
Abstract
Gliomas, including both glioblastoma multiforme (GBM) and lower-grade glioma (LGG), present a substantial challenge in neuro-oncology because of genetic heterogeneity and unsatisfactory prognosis. This study aimed to conduct a comprehensive multi-omics analysis of gliomas using various bioinformatics approaches to identify potential therapeutic targets and prognostic markers. A comprehensive analysis was conducted on 1327 sequencing data samples alongside their relevant clinical information sourced from The Cancer Genome Atlas (TCGA) pertaining to glioblastoma (GBM), low-grade glioma (LGG), the Chinese Glioma Genome Atlas (CCGA) and University of California Santa Cruz Xena (UCSC Xena) datasets. These tools were employed for gene expression profiling, survival analysis, and cell communication mapping. Spatial transcriptomics revealed the localization of mesenchymal (MES)-like malignant tumors, and drug sensitivity analysis was performed to evaluate responses to quinpirole and meropenem. Additionally, the Tumor Immune Dysfunction and Exclusion (TIDE) framework was utilized to gauge the responsiveness to immunotherapy. The MES-like malignant and monocyte/macrophage (mono/macro) cell subsets showed high hallmark scores, playing key roles in the tumor microenvironment. MES-like malignant marker gene scores correlated with overall survival across datasets, whereas mono/macro marker gene scores were significant in the TCGA-LGG and CCGA datasets. Key interactions between these cell types were found, especially with CD14-ITGB2, LGALS1-CD69, and APOE-TREM2. The mono/macro cell subset demonstrated better immune therapy responsiveness, as indicated by lower TIDE scores. Spatial transcriptomics revealed that MES-like malignant tumors are predominantly localized in four distinct regions, with the marker genes CHI3L1 and ADM confirming these locations. Drug sensitivity analysis revealed differential responses of the MES-like malignant cell subset to quinpirole and meropenem. Our results offer fresh perspectives on the differential roles of MES-like malignant and monocyte/macrophage cell subsets in tumor progression and immune modulation, providing novel insights into glioma biology.
Similar content being viewed by others
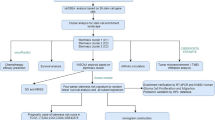


Introduction
Gliomas represent the most prevalent primary brain tumors, constituting around 30% of all tumors within the brain and central nervous system and 80% of all malignant brain tumors1. GBM is the most aggressive and lethal subtype, with a median survival duration of merely 14–16 months despite aggressive treatment2. Current therapeutic strategies for gliomas, including surgical resection, radiation therapy, and chemotherapy, exhibit limited efficacy and often cause significant side effects3. The heterogeneity of these tumors and their complex microenvironments limit the effectiveness of current diagnostic and therapeutic approaches, emphasizing the urgent need for innovative strategies to better understand and treat gliomas.
The heterogeneity of gliomas impedes the development of universal therapeutic strategies because different subtypes show distinct genetic, epigenetic, and transcriptomic profiles. Studies have demonstrated that specific gene expression patterns and molecular alterations can predict patient prognosis and therapeutic responses in various cancers, including gliomas. For example, mutations in the IDH1 and IDH2 are observed in LGGs and are related with a more satisfactory prognosis4. In contrast, GBMs often harbor alterations in the EGFR and PTEN genes5, contributing to their aggressive behavior, indicating that molecular profiling can offer critical insights into tumor behavior and patient outcomes. Mesenchymal-like cells are implicated in tumor invasiveness and drug resistance in gliomas. These cells exhibit distinct phenotypes and functions, and they can modulate the tumor microenvironment through cell communication and the release of extracellular vesicles, thereby influencing tumor progression. Moreover, Monocytes and macrophages play a crucial role in immune regulation within the tumor microenvironment. They can influence tumor progression and treatment response through both pro-inflammatory and anti-inflammatory mechanisms. Understanding the characteristics of these immune cells not only aids in uncovering the dynamic changes within the tumor microenvironment but also provides potential targets for immunotherapy. Consequently, investigating the transcriptomic characteristics of these cells is essential for understanding their role in glioma pathogenesis. Furthermore, the tumor microenvironment (TME), which includes immune cell infiltration and stromal interactions, is critical in glioma development and effectiveness of therapy6. By employing extensive multi-omics analyses to comprehend these intricate interactions, it is possible to uncover new therapeutic targets, refine patient stratification, and advance personalized treatment methods.
Extracellular vesicles (EVs), are small membrane-bound vesicles released by cells, capable of transferring information and substances between cells. They have become a major focus in cell communication research because of their release from cancer cells and their role in carrying functional proteins, small RNAs, and DNA from donor cells. Among these, exosomes measuring 30–100 nm are a key subtype7. These vesicles encapsulate bioactive molecules and can travel through various bodily fluids to distant sites, playing a pivotal role in intercellular communication and the TME8. However, comprehensive and precise studies of exosomes in gliomas are still lacking, particularly regarding single-cell sequencing, immune-related inquiries, and spatial distribution analyses. A deeper understanding of exosome roles in gliomas is essential for discovering and developing novel diagnostic and therapeutic plans.
Recent improvements in high-throughput sequencing and bioinformatics have facilitated compositive investigation of gene expression, mutations as well as cellular heterogeneity in gliomas. Single-cell RNA sequencing (scRNA-seq) has transformed our perception of tumor heterogeneity by allowing the detailed examination of complex tissues, into their constituent cell types and states9. Studies have revealed that gliomas exhibit significant intratumoral heterogeneity, with distinct cellular subpopulations driving tumor growth, invasion, and resistance to therapy10. Thus, scRNA-seq has uncovered the diversity of tumor and immune cells, offering insights into tumor evolution and resistance mechanisms in gliomas11. Research on other cancer types, such as melanoma12 and breast cancer13, has highlighted the utility of scRNA-seq in identifying novel therapeutic targets and elucidating TME interactions. Despite its advantages, scRNA-seq still has certain limitations. One primary challenge is the lack of spatial context resulted from the dissociation of tissues into single cells, which hinders the study of intercellular interactions and tissue spatial organization. Thus, a comprehensive multi-omics approach integrating single-cell analysis with other bioinformatic methods is necessary to fully elucidate the complex biology of gliomas. Spatial transcriptomics has became as a strong tool for mapping gene expression within tissue architecture, offering critical insights into the spatial organization of tumor cells and their microenvironments14. Combining spatial transcriptomics with scRNA-seq has emerged as a powerful approach, which hold significant promise for revealing new molecular mechanisms and therapeutic targets in gliomas.
This research adopted a multifaceted bioinformatics strategy to uncover the molecular underpinnings of gliomas. By integrating data from TCGA, UCSC Xena, and CCGA databases, we mapped the molecular and cellular landscapes of GBM and LGG, revealing the underlying mechanisms and spatial characteristics of MES-like malignant and mono/macro cell subsets in gliomas, along with the identification of potential therapeutic targets. By leveraging these novel insights into the cellular complexity of gliomas, we identified new potential therapeutic targets and biomarkers that could improve patient diagnosis and treatment.
Methods
Data download
We used the TCGAbiolinks package15 to download glioma (glioblastoma multiforme, GBM) data through The Cancer Genome Project (TCGA; https://portal.gdc.cancer.gov/; TCGA-GBM). Excluding samples without clinical information, we obtained count sequencing data from 156 glioma samples with clinical information available for prognostic overall survival (OS). The glioma count sequencing data were standardized to the fragments per kilobase million format. Relevant clinical information was gathered from the UCSC Xena database (http://genome.ucsc.edu)16. We employed the same method to download data for TCGA-LGG. Excluding samples without clinical information, we obtained sequencing data from 514 LGG samples with clinical information available for prognostic OS. We also downloaded the transcriptome sequencing dataset from the CCGA database17 (http://www.cgga.org.cn/) and excluded samples without clinical information. As a result, we acquired sequencing and corresponding clinical data for 657 glioma samples including prognostic OS (Table 1). Additionally, 121 exosome-related genes were identified from the literature18.
