
Abstract
X-linked sideroblastic anemia (XLSA) is a hereditary disorder affecting heme biosynthesis, caused by mutations in the ALAS2 gene, which encodes the erythroid-specific enzyme 5-aminolevulinate synthase. This enzyme, which requires pyridoxal 5’-phosphate (PLP) as a cofactor, catalyzes the first and rate-limiting step of heme synthesis in erythroid cells. XLSA is characterized by hypochromic microcytic anemia and ring sideroblasts in bone marrow, with most patients showing variable degrees of response to pyridoxine supplementation; however, female carriers of ALAS2 mutations often present a distinct clinical phenotype. A comprehensive review of the literature reveals over 100 distinct ALAS2 mutations linked to XLSA in more than 240 families. Here, we report seven new patients (four female cases) initially diagnosed with various conditions, later confirmed to have X-linked Sideroblastic Anemia due to ALAS2 mutations through genetic analysis. Among these, five represent novel ALAS2 mutations, including the first ever reported stop-loss mutation in ALAS2 associated with XLSA rather than X-linked dominant protoporphyria (XLDPP). Computational modelling of six reported cases revealed that four mutations significantly impact protein structure, conformation and cofactor interaction, consistent with our enzymatic assays demonstrating reduced ALAS2 activity. Furthermore, X-chromosome studies in female probands revealed a pronounced skewing of X-chromosome, which may provide an explanation for their distinct clinical manifestations in females.
Similar content being viewed by others


Introduction
Sideroblastic anemias (SAs) are a group of rare disorders, which are mainly characterized by the presence of ring sideroblasts, iron deposits in erythroblasts’ mitochondria. This trait is conserved in all forms of SAs, thus bone marrow aspirate smears aimed to identify these iron deposits are the most common standard of care test for an initial diagnosis1.
Sideroblastic anemias are primarily classified into two main groups: acquired and congenital (CSA) forms. CSAs are inherited disorders, but clinical manifestations may not appear immediately after birth, often becoming evident only in adulthood. Given that the prognosis and treatment options can differ significantly between acquired and congenital forms, it is essential to accurately differentiate late-onset CSA from acquired SA.
The underlying cause of CSA remains unidentified in over 40% of patients1. However, advancements in molecular genetics have increasingly enhanced the accuracy of clinical diagnosis. To date, several genes have been implicated in distinct forms of CSA, including SLC25A382,3 GLRX54, ABCB75, PUS16, YARS27,8, SLC19A29 and ALAS2 (erythroid-specific isoform of aminolaevulinic acid synthase)10. Among these, loss-of-function mutations in ALAS2 have been identified as the most prevalent and well-characterized cause of XLSA10,11. XLSA accounts for approximately 40% of all CSA cases reported in literature1.
Human ALAS2 gene is mapped on the X-Chromosome, Xp11.2112 and encodes the enzyme that catalyzes the first and rate-limiting step in heme biosynthesis. This process involves the conversion of glycine and succinyl-CoA into aminolaevulinic acid (ALA), a reaction that requires pyridoxal 5′-phosphate (pyridoxine or vitamin B6) as a cofactor. Loss-of-function mutations in ALAS2 impair heme synthesis, leading to reduced hemoglobin production11 and resulting in XLSA, as previously discussed. However, ALAS2 mutations are not solely associated with XLSA. Gain-of-function mutations, particularly those affecting the C-terminal domain of the protein, have been identified as the cause of X-Linked dominant protoporphyria, a distinct genetic disorder13.
XLSA primarily affects hemizygous men, while heterozygous female carriers are typically asymptomatic or may exhibit mild anemia14. Despite XLSA being an X-linked recessive disorder, women can also develop the condition. This occurs due to skewed X-chromosome inactivation in hematopoietic tissue, a phenomenon that becomes more pronounced with age15. Skewed inactivation disproportionately favors the mutated allele, resulting in clinical symptoms in affected women. Notably, one-third of all unrelated XLSA probands with identified ALAS2 mutations are women11.
Interestingly, the clinical presentation of XLSA differs between men and women. Men suffering from XLSA typically exhibit hypochromic microcytic sideroblastic anemia16, characterized by the “canonical” clinical features common to most CSAs: erythrocyte microcytosis (low mean corpuscular volume—MCV), hypochromia (low mean corpuscular hemoglobin—MCH), and elevated red cell distribution width (RDW)1,17. On the other hand, most females who express XLSA tend to present macrocytosis or even normocytosis as their primary hematological feature16. Age of onset is another distinguishing factor between genders. Due to the acquired skewing of X-chromosome inactivation discussed earlier, women tend to be diagnosed later than men. Men generally present their first symptoms during the first two decades of life, while women most often manifest symptoms in mid to late adulthood16,18. This difference underscores the impact of X-chromosome inactivation patterns on disease progression and clinical presentation.
The clinical symptoms of XLSA extend beyond the direct effects of impaired heme production. A hallmark feature of the condition is mitochondrial iron overload1, caused by ineffective erythropoiesis, which in turn leads to hepcidin suppression and parenchymal iron loading. This excess iron can cause damage to various organs and tissues, including the liver, heart, and pancreas. Symptoms of iron overload may include fatigue, abdominal and joint pain, hepatosplenomegaly (enlarged liver and spleen), and an increased risk of neurological complications19,20. These manifestations highlight the broader systemic impact of XLSA beyond anemia.
XLSA combines iron overload with varying degrees of anemia, and in XLSA the severity of anemia does not always correlate with the amount of iron stored in the liver. Interestingly, several cases of non-anemic subjects with XLSA have been reported to develop advanced liver damage, including well-established cirrhosis, usually identified in their third to fourth decade of life21,22,23,24.
There is no established “standard of care” for treating XLSA. However, several treatments are available to help manage its symptoms. For subjects with mild to moderate anemia, the standard initial approach typically involves vitamin B6 supplementation. This treatment targets the activity of ALAS2, the enzyme responsible for the rate-limiting step in heme biosynthesis, and can enhance hemoglobin production in approximately one-third of treated patients1. However, the effectiveness of pyridoxine is restricted to a subset of patients as its effect depends on the specific ALAS2 mutation and its location within the enzyme’s three-dimensional structure25. For patients with severe anemia unresponsive to pyridoxine, more invasive treatments may be required, including: Luspatercept injection26, blood transfusion, erythropoietin therapy, or even, in extreme cases, bone marrow transplantation. To address iron overload, a common complication in XLSA, treatment strategies include iron chelation therapy or controlled phlebotomies, chosen based on the patient’s anemia severity and overall clinical profile1. These interventions aim to prevent iron-related organ damage while balancing the patient’s hematological needs.
