AUC). Statistical significance will be assessed using a two-tailed test, with a P value below 0.05.
Evaluation and correlation analysis of infiltrationrelated immune cells
Utilizing the CIBERSORT website, we applied a filtering process to identify 22 categories of immune cell matrices. We initially employed the Pearson correlation coefficient to evaluate the degree of association among immune cells. A heatmap was created to show the relationships among immune cells, based on the resulting correlation matrix. In this heat map, blue signifies negative correlation, whereas red denotes positive correlation. Subsequently, to examine the disparities in immune cells, we utilized the Wilcoxon test. The results were then visualized with violin plots, which highlight the unique features of the observed variations. Furthermore, the Spearman correlation between distinct diagnostic markers and immune infiltrating cells was examined using a lollipop chart to depict the findings. A statistically significant result is indicated by a p-value below 0.0526.
Results
Screening of DEGs in pediatric septic shock
We observed 12 differentially expressed genes (DEGs), all of which were upregulated (Fig. 2, Supplementary Fig. 1).

Identified 12 upregulated differentially expressed genes between children with septic shock and healthy controls based on the criteria of P < 0.05 and |log2FC| > 2. logFC: log Fold Change; Sig: Significance; -log10(adj.P.Val): Negative Logarithm of the Adjusted P-Value.
GSEA
We conducted GSEA to examine the biological signaling pathway in pediatric patients with septic shock and healthy control subjects. The HALLMARK analysis results identified the five principal terms, as shown in Supplementary Fig. 2. Significantly, the complement and coagulation cascades, Fc gamma receptor-mediated phagocytosis, insulin signaling pathway, and regulation of the actin cytoskeleton were significantly enriched in pediatric septic shock patients (P adjusted < 0.05).
Functional enrichment analysis of DEGs
We conducted a functional analysis to enhance our comprehension of the biological roles of DEGs. The findings from the differential expression analysis indicated that these DEGs were linked to numerous diseases, including oral disease, coronary artery disease, pancreatitis, periodontitis, periodontitis, periodontal disease, dental disease, pancreatic disease, pre-eclampsia, myocardial infarction, obesity, overnutrition, nutritional disorders, dermatological conditions, and reproductive system disorders, among others (Supplementary Fig. 3). The Gene Ontology (Biological Process) enrichment analysis indicated that the DEGs participate in processes including the regulation of reactive oxygen species metabolism, positive regulation of reactive oxygen species metabolism, response to reactive oxygen species, regulation of neuroinflammatory response, collagen catabolism, endodermal cell differentiation, and acute-phase response. The differentially expressed genes (DEGs) are located in specific granules, tertiary granules, cytoplasmic vesicles, vesicles, granule lumens, and membrane compartments associated with endocytosis (Supplementary Fig. 4). The Gene Ontology (Molecular Function) enrichment analysis indicated that the DEGs display activities including serine-type endopeptidase activity, serine-type peptidase activity, serine hydrolase activity, endopeptidase activity, metalloendopeptidase activity, protease binding, iron ion binding, metallopeptidase activity, macrolide binding, and fatty acid synthase activity. The KEGG pathway analysis revealed that the DEGs are linked to multiple processes, including the regulation of reactive oxygen species metabolism, defense responses to bacteria, negative regulation of proteolysis, negative regulation of cytokine production, responses to oxidative stress, positive regulation of reactive oxygen species biosynthesis, regulation of neuroinflammatory responses, and collagen catabolism, among others (Supplementary Fig. 5).
Screening and validation of diagnostic markers
We utilized two machine-learning algorithms, SVM-RFE (Supplementary Fig. 6A) and LASSO regression analysis, to identify feature genes and select 10 predicted genes from statistically significant univariate variables (Supplementary Fig. 6B). Through the analysis of overlapping regions of feature genes via a Venn diagram, we identified CD177, MCEMP1, MMP8, and OLAH as the four overlapping feature genes (Supplementary Fig. 6C). These genes exhibited significant predictive accuracy, as evidenced by their ROC curves with AUCs of 0.957, 0.935, 0.957, and 0.941 (Supplementary Fig. 7). In the GSE26378 validation cohort, the expression levels of these genes were significantly elevated in the pediatric septic shock group relative to the control group (P < 0.01) (Supplementary Fig. 8). Furthermore, in the GSE26378 validation cohort, the ROC curves for CD177, MCEMP1, MMP8, and OLAH confirmed their efficacy as significant biomarkers, demonstrating AUCs of 0.998, 0.998, 0.978, and 0.966, respectively (Fig. 3).