Single-cell analysis
We utilized the “CreateSeuratObject” function from the R package Seurat v4.0 (Manufacturer: R Foundation for Statistical Computing)19 to import the count matrix of the GSM5833536 sample from the single-cell dataset GSE194329 and created a Seurat object. Mitochondrial genes were removed, and the sequencing depth of the GSM5833536 sample underwent normalization via the “SCTransform” function. Subsequently, Principal Component Analysis (PCA) was performed to discern the principal components (PCs) of significance, while the “Elbowplot” function facilitated the visualization of the variance attributed to each component. Finally, 20 PCs were selected for dimensionality reduction by Uniform Manifold Approximation and Projection, as well as t-distributed stochastic neighbor embedding analyses. The “FindNeighbors” function was employed with default settings alongside the 20 PCs to establish k-nearest neighbors based on Euclidean distance within the primary PCA framework. To categorize the cells into distinct clusters, the “FindClusters” function was applied with a resolution parameter set to 0.5. Ultimately, the “RunTSNE” function was utilized for dimensionality reduction, enabling visualization and exploration of the dataset.
We obtained the glioma-related single-cell dataset GSE131928, along with the corresponding cell information, from the Tumor Immune Single-cell Hub (TISCH) database (http://tisch.comp-genomics.org/home/)20*. The single-cell matrix was read using the “Read10X_h5” function, and a Seurat object was created using “CreateSeuratObject” for subsequent analyses.
Cell type annotation and single-cell group differential genes
We identified cell types using cell marker genes from the literature21 for annotating the single-cell dataset GSM5833536. The “DotPlot” function was employed to visualize the expression of model genes across the distinct cell types. To discern differentially expressed genes among cell clusters, we applied the “FindAllMarkers” function to the single-cell dataset GSE131928. This function compared gene expression in every cell cluster with other cells, employing the Wilcoxon rank-sum test. Differentially expressed genes with a log fold change (logFC) of > 0.5 and an adjusted p-value (adj. p) of < 0.05 were retained as markers for further analyses.
Gene set variation analysis (GSVA) of different cell subsets
GSVA is an unsupervised, non-parametric approach employed to assess gene set enrichment in microarray nuclear transcriptomes22. This methodology converts the gene expression matrix from various samples into a gene expression matrix across samples, facilitating the evaluation of pathway enrichment in different contexts. The human hallmark gene set was retrieved using the R package “msigdbr.” GSVA was applied to all genes present in the single-cell dataset to calculate differences in functional enrichment among distinct cell subsets. The screening criterion for GSVA was an adj. p-value of < 0.05.
Functional and pathway enrichment analysis
Gene Ontology (GO) analysis serves as a prevalent approach for conducting extensive functional enrichment examinations, which encompass Biological Processes (BPs), molecular functions, and cellular components. The Kyoto Encyclopedia of Genes and Genomes (KEGG) constitutes a widely-utilized repository that compiles data regarding genomes, biological pathways, diseases, and pharmacological agents23. We employed the R package clusterProfiler to execute GO and KEGG analyses of differentially expressed genes24. Items with an adj. p-value of < 0.05 and a false discovery rate value of < 0.25 were deemed as statistically significant. The p-value correction method used is the Benjamini–Hochberg (BH) procedure25, which controls the false discovery rate (FDR). This technique regulates the anticipated ratio of false discoveries among the rejected hypotheses by assigning adjusted p-values to each individual p-value.
Survival analysis
The single-sample gene set enrichment analysis algorithm allows for the quantification of the relative representation of each gene within a specified sample dataset. We employed the R package GSVA to compute the cell correlation score for each sample in both the TCGA and CCGA datasets by the single-sample gene set enrichment analysis algorithm derived from the expression matrix of each sample. Subsequently, we assimilate the OS information from the TCGA-GBM, TCGA-LGG, and CCGA datasets to ascertain the optimal grouping of cell scores by generating prognostic Kaplan–Meier (KM) survival curves. After these analyses, patients with glioma within the TCGA-LGG and CCGA datasets were stratified into high-risk and low-risk groups, respectively, based on the median gene scores.
Somatic mutation analysis of glioma subtypes
The “Masked Somatic Mutation” data was chosen as the somatic mutation data for glioma (GBM) samples from the TCGA website, and the VarScan software was used for data processing. Finally, the Maftools R package was applied for visualizing somatic mutations across various subtypes of GBM26.
Immune-related analysis
We used the Tumor Immune Dysfunction and Exclusion (TIDE)27 (http://tide.dfci.harvard.edu) score, a computational approach that predicts the underlying response to immune checkpoint blockade therapy. TIDE models the two fundamental mechanisms of tumor immune evasion: induction of T-cell dysfunction in tumors with high cytotoxic T lymphocyte (CTL) infiltration and prevention of T-cell infiltration in tumors with low CTL levels. We characterized T-cell dysfunction in large tumor populations by evaluating how the expression of individual gene in the tumor interacts with the degree of CTL infiltration to affect patient survival. Based on TIDE analysis results, we calculated and compared the TIDE grouping differences between the high and low score groups of the TCGA-GBM, TCGA-LGG, and CCGA datasets utilizing the Mann–Whitney U-test (Wilcoxon rank-sum test). In addition, we calculated the tumor mutation burden (TMB) differences between the high and low TCGA-GBM, TCGA-LGG, and CCGA groups by the Mann–Whitney U-test (Wilcoxon rank-sum test). Statistical significance was set at a p-value of < 0.05.
Spatial transcriptome analysis
We employed the “Load10X_Spatial” function from the R package Seurat v4.019 to import the GSM5833536 sample from the single-cell spatial transcriptome dataset GSE194329, subsequently creating a Seurat object. The “SCTransform” function was applied to normalize the GSM5833536 sample data. PCA was applied for identifying significant PCs, and 20 PCs were selected for Uniform Manifold Approximation and Projection for dimensionality reduction. The “FindNeighbors” function, using default parameters and 20 PC dimensions, was used to establish k-nearest neighbors based on Euclidean distance in the base PCA space. Cells were divided into different clusters using the “FindClusters” function. For the integration of single-cell and spatial datasets, we used “FindTransferAnchors” and “TransferData” and visualized gene expression across cell types using the “SpatialDimPlot” function.
Cell communication analysis
Multicellular organisms regulate vital processes and maintain orderly function through intercellular communication involving cytokines and membrane proteins. Receptor–ligand-mediated intercellular signaling is crucial for coordinating various BPs, such as development, differentiation, and disease. Cell communication analysis enables the recognition of interactions between various cell types by examining the expression levels and associations of receptors and ligands across diverse cell populations.
To enhance confidence in identifying potential ligand–receptor interactions involved in intercellular communication, we implemented a consensus-based strategy by comparing results from multiple inference methods. We used the “liana_wrap” and “liana_aggregate” functions from the R package LIANA for cell communication analysis, which essentially runs other methods in the background and then generates a consensus. This approach could enhance our ability to identify highly significant ligand–receptor interactions across different methods, including CellPhoneDB. To further deepen our understanding of intercellular communication, we employed a network analysis-based approach called Connectome, which infers functional connections between cells derived from single-cell transcriptome data. Moreover, we used various methods to identify ligand–receptor pairs with differential expression under different conditions in single-cell transcriptome data. We used log2FC to analyze gene expression variations. Additionally, we applied NATMI, a machine-learning-based method for predicting cell-to-cell ligand–receptor interactions. We also used SingleCellSignalR, a pathway-based approach for reconstructing cell-to-cell signaling networks from the single-cell transcriptome dataset. Finally, we implemented CellChat, a probabilistic graph model-based approach, to infer intercellular communication patterns within the same dataset.
Single-cell drug susceptibility analysis
Beyondcell is a method for single-cell drug sensitivity analysis that identifies subsets of tumor cells with varying drug responses and suggests cancer-specific therapies28. The corresponding R package was used for this analysis. Beyondcell operates as follows: first, it assesses the sensitivity of each individual cell to the drug by analyzing the expression of genes associated with the drug. Cells with similar drug sensitivity are then grouped using a clustering algorithm to form therapeutic clusters (TCSs). Differences in drug sensitivity among TCSs were analyzed to generate sensitization-based prioritization, guiding drug selection. Beyondcell was validated in five single-cell datasets, demonstrating that TCSs may effectively target malignant cells.