In this study, we report and characterize seven new unrelated cases of XLSA due to ALAS2 mutations—four women and three men—five of which involved previously unreported mutations. Structural modeling of six of the identified cases showed that four mutations substantially disrupted the protein’s architecture, altering its conformation and/or interaction with the cofactor. These findings were supported by enzymatic assays, which confirmed reduced ALAS2 activity. Additionally, analysis of X-chromosome inactivation patterns in female probands revealed significant skewing, offering a potential explanation for their unique clinical features. We also underscore the importance of integrating genetic studies into conventional diagnostic workflows to enhance diagnostic accuracy and reduce the likelihood of misdiagnosis. Altogether, we aim to provide a deep review of known ALAS2 cases while providing valuable molecular insights of seven new cases, potentially contributing to improve diagnostic accuracy, reduce misdiagnosis, and improve treatment approaches.
Results
Revision of the mutational spectrum in ALAS2 gene related to XLSA
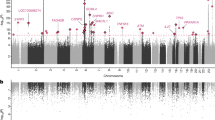
Although XLSA was first described over 70 years ago, comprehensive compilations of ALAS2 mutations remain scarce and outdated. In this study, we provide a systematic review of published ALAS2 mutations reported between 1992 and 2023, identifying over 100 unique variants from more than 240 unrelated families (Fig. 1, Supplementary Table 1). This analysis establishes a valuable resource for clinical diagnosis and molecular understanding of XLSA. Furthermore, our findings contribute to improving genetic counseling and offer valuable insights to guide development of future therapeutic strategies for XLSA patients.

Overview of ALAS2 mutational spectrum in XLSA. Reported mutations in this work are highlighted in bold, while newly reported mutations are also underlined. *The synonymous mutation R479 = alters the splicing site of exon 9. For extended information and references, see Supplementary Table 1.
Our systematic literature review, combined with newly identified mutations, confirms that missense mutations are the most prevalent type, accounting for approximately 90% of all reported ALAS2 variants. The remaining 10% consist of mutations in regulatory region27,28,29,30, splicing sites31, frameshifts32,33,34,35,36,37, nonsense mutations38,39, synonymous changes31, and a stop-loss (or nonstop) mutation (e.g., p.Ter588ArgextTer120 identified in this study).
From a geographical perspective, ALAS2 mutations causing XLSA have been reported globally, with a notably high prevalence in East Asian populations. While cases have been documented across all continents, Japan and China account for a significant proportion, possibly reflecting either a higher disease frequency in these regions or more comprehensive genetic screening efforts in these regions (Supplementary Table 1).
Genetic characterization and clinical overview of seven new XLSA cases: insights from next-generation sequencing and initial misdiagnoses
In this study, we report and characterize seven new cases of X-linked sideroblastic anemia that were definitively diagnosed through genetic testing and molecular analysis. Notably, all seven patients were initially misdiagnosed with other conditions, including three as myelodysplastic syndrome with ring sideroblasts (MDS-RS), one as congenital dyserythropoietic anemia (CDA), and three as unclassified anemia. Out of these seven cases, five of them present with new unreported mutations. All patients have undergone genetic testing using Next-Generation Sequencing (NGS) panels to exclude other potential causes of hereditary and acquired anemia. Available clinical information and treatment is described below, and clinical parameters are summarized in Table 1.
Proband 1 is a Spanish female, first time referred to the hematology department at Hospital La Paz (Madrid, Spain) due to macrocytic anemia when she was 29 years old. An initial diagnosis of CDA type I was suggested. She had no prior transfusion history or familial cases of anemia. At the initial visit, moderate anemia (Hb 10.5–11.5 g/dL) with macrocytosis (MCV 105–110 fL) and elevated RDW (15%) was observed. Reticulocyte counts were normal but inappropriately low given the severity of the anemia (0.80%; 23,700). Bone marrow aspiration revealed 10% ring sideroblasts, erythroid hyperplasia, macrocytosis, and megaloblastosis (Table 1). Twenty-two years later, repeat bone marrow aspiration showed an increase in ring sideroblasts to 26% (detected by Perls’ stain). Although the patient did not require blood transfusions and tolerated anemia well, she developed liver iron overload, likely due to ineffective erythropoiesis, which was treated with Deferasirox until July 2017, when iron levels normalized. Vitamin B6 treatment was initiated but discontinued after six months due to lack of response. Initial genetic tests were negative for HFE hemochromatosis and CDA. A genetic NGS panel identified the previously unreported ALAS2 variant c.1115T > C (p.Ile372Thr) in a heterozygous state.
Proband 2 is a Spanish 18-year-old male from ICO Badalona Hospital (Badalona, Spain) with a history of allergic asthma and phimosis surgery. His family history included maternal mild anemia without iron deficiency and a maternal grandmother with iron overload. He was referred to the hematology department due to mild anemia characterized by persistent microcytosis and hypochromia (Hb 12.2 g/dL, MCV 64.3 fL, MCH 20.3 pg) and a persistently elevated RDW (28.5%) (Table 1). A blood smear revealed a dual erythrocyte population with marked hypochromia, microcytosis, poikilocytosis, and some dacrocytes and spherocytes. Ferritin levels were within the upper-normal range at 298 ng/mL (normal range: 30–300), while the transferrin saturation index was notably elevated (72%). Reticulocyte counts were normal (55 × 10⁹/L). Copper, zinc, and lead levels were within normal ranges, and no systemic iron overload was detected. Ceruloplasmin was slightly decreased (17.9 mg/dL, reference range 20–60). Bone marrow aspirate demonstrated unilinear dysplasia of the erythroblastic series, with erythroid hyperplasia, severe dyserythropoiesis (80%), and 55% ring sideroblasts, along with marrow iron deficiency. Although the patient remained asymptomatic, vitamin B6 treatment was recommended but poorly adhered to, with no improvement in baseline hemoglobin. A liver MRI for iron quantification ruled out organ iron overload. Genetic testing ruled out HFE hemochromatosis, as well as the SF3B1 mutation linked to MDS-RS. NGS analysis identified a hemizygous missense variant in the ALAS2 gene, c.1354C > T (p.Arg452Cys), which was previously associated with XLSA40.