The ROC curve of the diagnostic efficacy verification in validation set.
Infiltration of immune cells results
Pediatric septic shock samples exhibited a greater prevalence of diverse immune cell types in comparison to normal samples, as determined by the CIBERSORT algorithm. The list comprises resting CD4 memory T cells, gamma delta T cells, naïve B cells, follicular helper T cells, activated NK cells, CD8 T cells, resting NK cells, activated dendritic cells, eosinophils, memory B cells, M1 macrophages, activated CD4 memory T cells, resting dendritic cells, and resting mast cells. Conversely, the proportions of plasma cells, regulatory T cells (Tregs), M0 macrophages, neutrophils, activated mast cells, naïve CD4 T cells, monocytes, and M2 macrophages were significantly lower (P < 0.05) (Fig. 4, Supplementary Fig. 9). Furthermore, the correlation analysis demonstrated that CD177, MCEMP1, MMP8, and OLAH displayed significant associations with various immune cells (Fig. 5), corroborating the study’s findings.

The differential expression of immune cells in pediatric patients with septic shock relative to a control group of healthy individuals. Con: healthy control group; Treat: pediatric septic shock group.

The Spearman correlation analysis between diagnostic markers and immune infiltrating cells. Abs(cor): Absolute value of correlation coefficient.
Discussion
Septic shock in children exhibits unique characteristics relative to adults1,27, significantly modifying the host’s immune status and resulting in an unfavorable prognosis. Although children generally demonstrate greater compensatory capacity than adults, allowing them to endure specific physiological and pathological challenges, the emergence of shock indicators denotes insufficient compensation28. Hence, prompt diagnosis and intervention are essential in these instances to avert the advancement of organ dysfunction.
This study aimed to identify characteristic biomarkers of septic shock in children to facilitate early intervention for multiple organ dysfunction caused by sepsis. Three datasets from the GEO database were examined: GSE8121 and GSE13904 as the analytical sets, and GSE26378 as the validation set. The samples from the three datasets were obtained from children aged 10 years or younger in the Pediatric Intensive Care Unit (PICU) at Cincinnati Children’s Hospital. Blood samples were collected at 24 h and on the third day post-admission. Total RNA was extracted from whole blood utilizing the PaxGene Blood RNA System (PreAnalytiX, Qiagen/Becton Dickinson, Valencia, CA), and sequencing was conducted with the Affymetrix GeneChip Human Genome U133 Plus 2.0 Array (Affymetrix, Santa Clara, CA)2,3,12,13,29,30. Each dataset underwent stringent quality control and standardization procedures, guaranteeing uniformity in cohort attributes, sampling techniques, and sequencing technologies. These measures mitigated potential biases arising from discrepancies in data processing, thereby improving the validity of the analysis. We recognized the potential for residual batch effects among datasets. We utilized the ComBat algorithm to execute batch effect correction during the integration of the GSE8121 and GSE13904 datasets, thereby enhancing the reliability and robustness of the combined analysis.
The research examined 12 differentially expressed genes (DEGs) between the septic shock cohort and the control group, specifically MCEMP1, CD177, MMP8, HP, IL1R2, RETN, MMP9, LTF, LCN2, OLFM4, CEACAM8, and OLAH. Following the identification of these differentially expressed genes (DEGs), they were analyzed through gene set enrichment analysis (GSEA), disease ontology (DO), gene ontology (GO), and Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analysis. Subsequently, LASSO31 and random forest analyses32 were utilized to identify characteristic genes. LASSO is an interpretable model that identifies significant features but excels primarily in linear scenarios, whereas random forest effectively addresses both linear and nonlinear challenges, offering robust predictive capabilities. However, it may exhibit suboptimal performance on high-dimensional sparse datasets and may be prone to overfitting complications. The convergence of the two methodologies produced four biomarkers: CD177, MCEMP1, MMP8, and OLAH, which partially mitigated each other’s deficiencies. The area under the receiver operating characteristic (ROC) curve (AUC) was computed to evaluate and confirm the diagnostic and predictive efficacy of the four biomarkers. An AUC value exceeding 0.9 signifies robust predictive efficacy. The ROC curve for the validation cohort exhibited elevated AUC values, further substantiating the dependability of these genes as prospective biomarkers for pediatric septic shock.