AUCell
AUCell identifies cells exhibiting active gene sets within scRNA-seq data and employs the area under the curve (AUC) to compute whether a critical subset of the input gene set is enriched in the genes expressed in every cell. The dispersion of AUC scores across entire cells facilitates the exploration of feature expression. As the AUCell scoring method is based on rank, it remains not influenced by gene expression units or standardization procedures. Additionally, since cells are evaluated individually, AUCell is well-suited for analyzing large datasets and can handle expression matrices grouped as needed. We selected specific cell marker genes for AUCell scoring to identify cell populations that exhibit elevated scores.
Statistical analysis
All data processing and analyses were conducted utilizing R version 4.2.0 (Manufacturer: R Foundation for Statistical Computing). Continuous variables are represented as mean ± standard deviation. The Wilcoxon rank-sum test was performed to compare two groups. Unless otherwise indicated, correlations among various molecules were assessed using Spearman’s correlation analysis. Statistical significance was set at an adj. p-value of < 0.05.
Results
Cell type annotation and GSVA
We downloaded the glioma-related dataset GSE131928 from TISCH and annotated the cells based on the TISCH-provided annotation information. Figure 1A,B illustrate the clustering and cell type annotations within GSE131928. The cell populations were classified into eight types: AC malignant, CD8Tex, MES-like malignant, mono/macro, neural progenitor cells (NPCs) malignant, oligodendrocyte, and oligodendrocyte precursor (OPC)-like malignant cells. To examine differences in hallmark gene set enrichment across these cell types, GSVA was applied to all genes. The results (Fig. 1C) revealed that most hallmark gene sets had elevated scores in the CD8Tex, MES-like malignant, and mono/macro cell subsets. Conversely, NPC malignant cells and oligodendrocytes exhibited lower scores across most hallmark gene sets.

Cell type annotation and GSVA for GSE131928. (a,b) Cell clustering and cell annotation distribution map of the GSE131928 dataset. Different colors represent different clusters or cells. (c) GSVA between different cells, cells on the abscissa, and different hallmark pathways on the ordinate, with red indicating higher scores and blue indicating lower scores. GSVA gene set variation analysis.
AUCell and survival analysis
AUCell was applied for scoring exosome-related genes derived from their expression in the single-cell dataset GSE131928. The scoring results were visualized through scatter plots (Fig. 2A) and group comparison plots (Fig. 2B). Our analysis revealed that the MES-like malignant and mono/macro cell subsets had the highest AUC scores and were associated with most hallmark pathways. Additionally, we calculated the correlation between different cell types and found strong correlations between the MES-like malignant and mono/macro cell subsets (Fig. 2C).

AU cell analysis of exosome-related genes and survival analysis. (a,b) Visualization of AUC scores of exosome-related genes between different cell clusters in a scatter plot (a) and group comparison plot (b). (c) Heat map of the correlation analysis of different cells: Red indicates a stronger correlation. d. Relationship between different cell marker genes and OS. OS overall survival, AUC area under the curve.
We applied the “FindAllMarkers” function to compare the gene expression of each cell against other cells using the Wilcoxon rank-sum test. Differentially expressed genes with logFC > 0.5 and adj. p < 0.05 for each cell cluster were identified and retained as cell marker genes. To further investigate the significant effects of the MES-like malignant and mono/macro cell subsets on antitumor activity in glioma, we performed a relation analysis between OS and diverse cell marker genes; KM curve analysis was also performed using the TCGA-GBM, TCGA-LGG, and CCGA datasets. Correlation analysis (Fig. 2D) revealed that the mono/macro cell subset was related with OS in the TCGA-LGG and CCGA datasets but not in the TCGA-GBM dataset. The MES-like malignant cell subset was relevant to OS in patients from the TCGA-GBM, TCGA-LGG, and CCGA datasets. Simultaneously, KM curves for OS were plotted separately for patient groups across these datasets (Fig. 3A–F). The results indicated that only the mono/macro cell subset was not associated with OS in the TCGA-GBM (p > 0.05). However, there was significant association between the mono/macro cell subset and OS in the other datasets (p < 0.05). A notable distinction was observed between the MES-like malignant marker gene subset and OS, with the high-scoring group presenting a poorer prognosis. Based on the combined results of exosome gene association and survival analysis for the TCGA-GBM, TCGA-LGG, and CCGA datasets, we selected the MES-like malignant and mono/macro cell subsets for further analyses. These cell types demonstrated a strong correlation with exosomes and a significant association with OS, making them key targets for subsequent analyses.

Survival analysis of MES-like malignant cells and mono/macro cells. (a,b) KM curves of the MES-like malignant cell subset (a), mono/macro cell subset (b), and OS in the TCGA-GBM dataset. (c,d) KM curves of the MES-like malignant cell subset (c), mono/macro cell subset (d), and OS in the TCGA-LGG dataset. (e,f) KM curves of the MES-like malignant cell subset (e), mono/macro cell subset (f), and OS in the CCGA dataset. KM Kaplan–Meier analysis, OS overall survival, MES mesenchymal, mono/macro monocyte/macrophage.
Gene set enrichment analysis of MES-like malignant and mono/macro cell subsets
GO and KEGG analyses were conducted to obtain deeper observations into BPs, cellular components, molecular functions, and pathways related to distinct gene subsets. The “FindAllMarkers” function, along with the Wilcoxon rank, was used for the comparison of the differences in gene expression between individual cell subsets and other subsets. Differentially expressed genes with logFC > 0.5 and adj. p-value < 0.05 for each cell cluster were retained as subset-specific genes for further analyses. GO analysis revealed that the top five enrichment results for the MES-like malignant cell subsets were distinct from one another. Cell subset 3 showed significant associations with functions such as antioxidant activity, long-chain fatty acid binding, IgG binding, and anti-inflammatory activity. Additionally, terms such as RAGE receptor binding and calcium-dependent protein binding were notably enriched in cell subset 3. GO analysis revealed specific functional enrichments across different cell subsets. Cell subset 16 was primarily associated with extracellular matrix binding, cytokine activity, and growth factor activity. Cell subset 12 was mainly enriched for terms related to collagen binding, and protein tyrosine kinase activity. Similarly, cell subset 8 showed associations with peptidase inhibitor activity, and extracellular matrix structural components, including enzyme inhibitor and peptidase regulator activities. Cell subset 0 essentially exhibited enrichment for laminin-binding, insulin-like growth factor binding, and growth factor binding activities. In contrast, cell subset 10 majorly demonstrated kinase activator activity, kinase regulator activity, and protein kinase regulator activity. Finally, cell subset 7 was primarily associated with receptor antagonist activity, signaling receptor inhibitor activity, and other terms (Fig. 4A).

GO/KEGG enrichment analysis of gene sets in the MES-like malignant and mono/macro cell subsets. (a) Bubble plot representation of Gene Ontology (GO) enrichment analysis results for different subsets of the MES-like malignant cell subsets. (b) Bubble diagram displaying the pathway (KEGG) enrichment analysis results for different cell subsets of the MES-like malignant cell subset. (c) Bubble plot displaying the GO enrichment analysis results of the different mono/macro cell subsets. (d) Bubble diagram displaying the pathway (KEGG) enrichment analysis results for the different mono/macro cell subsets. GO Gene ontology, KEGG kyoto encyclopedia of genes and genomes, MES mesenchymal, mono/macro monocyte/macrophage.
KEGG results showed that MES-like malignant cell subset 16 was related to the pancreatic cancer pathway and cell subset 8 was related to complement and coagulation cascades, pertussis, longevity regulating pathway-multiple species, and other pathways. Cell subset 0 was related to proteoglycans in cancer and other pathways, and cell subset 10 was related to the p53 signaling pathway, cellular senescence, cell cycle, and other pathways (Fig. 4B).