Proband 3 is a Spanish 43-year-old woman from ICO Badalona (Badalona, Spain) with no related family history and with a history of chronic restrictive anorexia nervosa and complications including vitamin B12, copper, and zinc deficiencies, hypogonadotropic hypogonadism, gelatinous bone marrow degeneration, and osteoporosis. She presented with bicytopenia characterized by neutropenia (ANC 0.6 × 10⁹/L), mild lymphopenia (Lymphocytes 1.0 × 10⁹/L), and hyporegenerative anemia (Hb 10.4 g/L, MCV 102.2 fL, MCH 35.7 pg, RDW 13.7%, reticulocytes 30.4 × 10⁹/L). Ferritin levels were within the normal range (125 ng/mL), but the transferrin saturation index was elevated (57%). Hepatic MRI excluded organ iron overload. A bone marrow aspirate revealed gelatinous degeneration with marrow iron deficiency and no significant dysplasia. Patient was treated with thiamine, erythropoietin and cobalamin without improvement. After iron treatment, a second aspirate showed marked dyserythropoiesis and 37% ring sideroblasts. Due to refractory multifactorial anemia unresponsive to iron replacement, the patient was treated with vitamin B6 (75 mg three times per day) during 10 months, without any hemoglobin improvement. Genetic testing ruled out mutations associated with acquired myelodysplasia, and a novel missense heterozygous variant was detected in the ALAS2 gene, c.506A > T (p.Asn169Ile). Sanger sequencing of the proband’s father, mother and brother revealed that the affected allele came from maternal inheritance.
Proband 4, a female born in 1937, was diagnosed with mild sideroblastic anemia (Hb 11 g/dL, 20% ring sideroblasts) and initially classified as having acquired MDS. She was enrolled in the Düsseldorf MDS registry. Clinical information is limited, as the patient has been lost to follow-up since early 2023. However, it is known that she became transfusion-dependent and did not receive any additional treatments. Genetic analysis revealed a novel heterozygous mutation in the ALAS2 gene, c.1762T > C (p.Ter588ArgextTer120), which results in the loss of the stop codon and a predicted elongation of the protein by 119 amino acids. C-terminal ALAS2 gain-of-function mutations cause erythroprotoporhyiria (EPP), but this patient had no reported clinical EPP features. The patient was also found to be heterozygous for the HFE C282Y mutation. Pathogenic mutations in the FECH gene, including the polymorphic IVS3-48C allele linked to EPP, were also excluded.
Proband 5, a German male born in 1937, was diagnosed with severe sideroblastic anemia characterized by microcytosis (Hb 8.8 g/dL, MCV 69.8 fL), elevated erythropoietin levels (52 mUI/L), and 25% ring sideroblasts. Initially classified as having MDS, he was also included in the Düsseldorf MDS registry. A bone marrow biopsy performed in 2013 revealed disease progression to RAEB-2 (> 10% marrow blasts). Unfortunately, limited clinical information is available, as the patient passed away in November 2014 due to infectious complications. Subsequent genetic testing using an NGS CSA panel identified a previously reported hemizygous missense mutation41 in the ALAS2 gene, c.1559C > T (p.Pro520Leu).
Proband 6, a German female born in 1950, was diagnosed with moderate sideroblastic anemia (Hb 9.8 g/dL), elevated erythropoietin (38 mUI/L), and ferritin levels (350 ng/mL), with 35% ring sideroblasts (Table 1). She was also included in the Düsseldorf MDS registry. The patient was initially treated with erythropoietin (EPO) without success. She was inadvertently included in a clinical trial for Luspatercept, where she showed a positive response, with her hemoglobin increasing from around 6 g/dL to approximately 10 g/dL after three injections over 9 weeks. However, she discontinued Luspatercept due to severe fatigue and resumed chronic transfusion therapy (approximately every 2 weeks), along with iron chelation therapy using Deferasirox. As per the last contact, the patient is still alive. Despite the initial diagnosis of MDS, an NGS panel later revealed a novel heterozygous mutation in the ALAS2 gene, c.1328G > A (p.Gly443Asp).
Proband 7 is a male born in 1992 from Alberta, Canada, with a family history suggesting a possibly affected uncle, though this was not confirmed. At 18, he presented with microcytic hypochromic anemia (Hb 9.0 g/dL, MCV 59 fL, MCHC 30.5 g/dL), low hematocrit (0.30 L/L), and high RDW (30.0%). At age 21, he continued to show persistent microcytic hypochromic anemia (Hb 8.1 g/dL, MCV 54 fL, MCHC 27.2 g/dL), with a further decrease in hematocrit (3.0%) and increased RDW (35.9%). Iron levels were elevated (234.55 µg/dL) and ferritin was high (593 ng/mL). A bone marrow aspirate was not performed, and thus the presence of ring sideroblasts could not be evaluated. Multiple diagnoses, including CDA types I and II, iron deficiency, thalassemia, Blackfan-Diamond anemia, Pearson syndrome, and lead poisoning were ruled out. The patient did not show clinical signs of iron overload or other associated complications like hepatomegaly, splenomegaly, or neurological issues but did experience muscle weakness and exacerbated anemia following a flu-like illness. Prior to the genetic diagnosis, he was treated with oral iron supplementation (Ferrous sulfate 100 mg twice daily for 4 weeks, followed by 12 mg daily) and did not require any transfusions. Following genetic diagnosis, pyridoxine and folic acid were recommended, along with periodic monitoring for iron overload via serum ferritin and liver MRI. The patient was referred for NGS analysis of a SA gene panel, which revealed a novel hemizygous missense mutation in the ALAS2 gene, c.497A > T (p.Lys166Met).
Computational structural modelling of the reported ALAS2 mutations
Computational structural modeling of the human ALAS2 protein was done in six out of the seven reported mutations (Fig. 2A). The modeling results revealed specific structural disruptions associated with most of these mutations.

Bioinformatic modelling of the p.Lys166Met (B), p.Asn169Ile (C), p.Ile372Thr (D), p.Gly443Asp (E), p.Arg452Cys (F), p.Pro520Leu (G) and p.Ter588ArgextTer120 (H) variants. Overview of the whole crystallized structure and the reported mutations location are shown in panel (A). Close view of the respective mutations compared with the wild-type are depicted in panels (B, C, D, E, F, G). Predicted aminoacidic sequence elongation for ALAS2 p.Ter588ArgextTer120 variant compared to wild-type ALAS2 shown in red (H).
For p.Lys166Met, located at a loop near the dimer interface, the mutation disrupts the interaction with Asp491 due to the loss of a positive charge, potentially destabilizing the dimer (Fig. 2B). p.Asn169Ile, at the homodimer interface, induces a steric clash that prevents homodimer formation, impairing protein function (Fig. 2C). p.Ile372Thr, in a loop near the succinyl binding site, disrupts an interaction with Ile337, likely altering loop conformation and affecting protein stability (Fig. 2D). p.Gly443Asp, located in an alpha-helix, introduces a steric clash with Arg447, potentially compromising structural integrity (Fig. 2E). The p.Arg452Cys variant, located on the protein surface, is not predicted to impact the substrate or the cofactor binding. However, given its presence in multiple patients and its location in a known hotspot (Supplementary Table 1), we cannot rule out potential effects on protein stability or half-life in vivo35 (Fig. 2F). Lastly, the p.Pro520Leu variant, located in a highly conserved loop, does not significantly alter the loop’s conformation or cause steric clashes. Family studies have previously indicated that Pro520Leu, either alone or in combination with the HFE C282Y mutation, is insufficient to cause X-linked sideroblastic anemia or significant iron overload, suggesting that additional genetic or environmental factors may play a role41. Considering its conserved location, Pro520Leu might contribute to pathogenesis through reduced protein stability or half-life rather than a direct enzymatic defect (Fig. 2G).