In this study, Gene Set Enrichment Analysis (GSEA) identified substantial gene set enrichment variations between the healthy control and septic shock cohorts, underscoring transitions from normal physiological conditions to a pathological state. In a healthy condition, immune system functions are oriented towards sustaining immune surveillance and tolerance mechanisms, thereby preventing excessive immune activation that could harm self-tissues. However, under pathological conditions, the expression of DEGs may excessively activate immune-related pathways, inflammation, and metabolism, resulting in the activation, migration, and proliferation of immune cells, which ultimately causes tissue and organ damage.
Moreover, the enrichment analysis of DO, GO, and KEGG revealed a significant correlation between the DEGs and multiple biological processes, such as immune cell activity, oxidative stress, neuroinflammation, and bacterial defense mechanisms. Immune response pathways emerged as a prevalent theme, underscoring the significance of immune dysregulation in the pathogenesis of septic shock. Consequently, the correlation between machine learning-validated biomarkers and immune cell infiltration in septic shock has become the focal point of our ongoing research.
To further investigate, the correlations between machine-learning-validated biomarkers and immune cell infiltration in septic shock were further examined using Pearson correlation coefficient analysis. Substantial positive correlations were identified between CD177, MCEMP1, and OLAH with neutrophils, as well as between MMP8 and M0 macrophages. Furthermore, CD177, MCEMP1, and MMP8 demonstrated substantial negative correlations with resting mast cells, whereas CD177 and OLAH revealed negative correlations with resting dendritic cells. Neutrophils, macrophages, mast cells, and dendritic cells are integral constituents of the innate immune system, functioning as the body’s primary defense against pathogens. CD17733,34,35,36,37,38 is a cell surface protein found on neutrophils, playing a role in chemotaxis and maturation. MCEMP139,40,41 is a protein expressed by mast cells that modulates the production of pro-inflammatory factors, whereas MMP838,41,42,43,44,45,46,47 promotes leukocyte adhesion. These biomarkers, in conjunction with OLAH36,48,49, are intricately linked to innate immunity. Prior research has indicated their elevated expression in sepsis, implying that suppressing their expression may offer viable therapeutic targets for sepsis management. The precise functions of these biomarkers in sepsis pathology and their interactions with developmental age are ambiguous, while their role in innate immunity is well-established.
Innate immunity offers a swift, non-specific reaction to infections, encompassing mechanisms such as dermal and mucosal barriers, inflammatory responses, and the activity of natural killer cells50. A proposed model of sepsis progression indicates that the initial inflammatory response evolves into a compensatory anti-inflammatory response syndrome51, a notion corroborated by our findings. This study revealed a reduction in innate immune cells, including macrophages, neutrophils, and mast cells, in children experiencing septic shock relative to healthy controls. Conversely, acquired immune cells, such as T cells and B cells, demonstrated elevated proportions. This transition indicates that a compensatory anti-inflammatory mechanism modulates the primary innate immune response, curtailing excessive inflammation while simultaneously inhibiting immune reactions to pathogens, thus exacerbating septic shock in pediatric patients.
This study utilized machine learning and bioinformatics to identify four biomarkers, CD177, MCEMP1, MMP8, and OLAH, and assessed their diagnostic significance and immune characteristics in pediatric septic shock. The results enhance our comprehension of immune dysregulation in these patients and offer significant direction for present and prospective precision therapies. This study has several limitations, including a limited sample size and dependence on a single database for validation, which may affect the generalizability and robustness of the results. Further research ought to prioritize augmenting the sample size and integrating multiple independent cohorts to bolster the validity of the findings. Additionally, exploring the potential interactions between genetic determinants and age-related factors in the innate immune response would yield a more thorough comprehension of their contributions to disease progression.
Data availability
The datasets used and analyzed during the current study are available from the corresponding author upon reasonable request.
References
Schlapbach, L. J. et al. International consensus criteria for pediatric sepsis and septic shock. Jama. 331 https://doi.org/10.1001/jama.2024.0179 (2024).