GO analysis of the mono/macro cell subset showed that the enrichment results of different cell subsets overlapped with each other, mainly with amyloid-beta binding, antigen binding, amide binding, immune receptor activity, major histocompatibility complex (MHC) class II receptor activity, peptide antigen binding, MHC protein complex binding, and others (Fig. 4C). KEGG enrichment analysis showed that the enrichment results among different cell subsets overlapped, mainly with lysosomes and osteoclast differentiation, pertussis, hematopoietic cell lineage, allograft rejection, phagosomes, antigen processing and presentation, leishmaniasis, tuberculosis, asthma, Staphylococcus aureus infection, and other pathways (Fig. 4D).
MES-like malignant and mono/macro cell communication analysis
We performed the LIANA package to implement consensus cell communication analysis based on different communication analysis methods. The intensity (Fig. 5A) and number (Fig. 5B) of cell communications were displayed using a bubble plot and heat map. It could be seen that the interaction patterns between the MES-like malignant and mono/macro cell subsets and other cells were relatively similar. The ligand–receptor combinations between the MES-like malignant and mono/macro cell subsets mainly included CD14-ITGB2, galectin-1 (LGALS1)-CD69, APOE-TREM2, S100A8-CD69, TGFB1-CXCR4, LGALS9-PTPRC, and LGALS1-PTPRC. The communication heat map showed that the MES-like malignant and AC malignant cell subsets had the highest communication frequency. Similarly, the mono/macro cell subset had the highest communication frequency. There was also high communication frequency between the MES-like malignant and mono/macro cell subsets.

Cell communication analysis and visualization of the MES-like malignant and mono/macro subsets. (a) Consensus-based cell communication analysis bubble map implemented with LIANA; the size of the dots represents the confidence level, and the color from dark to light represents a stronger communication effect. (b) Heat map of the number of interaction relationship pairs among the three cell types. A redder color indicates a greater number of interacting ligand–receptor pairs. MES, mesenchymal; mono/macro, monocyte/macrophage.
Immune correlation analysis of the MES-like malignant and mono/macro cell subsets in different datasets
Considering the current importance of immunotherapy in tumors, we also evaluated the sensitivity to immunotherapy of patients with glioma and different cell marker gene scores (MES-like malignant and mono/macro) in the TCGA-GBM, TCGA-LGG, CCGA datasets by the TIDE algorithm. The specific results were then analyzed using the Wilcoxon rank-sum test and presented as a group comparison plot. As shown in Fig. 6, the TIDE immunotherapy scores of the TCGA-GBM dataset were not markedly different between the high and low score groups of the MES-like malignant cell subset (p > 0.05, Fig. 6A). However, the TIDE immunotherapy score of patients from TCGA-GBM was greatly different between the high and low score groups of the mono/macro cell subset (p < 0.05, Fig. 6B) and was lower in the high score group than in the low score group, suggesting that the immunotherapy response of the high score group might outperform that of the low score group.

Immunological analysis of the MES-like malignant and mono/macro groups. (a,b) Group comparison of the TIDE immunoscore between the high and low score groups of the MES-like malignant cell subsets (a) and high and low score groups of the mono/macro cell subsets (b) in the TCGA-GBM dataset. (c,d) Group comparison of the TIDE immunoscores between the high and low score groups of the MES-like malignant cell subsets (c) and TIDE immunoscores between the high and low score groups of the mono/macro cell subsets (d) in the TCGA-LGG dataset. (e,f) Comparison of the TIDE immunoscores between the high and low score groups of the MES-like malignancy cell subsets (e) and TIDE immunoscores between the high and low score groups of the mono/macro cell subsets (f) in the CCGA dataset. The symbol ns indicates p < 0.05, no statistical significance. The symbol * indicates p < 0.05, ** indicates p < 0.01, and *** indicates p < 0.001. TIDE Tumor immune dysfunction and exclusion, TCGA the cancer genome atlas, GBM glioblastoma multiforme, LGG lower-grade glioma, CCGA Chinese Glioma Genome Atlas, MES mesenchymal; mono/macro, monocyte/macrophage.
Consistent with the results of TCGA-GBM, the TIDE immunotherapy scores of the TCGA-LGG dataset were not different between the high and low score groups of the MES-malignant cell subset (p > 0.05, Fig. 6C). However, the TIDE immunotherapy score of the TCGA-LGG dataset was markedly different between the high and low score groups of the mono/macro cell subset (p < 0.05, Fig. 6D) and was lower in the high score group than in the low score group, suggesting that the high score group had a better immunotherapy response than the low score group.
The TIDE immunotherapy score of the CCGA dataset was significantly different between the high and low score groups of the MES-like malignant cell subset (p < 0.05, Fig. 6E) and was higher in the high score group than in the low score group, suggesting that the low score group might have a better immunotherapy response than the high score group. Conversely, the TIDE immunotherapy score of the TCGA-GBM dataset was significantly different between the high and low score groups of the mono/macro cell subset (p < 0.05, Fig. 6F). It was lower in the high score group than in the low score group, suggesting that the high score group has a better immunotherapy response than the low score group.
In addition, we compared the differences in TMB scores between different score groups (Fig. 7A–D). The findings indicated that only the high and low score groups of the MES-malignant cell subset showed significant differences in the TCGA-LGG dataset (p < 0.05, Fig. 7C). The high score group of the MES-like malignant cell subsets had a higher TMB score.

Analysis of TMB differences between the MES-like malignant and mono/macro subsets. (a,b) Comparison plots of the TMB immunoscores between the high and low score groups of the MES-like malignant cell subsets (a) and between the high and low score groups of the mono/macro cell subsets (b) in the TCGA-GBM dataset. (c,d) Comparison of the TMB immunoscores between the high and low score groups of the MES-like malignant cell subsets (c) and between the high and low score groups of the mono/macro cell subsets (d) in the TCGA-LGG dataset. The symbol ns is equivalent to p < 0.05, not statistically significant. The symbol * indicates p < 0.05, ** indicates p < 0.01, and *** indicates p < 0.001. TMB tumor mutation burden, TCGA The Cancer Genome Atlas, GBM glioblastoma multiforme, LGG lower-grade glioma, MES mesenchymal, mono/macro monocyte/macrophage.
MES-like malignant and mono/macro grouping enrichment analysis in different datasets
We used the limma package to recognize differentially expressed genes between the MES-like malignant and mono/macro cell subsets in the different datasets. Differentially expressed genes with logFC > 1 and adj. p < 0.05 were used for gene set enrichment analysis, and the results are displayed in a bar chart (Fig. 8). GO enrichment analysis revealed that differentially expressed genes between the high and low score groups of the MES-like malignant cell subset were mainly associated with granulocyte chemotaxis, leukocyte migration, wound healing, and granulocyte chemotaxis in the TCGA-GBM dataset. Key terms included secretory granule lumen, extracellular matrix binding, and other related terms. KEGG analysis indicated that differentially expressed genes between the high and low score groups of the MES-like malignant cell subset were primarily related to the nuclear factor (NF)-kappa B signaling, interleukin-17 signaling, and p53 signaling pathways (Fig. 8A).

GO/KEGG enrichment analysis of the MES-like malignant and mono/macro groups in different datasets. (a) The enrichment analysis of differentially expressed genes in different groups of the MES-like malignant cell subsets in the TCGA-GBM dataset. (b) The enrichment analysis of differentially expressed genes in different groups of the mono/macro cell subsets in the TCGA-GBM dataset. (c) The enrichment analysis of differentially expressed genes in different groups of the MES-like malignant cell subsets in the TCGA-LGG dataset. (d) The enrichment analysis of differentially expressed genes in different groups of the mono/macro cell subsets in the TCGA-LGG dataset. (e) Bar chart of the enrichment analysis results of differentially expressed genes in different groups of the MES-like malignant cell subsets in the CCGAs dataset. (f) Bar chart of the enrichment analysis results of differentially expressed genes in different groups of the mono/macro cell subsets in the CCGAs dataset. TCGA The Cancer Genome Atlas, GBM glioblastoma multiforme, LGG brain lower-grade glioma, CCGA Chinese Glioma Genome Atlas, MES mesenchymal, mono/macro monocyte/macrophage.