The p.Ter588ArgextTer120 variant could not be modeled due to the absence of structural data for the predicted extension region. This mutation substitutes the stop codon (at position 588) with an Arginine residue, resulting in an elongation of the protein by an additional 119 amino acids (Fig. 2H). Previous studies have shown that ALAS2 C-terminal missense variants between residues 565–586 and truncating mutations between residues 557–563 result in a gain-of-function effect due to this region’s role in protein activity inhibition42. Even though the structure of this region remains unknown, the p.Ter588ArgextTer120 mutation is predicted (and confirmed in vitro; see below data) as a loss-of-function mutation, as it disrupts the inhibitory region by introducing a large, positively charged side chain.
Additional bioinformatic in silico prediction tools were assessed to support the clinical interpretation of the reported variants (Supplementary table 2). The seven reported mutations could be categorized into three main groups based on their classifications and prediction scores. The largest group consists of three variants (p.Lys166Met, p.Asn169Ile and p.Gly443Asp) initially classified as Variant of uncertain significance (VUS) but showing pathogenic features under ACMG guidelines, all with high CADD scores above 20 (26.4, 25.9 and 27.1 respectively). The second group includes three pathogenic or likely pathogenic variants (p.Ile372Thr, p.Arg452Cys, and p.Ter588ArgextTer120), supported by strong ACMG criteria and high CADD scores (25.1, 27.5 and 18.75 respectively). Finally, p.Pro520Leu represents an unusual case where despite being classified as benign, multiple prediction tools suggest pathogenicity, supported by a high CADD score of 26.6 (Supplementary Fig. 2).
In vitro functional characterization of ALAS2 variants: enzymatic activity and response to stress conditions
To explore the potential pathogenicity of the described variants, we conducted in vitro functional characterization of the seven reported ALAS2 variants through bacterial expression studies as described on Ducamp et al.43. Appropriate protein overexpression and equivalent expression levels between wild-type and mutant proteins were confirmed through immunoblot analysis (Supplementary Fig. 2). Enzymatic activity assays of bacterial lysates revealed that five of the reported variants (p.Ile372Thr, p.Pro520Leu, p.Gly443Asp, p.Asn169Ile and p.Ter588ArgextTer120), exhibited significant reduction in ALAS2 function (Fig. 3), with residual activities ranging from 0 to 50.56% compared to wild-type (data not shown). The remaining two variants (p.Arg452Cys and p.Lys166Met) demonstrated in-vitro enzymatic activity levels comparable to the wild-type construct under the conditions tested, as previously described for p.Arg452Cys mutation35.

Residual activity of ALAS2 variants. Residual activity is represented as a percentage of the wild-type enzymatic activity ± S.D. Enzymatic activity was measured on at least four independent bacterial cultures. Student’s two-tailed t-test was performed to establish statistical significance of comparisons between wild-type and all mutants (P value < 0.05). To ensure the reliability of the assay, we included one positive control mutant (p.Met567Glu-fs*2) and two negative control mutants (p.Asp263Asn and p.Ala539Ter), all of which have been previously tested and reported in ALAS2 enzymatic activity studies.13,43.
To further investigate the variants that showed normal activity under standard conditions (p.Arg452Cys and p.Lys166Met), we conducted additional enzymatic assays under stressed conditions. The variants were tested both in the absence of exogenous pyridoxal 5’-phosphate and for thermal stability by pre-incubating the samples at 37 °C for 30 or 60 min prior to enzymatic analysis. Neither the absence of exogenous PLP nor thermal challenge led to a significant reduction in activity for either of these variants (data not shown).
X-chromosome inactivation skewing in females
Although XLSA primarily affects males, several cases of heterozygous carrier women with clinical symptoms have been reported, albeit with different manifestations44. While XLSA has traditionally been considered as an X-linked recessive disease, the occurrence of affected heterozygous women, often related to skewed X-chromosome inactivation, has raised questions about its pathogenesis. Skewed X-inactivation can preferentially silence the X chromosome with the unaffected allele, allowing expression of the mutant allele and resulting in clinical symptoms16,45.
To test whether X-inactivation could explain the clinical presentation of the four female probands in this study, we conducted a methylation-based analysis, commonly known as HUMARA assay44, following the protocol outlined by Brancaleoni and collaborators46. This assay evaluates the methylation status of the androgen receptor (AR) and Zinc Finger MYM-Type Containing 3 (ZMYM3) genes to determine X-inactivation patterns.
For this analysis to be fully informative, male relatives of the female probands are required to identify which allele—the wild-type or mutant—was preferentially inactivated. Unfortunately, three out of the four female probands (Probands 1, 4, and 6) lacked available male relatives or had relatives who declined to participate, preventing us from definitively determining which allele was preferentially inactivated in these cases.
Unfortunately, for Proband 3, who had available relatives (mother, father and brother), PCR amplification of highly variable regions of the AR and ZMYM3 genes was inconclusive, as the proband exhibited identical allele profiles for both alleles, preventing their differentiation (Table 2). Interestingly, the proband’s unaffected brother inherited one maternal allele not shared with the proband (Supplementary Fig. 3) and the X paternal allele inherited by the proband is a wild-type allele. Sequencing of maternal gDNA confirmed that ALAS2 mutation is present on one of the mother’s alleles (data not shown). If there is skewed X-chromosome inactivation favoring the paternal allele, this could explain the proband’s clinical symptoms. Unfortunately, this hypothesis cannot be verified using the current methodology.
For Probands 1, 4, and 6, skewed X-inactivation was observed in both the AR and ZMYM3 genes (Table 2). However, due to the absence of data from male relatives, it was not possible to associate the skewing with either the wild-type or mutated allele. Furthermore, in Proband 6, analysis of the ZMYM3 gene was inconclusive, as the overlapping profiles of both alleles hindered accurate quantification.
Discussion
This study provides a comprehensive review of all reported ALAS2 mutations causing XLSA, including the presentation of seven new cases, five of which involve previously unreported variants. Among these novel mutations, four are missense (p.Lys166Met, p.Asn169Ile, p.Ile372Thr and p.Gly443Asp), while the fifth is the first-ever reported stop-loss mutation in ALAS2 gene (p.Ter588ArgextTer120). The remaining two cases involve previously described mutations (p.Arg452Cys35 and p.Pro520Leu41), which are among the most commonly reported, with at least 15 cases for p.Arg452Cys and 7 for p.Pro520Leu (Supplementary Table 1). Overall, six of our seven reported cases (85.7%) involve missense mutations, which is consistent with the 90% frequency of missense mutations across all reported cases.