Bode, C., Weis, S., Sauer, A., Wendel-Garcia, P. & David, S. Targeting the host response in sepsis: Current approaches and future evidence. Crit. Care 27, 478. https://doi.org/10.1186/s13054-023-04762-6 (2023).
Cao, M., Wang, G. & Xie, J. Immune dysregulation in sepsis: Experiences, lessons and perspectives. Cell. Death Discov. 9, 465. https://doi.org/10.1038/s41420-023-01766-7 (2023).
Angus, D. C. & van der Poll, T. Severe sepsis and septic shock. N. Engl. J. Med. 369, 840–851. https://doi.org/10.1056/NEJMra1208623 (2013).
Annane, D., Bellissant, E. & Cavaillon, J. M. Septic shock. Lancet 365, 63–78. https://doi.org/10.1016/s0140-6736(04)17667-8 (2005).
Muszynski, J. A. et al. Early immune function and duration of organ dysfunction in critically III children with sepsis. Am. J. Respir Crit. Care Med. 198, 361–369. https://doi.org/10.1164/rccm.201710-2006OC (2018).
Moins-Teisserenc, H. et al. Severe altered immune status after burn injury is associated with bacterial infection and septic shock. Front. Immunol. 12 https://doi.org/10.3389/fimmu.2021.586195 (2021).
De Backer, D. et al. Surviving sepsis campaign research priorities 2023. Crit. Care Med. 52, 268–296. https://doi.org/10.1097/ccm.0000000000006135 (2024).
Soon, W. W., Hariharan, M. & Snyder, M. P. High-throughput sequencing for biology and medicine. Mol. Syst. Biol. 9 https://doi.org/10.1038/msb.2012.61 (2013).
Clough, E. et al. NCBI GEO: archive for gene expression and epigenomics data sets: 23-year update. Nucleic Acids Res. 52, D138–d144. https://doi.org/10.1093/nar/gkad965 (2024).
Ray, M., Dharmarajan, S., Freudenberg, J., Zhang, W. & Patterson, G. A. Expression profiling of human donor lungs to understand primary graft dysfunction after lung transplantation. Am. J. Transpl. 7, 2396–2405. https://doi.org/10.1111/j.1600-6143.2007.01918.x (2007).
Wong, H. R. et al. Genomic expression profiling across the pediatric systemic inflammatory response syndrome, sepsis, and septic shock spectrum. Crit. Care Med. 37, 1558–1566. https://doi.org/10.1097/CCM.0b013e31819fcc08 (2009).
Wynn, J. L. et al. The influence of developmental age on the early transcriptomic response of children with septic shock. Mol. Med. 17, 1146–1156. https://doi.org/10.2119/molmed.2011.00169 (2011).
Grunwell, J. R. et al. Differential expression of the Nrf2-linked genes in pediatric septic shock. Crit. Care. 19, 327. https://doi.org/10.1186/s13054-015-1052-0 (2015).
Wong, H. R. et al. Corticosteroids are associated with repression of adaptive immunity gene programs in pediatric septic shock. Am. J. Respir Crit. Care Med. 189, 940–946. https://doi.org/10.1164/rccm.201401-0171OC (2014).
Leek, J. T., Johnson, W. E., Parker, H. S., Jaffe, A. E. & Storey, J. D. The Sva package for removing batch effects and other unwanted variation in high-throughput experiments. Bioinformatics 28, 882–883. https://doi.org/10.1093/bioinformatics/bts034 (2012).
Ritchie, M. E. et al. Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 43, e47. https://doi.org/10.1093/nar/gkv007 (2015).
Chang, L. Y. et al. Gene set correlation enrichment analysis for interpreting and annotating gene expression profiles. Nucleic Acids Res. 52, e17. https://doi.org/10.1093/nar/gkad1187 (2024).
Expansion of the Gene. Ontology knowledgebase and resources. Nucleic Acids Res. 45, D331–d338. https://doi.org/10.1093/nar/gkw1108 (2017).
Kanehisa, M., Furumichi, M., Tanabe, M., Sato, Y. & Morishima, K. KEGG: New perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. 45, D353–d361. https://doi.org/10.1093/nar/gkw1092 (2017).
Walter, W., Sánchez-Cabo, F. & Ricote, M. GOplot: an R package for visually combining expression data with functional analysis. Bioinformatics 31, 2912–2914. https://doi.org/10.1093/bioinformatics/btv300 (2015).