The GO enrichment results showed that differentially expressed genes between the high and low score groups of the mono/macro cell subset were mainly related to leukocyte cell–cell adhesion, leukocyte-mediated immunity, and leukocyte apoptosis in the TCGA-GBM dataset. Key terms included regulation of leukocyte proliferation, endocytic vesicle, secretory granule lumen, immune receptor activity, and other related terms. KEGG analysis showed that differentially expressed genes between the high and low score groups of the mono/macro cell subset were primarily associated with the NF-kappa B signaling pathway, nucleotide-binding oligomerization domain (NOD)-like receptor signaling pathway, and Toll-like receptor signaling pathway (Fig. 8B).
The GO enrichment results showed that the differentially expressed genes between the high and low score groups of the MES-like malignant cell subset were mainly related to external encapsulating structure organization, MHC protein complex, and external encapsulating structure organization in the TCGA-LGG dataset. Other terms included MHC class II protein complex, extracellular structure organization, and others. KEGG analysis indicated that the differentially expressed genes between the high and low score groups of the MES-like malignant cell subsets were primarily associated with the TNF signaling pathway, AGE/RAGE signaling pathway, diabetic complications, and intestinal immune network for IgA production (Fig. 8C).
The GO enrichment results showed that the differentially expressed genes between the high and low score groups of the mono/macro cell subset were primarily related to leukocyte-mediated immunity, leukocyte cell–cell adhesion, and regulation of leukocyte-mediated immunity in the TCGA-LGG dataset. Other related terms included MHC protein complex, endocytic vesicle, transport vesicle, immune receptor activity, and others. KEGG analysis showed that the differentially expressed genes between the high and low score groups of the mono/macro cell subset were mainly associated with the NF-kappa B signaling pathway, Toll-like receptor signaling pathway, and intestinal immune network for IgA production (Fig. 8D).
The GO enrichment results showed that the differentially expressed genes between the high and low score groups of the MES-like malignant cell subset were primarily associated with B cell-mediated immunity, immunoglobulin-mediated immune response, and extracellular matrix organization in the CCGA dataset. Other related terms included blood microparticles, endoplasmic reticulum lumen, and others. KEGG analysis revealed that the differentially expressed genes between the high and low score groups of the MES-like malignant cell subset were primarily associated with the TNF signaling pathway, PI3K-Akt signaling pathway, p53 signaling pathway, and other pathways (Fig. 8E).
The GO enrichment results showed that the differentially expressed genes between the high and low score groups of the mono/macro cell subset were mainly related to leukocyte-mediated immunity, myeloid leukocyte activation, and leukocyte activation in the CCGA dataset. Other relevant terms included adaptive immune response, cytoplasmic vesicle lumen, secretory granule lumen, and others. KEGG analysis showed that the differentially expressed genes between the high and low score groups of the mono/macro cell subset were mainly related to the TNF signaling pathway, NF-kappa B signaling pathway, TNF signaling pathway, and NOD-like receptor signaling (Fig. 8F).
Spatial transcriptome analysis
We used the Seurat pipeline to process the GSM5833536 sample in the spatial transcriptome GSE194329 dataset. After removing mitochondrial and ribosomal genes, we normalized the dataset using SCTransform and obtained 11 clusters (Fig. 9A). Following the integration of the single-cell and spatial transcriptome data using FindTransferAnchors and TransferData, we determined the spatial localization of different cell types (Fig. 9B). MES-like malignant tumors were mainly located in four different regions (Fig. 9B). The expression distribution of the MES-like malignant marker genes chitinase-3-like 1 (CHI3L1) and ADM further indicated the localization region of MES-like malignant tumors (Fig. 9C). In contrast, the mono/macro marker genes CD163 and CD68 showed no difference in expression between the different cell populations, and their expression levels were relatively low, resulting in no associated annotated cell population. In addition, the MES-like malignant tumor score was obtained with AUCell analysis based on the MES-like malignant marker genes from single-cell analysis (Fig. 9D). The score distribution showed high MES-like malignant scores in the localization region of MES-like malignant tumors, consistent with the above results.

Clustering of dataset GSE194329 and spatial analysis of the MES-like malignant and mono/macro groups. (a) UMAP and spatial localization maps of different cell subsets. (b) Map of the spatial localization of different cells, with darker colors indicating a higher likelihood. (c) Spatially localized CHI3L1 and ADM gene expression. (d) Distribution map of the AUCell scores for the MES-like malignant of mono/macro cell subsets obtained from the above single cells, with darker colors indicating higher scores. UMAP Uniform manifold approximation and projection, MES mesenchymal, mono/macro monocyte/macrophage.
Drug susceptibility analysis
We aggregated the drug rankings of the MES-like malignant cell subsets using the “bc4Squares” function in the Beyondcell package (Fig. 10A). The top left and bottom right corners contain all selected drugs to which the cells showed low and high sensitivity, respectively; the middle region includes the drugs with different drug sensitivity in the MES-like malignant cell subset compared with in other cells. Figure 10B,C represent the two drugs, quinpirole (Fig. 10B) and meropenem (Fig. 10C), which were significantly different in the MES-like malignant cell subset. Quinpirole acts as a dopamine agonist, and meropenem is an epoxide used in research trials for treating acquired immunodeficiency syndrome and Crohn’s, intestinal, gastrointestinal, and digestive system diseases.

Drug sensitivity analysis of the MES-like malignant subsets with the beyondcell package. (a) Summary plot of drug susceptibility ranking in the MES-like malignant cell subsets. (b,c) Scatter plot of drug sensitivity in the MES-like malignant cell subsets to drugs with large differences in drug sensitivity, such as quinpirole (b) and meropenem (c), with red indicating higher sensitivity; MES mesenchymal, mono/macro monocyte/macrophage.
Discussion
Gliomas are highly aggressive brain tumors that pose considerable challenges to clinical management owing to their rapid growth and invasive nature1. GBM is classified as a grade IV astrocytoma, constituting around 15% of intracranial neoplasms and 45–50% of malignant gliomas3. LGG, which is less aggressive than GBM, often progresses to higher-grade malignancies, further complicating treatment and management. Considering the complexity and heterogeneity of gliomas at the molecular and cellular levels, our study employs an integrative multi-omics approach to analyze glioma data, incorporating not only traditional transcriptomic analysis but also spatial transcriptomics and drug sensitivity analysis to provide new insights into the glioma microenvironment and its cellular interactions. This comprehensive approach enabled us to reveal differences in transcriptomic features, functions, and the roles of various cell subpopulations in tumor progression, which has been less addressed in the existing studies. Furthermore, considering that the GSE194329 single-cell dataset has not been thoroughly explored, our analysis of this novel dataset offers new perspectives on the cellular composition and functional characteristics of glioma, filling a gap in the current research in this field. Finally, we conducted an in-depth analysis of drug sensitivity, exploring the responses of different cell subpopulations to specific drugs and uncovering the potential therapeutic effects of new drugs.
In gliomas, scRNA-seq analysis has uncovered an intricate landscape of cells that contribute to tumor progression and therapy resistance. Our research used this technology to analyze the complex cellular composition of glioblastoma, identifying eight cell subtypes: astrocyte-like malignancies, CD8Tex, MES-like malignancies, mono/macro, NPC-like malignancies, oligodendrocytes, OPC-like malignancies, and oligodendrocyte progenitor cells. These results align with recent findings highlighting the diversity of tumor cells and their potential impact on patient outcomes29. GSVA enabled us to assign functional signatures to the identified cell populations. Notably, high expression scores were acquired for most hallmark gene sets in the CD8Tex, MES-like malignant, and mono/macro cell subsets. This indicated that these cell subtypes might be pivotal drivers of oncogenic pathways in glioblastomas. The observation that the MES-like malignant and mono/macro cell subsets have high AUC scores and are associated with multiple hallmark pathways aligns with previous findings. For example, Chen et al. demonstrated that single-cell molecular analysis could identify the pathways driving GBM relapse, emphasizing the significance of understanding subclonal evolution30. The strong association of the MES-like malignant and mono/macro cell subsets with OS underscores their potential as prognostic markers or therapeutic targets.