Computational modeling suggests that the p.Arg452Cys and p.Pro520Leu variants are unlikely to directly disrupt substrate or cofactor binding. However, several lines of evidence support their pathogenicity: both amino acids (Arg452 and Pro520) are fully conserved across vertebrates35,41, some bioinformatic analyses predict deleterious effects, and these mutations have been reported in multiple patients with XLSA (Supplementary Table 1). Considering these factors, along with comprehensive evaluation, all seven variants should be considered as likely pathogenic.
Since ALAS2 mutations associated with XLSA are typically loss-of-function mutations, enzymatic activity assays were conducted to assess the impact of the ALAS2 variants reported in this study and compare the results with computational modeling predictions. Significant reductions in enzymatic activity were observed for five of the seven mutations: p.Asn169Ile, p.Ile372Thr, p.Gly443Asp, p.Pro520Leu, and p.Ter588ArgextTer120. Despite in-silico predictions suggesting pathogenicity, the p.Arg452Cys and the p.Lys166Met variants showed no significant decrease in activity, consistent with previous studies for p.Arg452Cys and p.Arg452His35. Although p.Arg452Cys is frequently observed in XLSA patients, our results suggest that additional in-vivo factors may contribute to reduced ALAS2 activity in patients’ bone marrow, rather than the mutations directly affecting enzyme function. Notably, our study demonstrated a significant reduction in enzymatic activity for the Pro520Leu variant (Fig. 3), aligning with previous reports of reduced activity, though earlier findings did not reach statistical significance43. This discrepancy may be due to differences in experimental conditions, assay sensitivity, or protein expression systems, highlighting the complexity of in vitro functional assessments and the need for multiple lines of evidence when evaluating variant pathogenicity.
Gain-of-function mutations in the C-terminal region of ALAS2 are associated with XLDPP, characterized by protoporphyrin IX overproduction and severe photosensitivity13,42,47,48,49,50, while loss-of-function mutations lead to XLSA. In this study, we report for the first time a stop-loss mutation in the ALAS2 gene (p.Ter588ArgextTer120).
Although the p.Ter588ArgextTer120 variant could not be computationally modeled, our findings indicate that the predicted elongated protein is overproduced in E. coli (Supplementary Fig. 2) and significantly reduces ALAS2 activity (Fig. 3), likely due to impaired protein folding or stability. Therefore, the effect of this C-terminal mutation differs from previous ALAS2 mutations in the C-terminal, which are associated with a gain-of-function and XLDPP, as this mutation leads to a loss-of-function and XLSA. This mutation contributes to XLSA, as evidenced by the presence of ring sideroblasts (20%) in the patient’s bone marrow, a hallmark of the disease. These results underscore the critical role of mutation type and position in shaping the clinical outcome.
Although X-linked sideroblastic anemia is typically described as an X-linked recessive disorder, several heterozygous women have been reported with symptoms, often presenting with different clinical features from affected men11,15,44. Most ALAS2 mutations impair the rate-limiting step of heme biosynthesis in erythroid cells, leading to microcytic, hypochromic anemia in males. In contrast, symptomatic female carriers often show macrocytic anemia, likely due to skewed X-chromosome inactivation favoring the mutant allele in hematopoietic tissue45. This results in a small population of erythroid precursors expressing the normal allele, causing macrocytosis due to early release from the bone marrow in response to elevated erythropoietin levels16. We demonstrated that three out of four symptomatic female probands in this study exhibited skewed X-chromosome inactivation. However, the inability to unequivocally assign which allele was skewed stemmed from the lack of male relative samples (father or brother) for probands 1, 4, and 6. For Proband 3, despite having samples from her unaffected father and brother, the allele profiles were uninformative, preventing an assessment of X-chromosome skewing. Nevertheless, the available data confirms that the proband inherited the affected maternal allele.
The pattern of X-chromosome inactivation in hematopoietic cells and the functional effects of ALAS2 mutations contribute to the variable clinical presentation of female carriers15,16,25,44,45,51. Many symptomatic females with XLSA present with macrocytic, rather than microcytic, erythrocytes, which can overlap with other inherited anemias, complicating differential diagnosis. Genetic testing is crucial for accurate diagnosis, as seen in the seven cases reported here, where none were initially suspected of XLSA and were only confirmed through genetic analysis. Relying on biomarkers or clinical symptoms alone is outdated, as these may lead to misdiagnosis and inappropriate treatments. For instance, mutations in SLC25A38 cause a hypochromic microcytic anemia similar to XLSA but unresponsive to pyridoxine2, while hereditary hyperferritinemia with cataract syndrome (HHCS) may be confused with hemochromatosis52,53 based solely on elevated ferritin levels, leading to potentially harmful treatments like iron depletion. Genetic diagnosis in XLSA is particularly critical in females, where clinical features can be misleading, and it helps clarify genotype–phenotype relationships, enabling personalized treatment.
Our comprehensive revision of all mutations identified in ALAS2 has significant clinical implications for the care of XLSA patients. By summarizing mutation types, frequencies, and associated clinical presentations, it offers clinicians a valuable resource for variant interpretation and diagnosis. Furthermore, understanding the full spectrum of ALAS2 mutations and their effects can guide therapeutic decisions, improve prognostic assessments, and enhance genetic counseling for affected families.
Overall, this work provides a comprehensive review of over 100 ALAS2 mutations associated with X-linked sideroblastic anemia, reports seven new cases (including five novel mutations), and examines the impact of X-chromosome inactivation in female carriers. It emphasizes the importance of incorporating genetic studies into diagnostic workflows to improve diagnostic accuracy, minimize misdiagnoses, and advance personalized treatment approaches for affected individuals and families.
Methods
Study subjects
Seven subjects from seven unrelated families were included in this study. Three of the subjects were included from the German Myelodysplastic syndrome (MDS) registry (Dr. Norbert Gattermann). Subjects were diagnosed and followed-up in four independent institutions from three different countries: Hospital La Paz de Madrid, Spain; ICO-Hospital Germans Trias i Pujol, Spain; University Hospital Düsseldorf; and Alberta Children’s Hospital, Canada.
Written informed consent was obtained from all participants and their relatives. The study was conducted in accordance with the ethical principles of the Declaration of Helsinki, and the protocol was approved by the Ethics Committee of Hospital General de Catalunya on 30th November 2022. Blood from healthy donors used as controls for the studies was obtained from the Catalan Tissue and Blood Bank (BST) through an agreement.