Cheung-Lee, W. L. & Link, A. J. Genome mining for Lasso peptides: Past, present, and future. J. Ind. Microbiol. Biotechnol. 46, 1371–1379. https://doi.org/10.1007/s10295-019-02197-z (2019).
Fernández-Delgado, M. et al. An extensive experimental survey of regression methods. Neural Netw. 111, 11–34. https://doi.org/10.1016/j.neunet.2018.12.010 (2019).
Huang, S. et al. Applications of support vector machine (SVM) learning in cancer genomics. Cancer Genom. Proteom. 15, 41–51. https://doi.org/10.21873/cgp.20063 (2018).
Engebretsen, S. & Bohlin, J. Statistical predictions with Glmnet. Clin. Epigenet. 11, 123. https://doi.org/10.1186/s13148-019-0730-1 (2019).
Le, T., Aronow, R. A., Kirshtein, A. & Shahriyari, L. A review of digital cytometry methods: Estimating the relative abundance of cell types in a bulk of cells. Brief. Bioinform. 22 https://doi.org/10.1093/bib/bbaa219 (2021).
Sanchez-Pinto, L. N. et al. Development and validation of the Phoenix criteria for pediatric sepsis and septic shock. Jama 331 https://doi.org/10.1001/jama.2024.0196 (2024).
Hilarius, K. W. E., Skippen, P. W. & Kissoon, N. Early recognition and emergency treatment of sepsis and septic shock in children. Pediatr. Emerg. Care 36, 101–106. https://doi.org/10.1097/pec.0000000000002043 (2020).
Shanley, T. P. et al. Genome-level longitudinal expression of signaling pathways and gene networks in pediatric septic shock. Mol. Med. 13, 495–508. https://doi.org/10.2119/2007-00065.Shanley (2007).
Wong, H. R. et al. Genomic expression profiling across the pediatric systemic inflammatory response syndrome, sepsis, and septic shock spectrum. Crit Care Med 37, 1558–1566. https://doi.org/10.1097/CCM.0b013e31819fcc08 (2009).
Tang, G. et al. Evaluation and analysis of incidence and risk factors of lower extremity venous thrombosis after urologic surgeries: A prospective two-center cohort study using LASSO-logistic regression. Int. J. Surg. 89, 105948. https://doi.org/10.1016/j.ijsu.2021.105948 (2021).
Becker, T., Rousseau, A. J., Geubbelmans, M., Burzykowski, T. & Valkenborg, D. Decision trees and random forests. Am. J. Orthod. Dentofac. Orthop. 164, 894–897. https://doi.org/10.1016/j.ajodo.2023.09.011 (2023).
Demaret, J. et al. Identification of CD177 as the most dysregulated parameter in a microarray study of purified neutrophils from septic shock patients. Immunol. Lett. 178, 122–130. https://doi.org/10.1016/j.imlet.2016.08.011 (2016).
Miettinen, H. M., Gripentrog, J. M., Lord, C. I. & Nagy, J. O. CD177-mediated nanoparticle targeting of human and mouse neutrophils. PLoS One 13, e0200444. https://doi.org/10.1371/journal.pone.0200444 (2018).
Mulet, M. et al. Dysregulated neutrophil extracellular traps formation in sepsis. Immunology 170, 374–387. https://doi.org/10.1111/imm.13676 (2023).
Szakmany, T. et al. The ‘analysis of gene expression and biomarkers for point-of-care decision support in sepsis’ study; Temporal clinical parameter analysis and validation of early diagnostic biomarker signatures for severe inflammation andsepsis-SIRS discrimination. Front. Immunol. 14, 1308530. https://doi.org/10.3389/fimmu.2023.1308530 (2023).
Yi, C., Zhang, H., Yang, J., Chen, D. & Jiang, S. Elucidating common pathogenic transcriptional networks in infective endocarditis and sepsis: Integrated insights from biomarker discovery and single-cell RNA sequencing. Front. Immunol. 14, 1298041. https://doi.org/10.3389/fimmu.2023.1298041 (2023).
Zhang, W. Y. et al. Analysis and validation of diagnostic biomarkers and immune cell infiltration characteristics in pediatric sepsis by integrating bioinformatics and machine learning. World J. Pediatr. 19, 1094–1103. https://doi.org/10.1007/s12519-023-00717-7 (2023).