The MES-like malignant cell subsets represent highly aggressive and therapy-resistant subtypes of glioma cells31. Our study identified significant gene expression differences between these cells and other glioma subtypes, with high scores in key pathways related to cancer and immune evasion, including genes such as CHI3L1 and ADM. These different cells are located in distinct tumor regions, underscoring their role in tumor progression and heterogeneity. For example, the four developmental stages of GBM cells, including NPC-like, OPC-like, AC-like, and MES-like, are found in separate tumor regions, establishing individual niches32. The perinecrotic regions showed a higher level of immunosuppression than the endogenous GBM microenvironment, and the perivascular regions were associated with increased pro-inflammatory activity. Moreover, MES-like malignant cells contain multiple highly proliferative cells, demonstrating their aggressive nature33 and MES-like malignant cells are linked to elevated expression of a mesenchymal program in macrophages and enhanced T-cell cytotoxicity, underscoring significant changes in the immune microenvironment29. Additionally, we found that the MES-like malignant cell subset had a higher TMB, indicating a greater likelihood of neoantigen formation, possibly enhancing the responsiveness of tumors to immunotherapy, particularly to immune checkpoint blockade. The association between high TMB and improved outcomes in patients undergoing immune checkpoint blockade suggests that measuring the TMB alongside the MES-like cell subset scores could aid in stratifying patients and optimizing treatment strategies.
The mono/macro cell subsets, including monocytes and macrophages, are pivotal in the TME, influencing tumor progression, immune response, and therapy resistance. Our study identified specific markers for the mono/macro cell subsets that were related to OS in the TCGA-LGG and CCGA datasets but not in the TCGA-GBM dataset. This differential association suggests that the role of the mono/macro cell subsets may vary among glioma subtypes. Previous research34 has shown that tumor-associated macrophages can adopt either a pro-tumorigenic M2 phenotype or an anti-tumorigenic M1 phenotype, depending on the tumor context. The significant correlation between mono/macro cell marker expression and patient survival in LGG suggests that these cells might exert a more prominent impact on modulating the TME and influencing clinical outcomes in these patients. Targeting tumor-associated macrophages to shift their phenotype from M2 to M1 presents a promising intervention for enhancing the survival of patients with gliomas. Additionally, the mono/macro cell subsets had high AUCell scores for various hallmark pathways, indicating their active participation in the glioma microenvironment. Hence, the mono/macro cell subsets may contribute to tumor growth, immune suppression, and therapy resistance in GBM.
Additionally, intercellular communication analysis further demonstrated the dominance of the MES-like malignant and mono/macro cell subsets in the signaling networks within the GBM microenvironment. This finding aligns with previous results showing that macrophage-derived signals can influence tumor cell states and immune cell behaviors35. Understanding these communication networks is crucial for developing interventions that disrupt pro-tumor signaling pathways. Our analysis highlighted intricate networks of interactions involving ligand–receptor pairs, such as LGALS1-CD69 and APOE-TREM2, which are likely to affect key processes within the tumor milieu, including inflammation.
LGALS1 is a beta-galactoside-binding protein participating in diverse cellular processes. In gliomas, LGALS1 accelerates tumor progression by modulating the immune microenvironment and enhancing tumor cell survival36. CD69 is an early activation marker expressed on the surface of diverse immune cells, such as T, B, and natural killer cells. In gliomas, CD69 has been implicated in the regulation of immune responses in the TME37. However, interactions between LGALS1 and CD69 in gliomas have not been previously reported. Our cell communication analysis identified LGALS1 and CD69 as part of a significant ligand–receptor pair (LGALS1–CD69) crucial for intercellular interactions within glioma tumors. The high expression of LGALS1 in specific glioma subtypes suggests its role in promoting immune evasion and tumor growth. In contrast, CD69 involvement indicates the modulation of T-cell function and immune regulatory mechanisms employed by the tumor. Targeting the LGALS1 and CD69 pathways may disrupt these interactions, potentially enhancing antitumor immune responses and improving the efficacy of glioma immunotherapies.
APOE serves as a lipid-binding protein contributing to lipid metabolism, neurobiology, and immune regulation38. In gliomas, APOE influences tumor growth, invasion, and immune modulation39. TREM2, expressed on myeloid cells such as microglia and macrophages, exerts a crucial role in immune response modulation, inflammation, and phagocytosis40. In gliomas, TREM2 is associated with tumor-associated macrophages and an immunosuppressive microenvironment that supports tumor growth and progression41. One study found that TREM2 and APOE were upregulated in a specific subset of macrophages in renal tumors and were notably enriched in tumors from patients who relapsed after surgery42. This suggests that TREM2- and APOE-positive macrophage infiltration might exert as a promising prognostic biomarker in terms of clear cell renal cell carcinoma recurrence. Similarly, we identified APOE as a crucial ligand mediating communication between the MES-like malignant and mono/macro cell subsets, particularly through its interaction with the significant receptor TREM2. This interaction underscores the effects of APOE and TREM2 in modulating the immune microenvironment of gliomas, potentially facilitating the recruitment and activation of tumor-associated macrophages. Understanding the APOE-TREM2 interaction might provide insight into how glioma cells manipulate immune cell behavior to promote immune evasion and tumor progression.
Enrichment analyses elucidated the BPs and pathways associated with cancer development in the MES-like malignant and mono/macro cell subsets. For instance, these cells were implicated in pathways associated with pancreatic cancer and immunological processes, such as osteogenic differentiation, providing an enhanced insight into their functions in oncogenesis. Moreover, the TIDE scores suggested that patients with high mono/macro scores might exhibit improved responsiveness to immunotherapy than those with low scores. In contrast, high MES-like malignant scores were associated with poorer immune therapy responsiveness in certain datasets, such as CCGA.
To elucidate the molecular mechanisms driving GBM and LGG, we identified several key signaling pathways that were enriched in the MES-like malignant and mono/macro cell subsets. The NF-kappa B signaling pathway is fundamental to regulate the immune response and cell survival, as well as its dysregulation is related to diverse cancers, including gliomas43. Additionally, the interleukin-17 signaling pathway contributes to inflammation and has been implicated in tumor progression and metastasis across several cancer types44. The p53 signaling pathway, a critical regulator of the cell cycle and apoptosis, is frequently mutated in cancers, resulting in uncontrolled cell proliferation and tumor development45. The enrichment of these pathways in the MES-like malignant cell subsets suggests that these cells may have enhanced capabilities for survival, proliferation, and evasion of immune surveillance, influencing their aggressive behavior and unfavorable prognosis. Conversely, the enrichment of the NF-kappa B, NOD-like receptor, and Toll-like receptor signaling pathways in the mono/macro cell subsets highlights their potential role in modulating the TME through inflammatory and immune responses. These results align with earlier research demonstrating the involvement of these signaling pathways in glioma pathogenesis and their potential as therapeutic targets46. Understanding the differential activation of these pathways in distinct glioma cell types offers deeper insights into the heterogeneity of gliomas and the nuanced interaction between tumor cells and the immune microenvironment. It accelerates the advancement of targeted treatment to restrain tumor growth and optimize patient outcomes. For instance, targeting the NF-kappa B signaling pathway in the MES-like malignant cell subsets could potentially reduce their survival and proliferation. Meanwhile, modulating the NOD-like receptor and Toll-like receptor pathways in the mono/macro cell subsets could enhance antitumor immune responses.