NGS and Sanger sequencing analysis
Genomic DNA was extracted from peripheral blood samples of the patient and his relatives using the QIAamp DNA Mini Kit (Qiagen, Valencia, CA, USA) according to the manufacturer’s instructions. The proband genomic DNA was studied using a targeted NGS gene panel (BloodGenetics #10020 for subjects 1, 2, 3, 5 and 7. Patients 4 and 6 did not have NGS performed, but mutation was detected via Sanger sequencing) including known genes associated with Congenital sideroblastic anemias (ABCB7, ALAS2, GLRX5, HSCB, HSPA9, LARS2, MTATP6, NDUFB11, PUS1, SF3B1, SLC19A2, SLC25A38, STEAP3, TRNT1 and YARS2). Genomic DNA libraries were obtained using the HaloPlexHS 0.5–2.5 Mb ILM 48 reactions kit (Agilent, Santa Clara, CA, USA). Library sequencing was performed using Illumina MiniSeq Mid Output kit (300 cycles) and an Illumina MiniSeq sequencer (Illumina, San Diego, CA, USA). Patient 3 relatives (father and brother) were also confirmed to be wild-type via Sanger sequencing.
Genetic variants are reported following the official Human Genome Variation Society (HGVS) nomenclature and refer to NM_000032.5 for the Homo sapiens ALAS2 transcript variant and NP_000023.2 for the Homo sapiens ALAS2 protein.
New mutations reported in this study have been submitted to ClinVar (http://www.ncbi.nlm.nih.gov/clinvar) with the following SCV accession numbers: SCV005688786, SCV005688785, SCV005688784, SCV005849004, SCV005849006, SCV005849003 and SCV005849005. All mutations detected by NGS were confirmed by conventional Sanger sequencing.
DNA sequencing, mutation validation and analysis
Patient DNA was obtained from peripheral blood using the QIAamp DNA Mini Kit (Qiagen, Valencia, CA, USA) and analysed using NGS in BloodGenetics S.L. (Esplugues de Llobregat, Spain) as previously detailed in Material and Methods. Data were analysed with SureCall software (Agilent Technologies, Santa Clara, CA, USA) and Varsome Clinical software (Saphetor SA, Lausanne, Switzerland).
Mutations identified through NGS were validated by Sanger sequencing, by using specific primers designed to cover all regions of interest (Supplementary table 3). 50 ng of DNA were amplified by standard PCR and loaded into 2% agarose gels with ethidium bromide. PCR products were purified with NucleoSpin® Gel and PCR Clean-up Kit (Macherey-Nagel, GmbH & co KG, Düren, Germany) and sent for sequencing using the conventional Sanger method. Sanger sequencing results were analyzed using SnapGene® 5.2.5 (Dotmatics, Bishop’s Stortford, United Kingdom) software. Specific PCR conditions are available upon request.
Bioinformatic analysis
Human ALAS2 molecular model was constructed from 5QQQ X-ray crystal structure at 1.93 A of resolution54. Regions 39–43 and 403–415 were remodeled using Modeller55. Energy minimization was performed in the resulting model using GROMACS 556. Pymol57 was used to introduce the mutated residues in the initial molecular model. The UCSC Genome Browser58 was used to extract sequence conservation among vertebrates.
For in-silico assessments, the following versions were used: CADD Score (version GRCh38-v1.7)59, VarSome v16.6.060, MutationTaster2021 (version GRCh37, Ensembl 102)61, PROVEAN v1.162 and SIFT (Version from 31 March 2022 update)63. The rest of the prediction tools results were obtained from extended analysis through VarSome and Varsome Clinical.
X-chromosome inactivation
HUMARA assay for X-Chromosome Inactivation studies was performed based on protocol detailed in Brancaleoni V. and collaborators46, with some adjustments. As in the reference paper, both human androgen receptor (AR) and zinc-finger MYM type 3 (ZMYM3) genes were used.
Digestion of DNA obtained from the whole blood of the patients was performed using methylation-sensitive restriction enzymes HpaII and HhaI. 20U of each enzyme were used together with 500 ng of subject gDNA in a 20 µL reaction volume. Digestion of gDNA was performed for 15 min at 37 ºC using High Fidelity (HF®) restriction enzymes from NEB (New England Biolabs, Ipswich, MA, USA), followed by an enzyme inactivation at 80 ºC for 20 min.
For the amplification step, a 25 µL or 50 µL PCR reaction was performed mixing 50 ng of each DNA with 0.25 µM FAM and HEX-labelled forward and reverse primers listed below in Supplementary table 4, 1 mM MgCl2, 1 mM dNTPs, 1.2 M Betaine (Sigma-Aldrich, St. Louis, MO, USA), 0.6–1.2 µL PFU DNA Polymerase (EMBL Heidelberg core facility, Heidelberg, Germany) and 1 × PFU buffer (10X buffer includes: 200 mM Tris–HCL pH 8.8, 100 mM (NH4)2SO4, 100 mM KCl, 20 mM MgSO4 and 1% (v/v) Triton-X100).
The PCR conditions used for amplification of AR and ZMYM3 genes were as follows: 95 °C for 5 min followed by 35 cycles of 95 °C for 30 s, 30 s of a Tm gradient (68–55 °C) where each primer pair specific Tm was used (see Supplementary table 4) and 72 °C for 30 s to end with a final step at 72 °C for 5 min.
Fragment analysis was performed using ABI3500 Genetic Analyzer (Applied Biosystems, Waltham, MA, USA) at Servei de Genòmica i Espectroscòpia de Biomolècules (SGiEB) (Universitat Autònoma de Barcelona, Barcelona, Spain).
Quantification of percentage of the relative X-chromosome inactivation was also performed as detailed in Brancaleoni V. and collaborators46.
ALAS2 enzyme activity functional assay
ALAS2 activity assay was performed as previously described in Ducamp S. and collaborators43, with some adjustments.
Human ALAS2 ORF was cloned from a pCMV6-AN-Myc-DDK-ALAS2 plasmid (OriGene, custom order) into an empty pGEX-6P-1 plasmid, to get a recombinant fusion protein of a N-terminal Glutathione S-transferase (GST) of approximately 25 KDa (218 AA) plus our human ALAS2 (~ 64 KDa, 588 AA) using PasI and NotI restriction enzymes. A previous step of PasI restriction site was needed to perform in-frame cloning in pGEX-6P-1. Mutations under study were introduced by site-directed mutagenesis, transformed into DH5alpha and further confirmed by Sanger sequencing. For X588R mutation (p. Ter588ArgextTer120), ALAS2 ORF had to be enlarged with around 360 additional base pairs from 3’ gDNA region of ALAS2. Sequence of primers used for addition of PasI restriction site, cloning, mutagenesis of all mutants and mutation confirmation by sequencing are available in supplementary data (Supplementary table 5).
Since DH5alpha is not a suitable strain for protein expression, mutant constructs were later transformed into BL21-CodonPlus(DE3)-RIPL competent cells, an enhanced E. coli strain with modified codons to mimic eukaryotic codon usage.