Chen, H., Chen, E., Lu, Y. & Xu, Y. Identification of immune-related genes in diagnosing retinopathy of prematurity with sepsis through bioinformatics analysis and machine learning. Front. Genet. 14, 1264873. https://doi.org/10.3389/fgene.2023.1264873 (2023).
Chen, J. X., Xu, X. & Zhang, S. Silence of long noncoding RNA NEAT1 exerts suppressive effects on immunity during sepsis by promoting microRNA-125-dependent MCEMP1 downregulation. IUBMB Life. 71, 956–968. https://doi.org/10.1002/iub.2033 (2019).
Kong, C., Zhu, Y., Xie, X., Wu, J. & Qian, M. Six potential biomarkers in septic shock: A deep bioinformatics and prospective observational study. Front. Immunol. 14, 1184700. https://doi.org/10.3389/fimmu.2023.1184700 (2023).
Atkinson, S. J. et al. Matrix Metalloproteinase-8 augments bacterial clearance in a juvenile sepsis model. Mol. Med. 22, 455–463. https://doi.org/10.2119/molmed.2016.00058 (2016).
Cajander, S. et al. Profiling the dysregulated immune response in sepsis: Overcoming challenges to achieve the goal of precision medicine. Lancet Respir Med. 12, 305–322. https://doi.org/10.1016/s2213-2600(23)00330-2 (2024).
Dong, J., Wang, S., Hu, Z. & Gong, L. Extracellular proteins as potential biomarkers in Sepsis-related cerebral injury. Front. Immunol. 14, 1128476. https://doi.org/10.3389/fimmu.2023.1128476 (2023).
Fiotti, N. et al. Genetic variants of matrix metalloproteinase and sepsis: The need speed study. Biomolecules. 12 https://doi.org/10.3390/biom12020279 (2022).
Wang, M., Jiang, F., Zhang, L., Zhang, J. & Xie, H. Knockdown of P2Y4 ameliorates sepsis-induced acute kidney injury in mice via inhibiting the activation of the NF-κB/MMP8 axis. Front. Physiol. 13, 953977. https://doi.org/10.3389/fphys.2022.953977 (2022).
Yao, R. Q. et al. Combination of transcriptional biomarkers and clinical parameters for early prediction of sepsis indued acute respiratory distress syndrome. Front. Immunol. 13, 1084568. https://doi.org/10.3389/fimmu.2022.1084568 (2022).
Banerjee, S., Mohammed, A., Wong, H. R., Palaniyar, N. & Kamaleswaran, R. Machine learning identifies complicated sepsis course and subsequent mortality based on 20 genes in peripheral blood immune cells at 24 H Post-ICU admission. Front. Immunol. 12, 592303. https://doi.org/10.3389/fimmu.2021.592303 (2021).
Coletta, C. et al. Endothelial dysfunction is a potential contributor to multiple organ failure and mortality in aged mice subjected to septic shock: Preclinical studies in a murine model of cecal ligation and puncture. Crit. Care. 18, 511. https://doi.org/10.1186/s13054-014-0511-3 (2014).
Faenza, I. & Blalock, W. L. Innate Immunity: A Balance between Disease and Adaption to Stress. Biomolecules. 12 https://doi.org/10.3390/biom12050737 (2022).
Bone, R. C., Grodzin, C. J. & Balk, R. A. Sepsis: a new hypothesis for pathogenesis of the disease process. Chest 112, 235–243. https://doi.org/10.1378/chest.112.1.235 (1997).
Funding
This work was supported by the National Natural Science Foundation of China (NSFC) (8237141538 to Kai-liang Fan, 82074227 to Kai-liang Fan).
Author information
Authors and Affiliations
Contributions
P.L. and K-l.Fcontributed equally to this paper. P.L. and K-lF. designed the research, analyzed the data, and wrote the manuscript; N.X., X-p.S., S.X., X-y.W., L-y.D., X.Z., J-m.L., R-f.L., D.Z., W.L. prepared figures. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Lyu, P., Xie, N., Shao, Xp. et al. Integrating bioinformatics and machine learning for comprehensive analysis and validation of diagnostic biomarkers and immune cell infiltration characteristics in pediatric septic shock. Sci Rep 15, 10456 (2025). https://doi.org/10.1038/s41598-025-95028-4
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-95028-4