Spatial transcriptomics further localized the MES-like malignant cell subsets within specific tumor regions. Previous research has shown that the number of MES-like malignant cell subsets is notably elevated in GBM tissues than in normal brain tissues47. These cells express specific proteins such as CHI3L1, which is consistent with the findings of our spatial transcriptomics analysis48. CHI3L1, a secreted glycoprotein implicated in various cancers including lung, liver, and thyroid cancers49,50,51, is related to tumor aggressiveness and unfavorable patient outcomes in gliomas52. In our study, CHI3L1 was identified as a hallmark gene of the MES-like malignant cell subsets, with its expression being significantly higher in these cells. This elevated expression of CHI3L1 was related to worse OS in the TCGA-GBM and TCGA-LGG datasets. Spatial transcriptomic data revealed that CHI3L1 expression was localized to four distinct regions within the TME, suggesting a heterogeneous distribution that might reflect areas of differential tissue remodeling or inflammation. The high expression of CHI3L1 in the MES-like malignant cell subsets indicates its involvement in the TME, potentially modulating immune responses and enhancing cell invasiveness. The elevated AUCell scores for CHI3L1 in the MES-like malignant regions further support its involvement in promoting malignancy and therapy resistance. These findings align with these previous observations and emphasize the importance of CHI3L1 in glioma pathophysiology, especially in malignant MES-like cell subtypes. This suggests that CHI3L1 could act as a promising biomarker for glioma prognosis and a treatment target.
Finally, drug sensitivity analysis revealed the differential responses of glioma cell populations to various therapeutics. We identified significant differences in the response of the MES-like malignant cell subsets to quinpirole and meropenem, suggesting potential therapeutic implications. Our results demonstrated the unique responsiveness of the MES-like malignant cell subsets to quinpirole, aligning with recent findings indicating that dopamine receptor activation can regulate glioma cell proliferation and migration, thereby influencing the TME53. Meropenem, famed for its proficiency in penetrating the blood–brain barrier, showed notable efficacy against the MES-like malignant cell subsets in our analysis54. This underscores its potential as a repurposed option for gliomas.
Limitations
Despite its comprehensive analysis and significant findings, this article has several limitations. Initially, the research was primarily reliant on bioinformatics and computational methods, lacking the integration of lab experiments that could provide a more direct biological validation of the results. Moreover, while the sample size was substantial, it may still be insufficient to capture the full heterogeneity of GBM and LGG. Furthermore, the scarcity of clinical validation means that the prognostic and therapeutic implications derived from these data require further confirmation in medical contexts. Additionally, the use of various datasets from different sources may introduce batch effects, potentially impacting the consistency and reliability of the results.
Conclusion
This study successfully integrated multiple bioinformatics approaches to analyze glioma gene expression, cell types, functional enrichment, immune responses, somatic mutations, and drug sensitivity. Key findings include the identification of distinct cell clusters with differential gene expression, significant functional enrichment differences among cell subpopulations, and promising prognostic markers and treatment. The research elucidated spatial expression patterns of specific genes and intricate intercellular communication within the TME. These insights enhance our insight into glioma biology and support the advancement of personalized therapeutic strategies. Future research incorporating clinical validation and addressing current limitations will be key to converting these findings into effective clinical applications.
Data availability
The datasets of GBM and LGG are available from TCGA, UCSC Xena database and CCGA database. The count matrix of the GSM5833536 from the scRNA-seq dataset GSE194329 can be obtained by the CreateSeuratObject function from the R package. GSE131928 and corresponding cell information are available from the TISCH database. For more details, please visit: https://github.com/MD-zzZ/spatial-GBM.
Abbreviations
- AUC:
-
Area under the curve
- AUCell:
-
Area under the curve cell scoring
- BP:
-
Biological process
- CCGA:
-
Chinese glioma genome atlas
- CHI3L1 :
-
Chitinase-3-like 1
- CD8Tex:
-
CD8 T-cell exhausted
- GBM:
-
Glioblastoma multiforme
- GEO:
-
Gene expression omnibus
- ICB:
-
Immune checkpoint blockade
- LGALS1 :
-
Galectin-1
- LGG:
-
Lower-grade glioma
- MES-like:
-
Mesenchymal-like
- mono/macro:
-
Monocyte/macrophage
- NF-kappa B:
-
Nuclear factor kappa-light-chain-enhancer of activated B cells
- NOD-like receptor:
-
Nucleotide-binding oligomerization domain-like receptor
- NPC:
-
Neural progenitor cell
- OS:
-
Overall survival
- scRNA-seq:
-
Single-cell RNA sequencing
- TCGA:
-
The cancer genome atlas
- TIDE:
-
Tumor immune dysfunction and exclusion
- TME:
-
Tumor microenvironment
- TREM2 :
-
Triggering receptor expressed on myeloid cells 2
References
van den Bent, M. J. et al. Primary brain tumours in adults. Lancet. 402, 1564–1579. https://doi.org/10.1016/S0140-6736(23)01054-1 (2023).
Lim, M., Xia, Y., Bettegowda, C. & Weller, M. Current state of immunotherapy for glioblastoma. Nat. Rev. Clin. Oncol. 15, 422–442 (2018).
Stitzlein, L. M., Adams, J. T., Stitzlein, E. N., Dudley, R. W. & Chandra, J. Current and future therapeutic strategies for high-grade gliomas leveraging the interplay between epigenetic regulators and kinase signaling networks. J. Exp. Clin. Cancer Res. 43, 12 (2024).
Waitkus, M. S., Diplas, B. H. & Yan, H. Isocitrate dehydrogenase mutations in gliomas. Neuro Oncol. 18, 16–26 (2016).
Nam, Y. et al. Pharmacogenomic profiling reveals molecular features of chemotherapy resistance in IDH wild-type primary glioblastoma. Genome Med. 15, 16 (2023).
Sai Krishna, A. V. S., Ramu, A., Hariharan, S., Sinha, S. & Donakonda, S. Characterization of tumor microenvironment in glioblastoma multiforme identifies ITGB2 as a key immune and stromal related regulator in glial cell types. Comput. Biol. Med. 165, 107433 (2023).
van Niel, G., D’Angelo, G. & Raposo, G. Shedding light on the cell biology of extracellular vesicles. Nat. Rev. Mol. Cell Biol. 19, 213–228 (2018).
Yang, E. et al. Exosome-mediated metabolic reprogramming: the emerging role in tumor microenvironment remodeling and its influence on cancer progression. Signal Transduct. Target Ther. 5, 242 (2020).
Su, M. et al. Data analysis guidelines for single-cell RNA-seq in biomedical studies and clinical applications. Mil. Med. Res. 9, 68 (2022).
Janjua, T. I. et al. Frontiers in the treatment of glioblastoma: past, present and emerging. Adv. Drug Deliv. Rev. 171, 108–138 (2021).
Hernández Martínez, A., Madurga, R., García-Romero, N. & Ayuso-Sacido, Á. Unravelling glioblastoma heterogeneity by means of single-cell RNA sequencing. Cancer Lett. 527, 66–79 (2022).
Helmink, B. A. et al. B cells and tertiary lymphoid structures promote immunotherapy response. Nature 577, 549–555 (2020).
Bassez, A. et al. A single-cell map of intratumoral changes during anti-PD1 treatment of patients with breast cancer. Nat. Med. 27, 820–832 (2021).
Longo, S. K., Guo, M. G., Ji, A. L. & Khavari, P. A. Integrating single-cell and spatial transcriptomics to elucidate intercellular tissue dynamics. Nat. Rev. Genet. 22, 627–644 (2021).
Colaprico, A. et al. TCGAbiolinks: an R/Bioconductor package for integrative analysis of TCGA data. Nucleic Acids Res. 44, e71. https://doi.org/10.1093/nar/gkv1507 (2016).
Goldman, M. J. et al. Visualizing and interpreting cancer genomics data via the Xena platform. Nat. Biotechnol. 38, 675–678 (2020).
Zhao, Z. et al. Chinese glioma genome atlas (CGGA): A comprehensive resource with functional genomic data from Chinese glioma patients. Genom. Proteom. Bioinform. 19, 1–12 (2021).
Qiu, P., Guo, Q., Yao, Q., Chen, J. & Lin, J. Characterization of exosome-related gene risk model to evaluate the tumor immune microenvironment and predict prognosis in triple-negative breast cancer. Front. Immunol. 12, 736030. https://doi.org/10.3389/fimmu.2021.736030 (2021).