Overnight cultures were started from glycerinate stocks in 5 ml Luria Bertani broth (LB) supplemented with 50 mg/ml Ampicillin and 25 mg/ml Chloramphenicol. The day after, exponential growth of the culture was performed during 1.5 h in 20 ml LB + Amp + CHL cultures from the overnight culture, until an OD600nm of 0.9–1.2 was reached. Induction of ALAS2 protein expression was done by adding 0.5 mM isopropyl b-D-thiogalactopyranoside (IPTG) and the presence of a strong ALA dehydratase inhibitor—0.5 mM 4,6-dioxoheptanoic acid (succinyl acetone). Incubation was carried out in LB + Amp + CHL medium for 5 h at 25 °C. Activity assay under standard conditions was performed on bacterial lysates. Pellets were sonicated in 300 ul of HEPES 50 mM pH 7.5 while maintained on ice. Total protein concentration was measured by a standard Bradford assay (Bio-Rad Laboratories GmBh, Hercules, CA). 600 ul of sample, adjusted to a final concentration of 0.2 mg of total protein per ml, were preincubated for 5 min at 37 °C before the assay. After this step, 100 ul of a solution consisting of 1 mM Succinyl-CoA, 10 mM Glycine, 50 mM HEPES, and 0.5 mM PLP (Sigma-Aldrich, St. Louis, MO) was added to the pre-incubated samples. Mixture was incubated at 37 °C for 20 min, and reaction was stopped by adding 60 ul of trichloroacetic acid (TCA). After incubation on ice for 5 min, reactions were centrifuged at 12,000 g for 5 min. 600 ul of the assay reaction was then mixed with 600 ul of Acetate buffer 1 M pH 4.6 and 100 ul acetylacetone. Samples were incubated at 10 min at 100 °C, and after sample cool down, the reaction product 5-aminolevulinate (ALA) was quantified by colorimetry through the Ehrlich’s reaction. To do so, 1200 ul of assay reaction (once cooled down) were mixed with 1200 ul Ehrlich’s reagent. Mixtures were incubated for 15 min at room temperature and ALA synthesized was measured at OD 553 nm.
The specific activity (SA) was expressed in pmol of ALA/mg total protein at 37 °C. The residual activity (%) was determined by expressing the specific activity of mutants relative to that of the wild-type ALAS2.
Western blotting
Cell pellets after IPTG incubation were resuspended in 300 µL HEPES 50 mM pH 7.5 and sonicated at 40A. After sonication, samples were centrifuged 5 min at 15,000 rpm, 4 °C. Protein concentration was quantified using a standard Bradford assay (Bio-Rad, Hercules, CA, USA). Samples were incubated for 5 min at 95 °C in Laemmli buffer (containing 125 mM Tris–HCl pH 6.8, 10% SDS, 25% glycerol, 1% beta-mercaptoethanol, and bromophenol blue). Then, 15 µg of total protein lysate was separated by sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) (10%) and transferred to polyvinylidene fluoride (PVDF) membranes using the iBlotTM 2 Transfer Device (ThermoFisher Scientific, Waltham, MA, USA). Correct loading and transfer were checked by Ponceau S staining (Sigma-Aldrich, St. Louis, MO, USA). Immunoblots were incubated with blocking solution (5% skim milk, 0.1% Tween 20, in TBS/TBS-T) for 1 h at room temperature before incubation with primary antibody. Membrane was incubated with primary antibody anti GST (1/1000, G7781, Sigma-Aldrich, St. Louis, MO, USA) overnight at 4 °C. The blots were then washed with TBST and incubated during one hour at room temperature with a secondary anti rabbit IgG (H + L), labeled with Horseradish peroxidase (HRP) (1:5000, Jackson ImmunoResearch). GST-fusion proteins were immunodetected with Immobilon(R) Forte Western HRP Substrate (Merck Millipore, Burlington, MA, USA) and visualized with ChemiDocTM (Bio-Rad, Hercules, CA, USA) both according to the manufacturer’s instructions.
Statistical analysis
Statistical analyses were performed using Prism 9.0 (GraphPad Software Inc, San Diego, CA, USA). Data are shown as mean ± S.D. and Student’s two-tailed t-test for WT and mutations comparisons were calculated. Tests for normal distribution was performed for all data including Anderson–Darling test, D’Agostino & Pearson test, Shapiro–Wilk test and Kolmogorov–Smirnov test. Mann–Whitney test was calculated for those samples that did not meet normality. p values < 0.05 (*), < 0.01 (**), < 0.001 (***), < 0.0001 (****) are indicated.
Data availability
The datasets generated during and/or analysed during the current study are available from the corresponding author on reasonable request.
References
Bottomley, S. S. & Fleming, M. D. Sideroblastic anemia. Hematol. Oncol. Clin. N. Am. 28, 653–670 (2014).
Guernsey, D. L. et al. Mutations in mitochondrial carrier family gene SLC25A38 cause nonsyndromic autosomal recessive congenital sideroblastic anemia. Nat. Genet. 41, 651–653 (2009).
Kannengiesser, C. et al. Missense SLC25A38 variations play an important role in autosomal recessive inherited sideroblastic anemia. Haematologica 96, 808–813 (2011).
Camaschella, C. et al. The human counterpart of zebrafish shiraz shows sideroblastic-like microcytic anemia and iron overload. Blood 110, 1353–1358 (2007).
Allikmets, R. et al. Mutation of a putative mitochondrial iron transporter gene (ABC7) in X-linked sideroblastic anemia and ataxia (XLSA/A). Hum. Mol. Genet. 8, 743–749 (1999).
Bykhovskaya, Y., Casas, K., Mengesha, E., Inbal, A. & Fischel-Ghodsian, N. Missense mutation in pseudouridine synthase 1 (PUS1) causes mitochondrial myopathy and sideroblastic anemia (MLASA). Am. J. Hum. Genet. 74, 1303–1308 (2004).
Riley, L. G. et al. Mutation of the mitochondrial Tyrosyl-tRNA synthetase gene, YARS2, Causes Myopathy, Lactic Acidosis, And Sideroblastic Anemia—MLASA syndrome. Am. J. Hum. Genet. 87, 52–59 (2010).
Riley, L. G. et al. The phenotypic spectrum of germline YARS2 variants: From isolated sideroblastic anemia to mitochondrial myopathy, lactic acidosis and sideroblastic anemia 2. Haematologica 103, 2008–2015 (2018).
Labay, V. et al. Mutations in SLC19A2 cause thiamine-responsive megaloblastic anaemia associated with diabetes mellitus and deafness. Nat. Genet. 22, 300–304 (1999).
Cotter, P. D., Baumann, M. & Bishop, D. F. Enzymatic defect in ‘X-linked’ sideroblastic anemia: Molecular evidence for erythroid δ-aminolevulinate synthase deficiency. Proc. Natl. Acad. Sci. U. S. A. 89, 4028–4032 (1992).