Hao, Y. et al. Integrated analysis of multimodal single-cell data. Cell 184, 3573-3587.e29. https://doi.org/10.1016/j.cell.2021.04.048 (2021).
Sun, D. et al. TISCH: a comprehensive web resource enabling interactive single-cell transcriptome visualization of tumor microenvironment. Nucleic Acids Res. 49, D1420–D1430 (2021).
Xiong, A., Zhang, J., Chen, Y., Zhang, Y. & Yang, F. Integrated single-cell transcriptomic analyses reveal that GPNMB-high macrophages promote PN-MES transition and impede T cell activation in GBM. EBioMedicine 83, 104239. https://doi.org/10.1016/j.ebiom.2022.104239 (2022).
Hänzelmann, S., Castelo, R. & Guinney, J. GSVA: gene set variation analysis for microarray and RNA-seq data. BMC Bioinform. 14, 7 (2013).
Kanehisa, M. & Goto, S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 28, 27–30 (2000).
Yu, G., Wang, L. G., Han, Y. & He, Q. Y. clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS 16, 284–287 (2012).
Benjamini, Y. & Hochberg, Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B (Methodol.) 57, 289–300 (2018).
Mayakonda, A., Lin, D. C., Assenov, Y., Plass, C. & Koeffler, H. P. Maftools: efficient and comprehensive analysis of somatic variants in cancer. Genome Res. 28, 1747–1756 (2018).
Jiang, P. et al. Signatures of T cell dysfunction and exclusion predict cancer immunotherapy response. Nat. Med. 24, 1550–1558 (2018).
Fustero-Torre, C. et al. Beyondcell: targeting cancer therapeutic heterogeneity in single-cell RNA-seq data. Genome Med. 13, 187 (2021).
Hara, T. et al. Interactions between cancer cells and immune cells drive transitions to mesenchymal-like states in glioblastoma. Cancer Cell 39, 779-792.e11. https://doi.org/10.1016/j.ccell.2021.05.002 (2021).
Chen, X. et al. Relapse pathway of glioblastoma revealed by single-cell molecular analysis. Carcinogenesis 39, 931–936 (2018).
Xiao, Y. et al. Single-cell transcriptomics revealed subtype-specific tumor immune microenvironments in human glioblastomas. Front. Immunol. 13, 914236 (2022).
Liu, M. et al. Spatial transcriptomics reveals segregation of tumor cell states in glioblastoma and marked immunosuppression within the perinecrotic niche. Acta Neuropathol. Commun. 12, 64 (2024).
Neftel, C. et al. An integrative model of cellular states, plasticity, and genetics for glioblastoma. Cell. 178, 835–849.e21.
Li, M. et al. Metabolism, metabolites, and macrophages in cancer. J. Hematol. Oncol. 16, 80 (2023).
Wang, Z. et al. Tumor-secreted lactate contributes to an immunosuppressive microenvironment and affects CD8 T-cell infiltration in glioblastoma. Front. Immunol. 14, 894853 (2023).
Le Mercier, M., Fortin, S., Mathieu, V., Kiss, R. & Lefranc, F. Galectins and gliomas. Brain Pathol. 20, 17–27 (2010).
Rafei, M., Rouette, A., Brochu, S., Vanegas, J. R. & Perreault, C. Differential effects of γc cytokines on postselection differentiation of CD8 thymocytes. Blood 121, 107–117 (2013).
Krasemann, S. et al. The TREM2-AOE pathway drives the transcriptional phenotype of dysfunctional microglia in neurodegenerative diseases. Immunity 47, 566-581.e9. https://doi.org/10.1016/j.immuni.2017.08.008 (2017).
Mahley, R. W. Apolipoprotein E: from cardiovascular disease to neurodegenerative disorders. J. Mol. Med. (Berl.) 94, 739–746. https://doi.org/10.1007/s00109-016-1427-y (2016).
Colonna, M. The biology of TREM receptors. Nat. Rev. Immunol. 23, 580–594. https://doi.org/10.1038/s41577-023-00837-1 (2023).
Ulland, T. K. et al. TREM2 maintains microglial metabolic fitness in Alzheimer’s disease. Cell 170, 649-663.e13. https://doi.org/10.1016/j.cell.2017.07.023 (2017).
Obradovic, A. et al. Single-cell protein activity analysis identifies recurrence-associated renal tumor macrophages. Cell 184, 2988-3005.e16. https://doi.org/10.1016/j.cell.2021.04.038 (2021).
Karin, M. & Greten, F. R. NF-kappaB: linking inflammation and immunity to cancer development and progression. Nat. Rev. Immunol. 5, 749–759 (2005).
Huangfu, L., Li, R., Huang, Y. & Wang, S. The IL-17 family in diseases: from bench to bedside. Signal Transduct. Target Ther. 8, 402 (2023).
Vousden, K. H. & Prives, C. Blinded by the light: the growing complexity of p53. Cell 137, 413–431 (2009).
Kawai, T. & Akira, S. The role of pattern-recognition receptors in innate immunity: update on toll-like receptors. Nat. Immunol. 11, 373–384 (2010).
Niklasson, M. et al. Mesenchymal transition and increased therapy resistance of glioblastoma cells is related to astrocyte reactivity. J. Pathol. 249, 295–307 (2019).
Guetta-Terrier, C. et al. CHI3L1 is a modulator of glioma stem cell states and a therapeutic target in glioblastoma. Cancer Res. 83, 1984–1999 (2023).
Kim, K. C. et al. Suppression of metastasis through inhibition of chitinase 3-like 1 expression by miR-125a-3p-mediated up-regulation of USF1. Theranostics. 8, 4409–4428 (2018).
Qiu, Q. C. et al. CHI3L1 promotes tumor progression by activating TGF-β signaling pathway in hepatocellular carcinoma. Sci. Rep. 8, 15029 (2018).
Cheng, S. P. et al. Overexpression of chitinase-3-like protein 1 is associated with structural recurrence in patients with differentiated thyroid cancer. J. Pathol. 252, 114–124 (2020).
Tanwar, M. K., Gilbert, M. R. & Holland, E. C. Gene expression microarray analysis reveals YKL-40 to be a potential serum marker for malignant character in human glioma. Cancer Res. 62, 4364–4368 (2002).
Yang, K. et al. Activation of dopamine receptor D1 inhibits glioblastoma tumorigenicity by regulating autophagic activity. Cell Oncol. 43, 1175–1190 (2020).
Schneider, F., Gessner, A. & El-Najjar, N. Efficacy of vancomycin and meropenem in central nervous system infections in children and adults: current update. Antibiotics. 11, 173 (2022).
Acknowledgements
We are grateful for the financial support from the National Natural Science Foundation of China, Shenzhen Science and Technology Program and the Key Project of Department of Education of Guangdong Province. We appreciate the assistance from Peng Huang at School of Biomedical Engineering in Shenzhen University.
Funding
This study was supported by the National Natural Science Foundation of China (No. 82272885, No. 82102706 and No. 82403810), Shenzhen Science and Technology Program (No. JCYJ20210324100001004, No. JCYJ20230808105205011) and the Key Project of Department of Education of Guangdong Province (No. 2021ZDZX2020).
Author information
Authors and Affiliations
Contributions
B.Y.Z., X.L.D and S.G.Z conceived and designed experiments. B.Y.Z, J.N.W and T.H.Z performed the majority of data and statistical analysis. C.Z., J.J. and S.Y.L. assisted with the data colection. MY.H and X.R provided technical assistance. F.P., R.T.Z. and X.C. contributed to the design of the interface and data views. B.Y.Z. wrote the manufacture. X.L.D. and S.G.Z. directed and oversaw the study. All the authors have read and approved the final version of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Consent for publication
The consents for publication from all authors were obtained.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Zhao, B., Wu, J., Zhang, T. et al. A spatial transcriptomics study of MES-like and mono/macro cells in gliomas. Sci Rep 15, 12730 (2025). https://doi.org/10.1038/s41598-025-95277-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-025-95277-3