Bottomley, S. S. & Fleming, M. D. Sideroblastic Anemias. In Wintrobe’s Clinical Hematology 14th edition 2078–2118 (2019).
Cotter, P. D., Willard, H. F., Gorski, J. L. & Bishop, D. F. Assignment of human erythroid δ-aminolevulinate synthase (ALAS2) to a distal subregion of band Xp11.21 by PCR analysis of somatic cell hybrids containing X; Autosome translocations. Genomics 13, 211–212 (1992).
Ducamp, S. et al. Molecular and functional analysis of the C-terminal region of human erythroid-specific 5-aminolevulinic synthase associated with X-linked dominant protoporphyria (XLDPP). Hum. Mol. Genet. 22, 1280–1288 (2013).
Nzelu, D. et al. X-linked sideroblastic anaemia in a female fetus: A case report and a literature review. BMC Med. Genom. 14, 1–6 (2021).
Busque, L. et al. Nonrandom X-inactivation patterns in normal females: Lyonization ratios vary with age. Blood 88, 59–65 (1996).
Aivado, M. et al. X-linked sideroblastic anemia associated with a novel ALAS2 mutation and unfortunate skewed X-chromosome inactivation patterns. Blood Cells Mol. Dis. 37, 40–45 (2006).
Ducamp, S. & Fleming, M. D. The molecular genetics of sideroblastic anemia. Blood 133, 59–69 (2019).
Aivado, M., Gattermann, N. & Bottomley, S. X chromosome inactivation ratios in female carriers of X-linked sideroblastic anemia. Blood 97, 4000–4002 (2001).
McLaren, G. D., Muir, W. A., Kellermeyer, R. W. & Jacobs, A. Iron overload disorders: Natural history, pathogenesis, diagnosis, and therapy. CRC Crit. Rev. Clin. Lab. Sci. 19, 205–266 (1983).
Gattermann, N., Muckenthaler, M. U., Kulozik, A. E., Metzgeroth, G. & Hastka, J. The evaluation of iron deficiency and iron overload. Dtsch. Arztebl. Int. 118, 847–856 (2021).
Byrd, R. B. & Cooper, T. Hereditary iron-loading anemia with secondary hemochromatosis. Ann. Intern. Med. 55, 103 (1961).
Peto, T. E. ., Pippard, M. & Weatherall, D. Iron overload in mild sideroblastic anaemias. In The Lancet 375–378 (1983).
Bottomley, S. S. Sideroblastic anemia: Death from iron overload. Hosp. Pract. 26, 55–56 (1991).
Fairbanks, V. F., Dickson, E. R. & Thompson, M. E. Hereditary sideroblastic anemia. Hosp. Pract. 26, 53–55 (1991).
Shoolingin-Jordan, P. M. et al. 5-Aminolevulinic acid synthase: Mechanism, mutations and medicine. Biochim. Biophys. Acta Proteins Proteom. 1647, 361–366 (2003).
Van Dijck, R., Goncalves Silva, A. M. & Rijneveld, A. W. Luspatercept as potential treatment for congenital sideroblastic anemia. N. Engl. J. Med. 388, 1435–1436 (2023).
May, A. & Bishop, D. F. The molecular biology and pyridoxine responsiveness of X-linked sideroblastic anaemia. Haematologica 83, 56–70 (1998).
Harigae, H. et al. A novel mutation of the erythroid-specific δ-aminolevulinate synthase gene in a patient with non-inherited pyridoxine-responsive sideroblastic anemia. Am. J. Hematol. 62, 112–114 (1999).
Huang, J. et al. A hemizygous pR204Q mutation in the ALAS2 gene underlies X-linked sideroblastic anemia in an adult Chinese Han man. BMC Med. Genom. 14, 1–5 (2021).
Susanto, T. A. K. & Bhattacharyya, R. X-linked sideroblastic anemia in a Malay Boy with ALAS2 S568G mutation. J. Pediatr. Hematol. Oncol. 39, 408–409 (2017).
Edgar, A. J., Losowsky, M. S., Noble, J. S. & Wickramasinghe, S. N. Identification of an arginine452 to histidine substitution in the erythroid 5-aminolaevulinate synthetase gene in a large pedigree with X-linked hereditary sideroblastic anaemia. Eur. J. Haematol. 58, 1–4 (1997).
Bekri, S. et al. A promoter mutation in the erythroid-specific 5-aminolevulinate synthase (ALAS2) gene causes X-linked sideroblastic anemia. Blood 102, 698–704 (2003).
Ravindra, N. et al. Novel frameshift variant (c.409dupG) in SLC25A38 is a common cause of congenital sideroblastic anaemia in the Indian subcontinent. J. Clin. Pathol. 74, 157–162 (2021).
Zhang, Y. et al. Intron 1 GATA site enhances ALAS2 expression indispensably during erythroid differentiation. Nucleic Acids Res. 45, 657–671 (2017).
Furuyama, K. et al. Arg452 substitution of the erythroid-specific 5-aminolaevulinate synthase, a hot spot mutation in X-linked sideroblastic anaemia, does not itself affect enzyme activity. Eur. J. Haematol. 76, 33–41 (2006).
Oakley, J. H. et al. A synonymous coding variant that alters ALAS2 splicing and causes X-linked sideroblastic anemia. Pediatr. Blood Cancer 69, 3–4 (2022).
Bhatia, P., Singh, A. & Hedge, A. A novel g.55040074delT in ALAS2 gene resulting in a monomeric protein and severe sideroblastic anemia phenotype. J. Pediatr. Hematol. Oncol. 39, 463–465 (2017).
Xu, W. & Zhu, P. G514A mutation in the erythroid-specific 5-aminolevulinate synthase gene leading to X-linked sideroblastic anemia in a family. Blood 100(Suppl), 15B (2002).
Cui, R., Xu, Z., Qin, T., Zhang, Y. & Xiao, Z. Congenital sideroblastic anemia-a new family with identification of K156E mutation of ALAS2 gene and literature review. Zhonghua Xue Ye Xue Za Zhi 35, 142–146 (2014).
Bottomley, S. S., May, B. K., Cox, T. C., Cotter, P. D. & Bishop, D. F. Molecular defects of erythroid 5-aminolevulinate synthase in X-linked sideroblastic anemia. J. Bioenerg. Biomembr. 27, 161–168 (1995).
Lee, P. L. et al. Three kinships with ALAS2 P520L (c. 1559 C → T) mutation, two in association with severe iron overload, and one with sideroblastic anemia and severe iron overload. Blood Cells Mol. Dis. 36, 292–297 (2006).
Tchaikovskii, V., Desnick, R. J. & Bishop, D. F. Molecular expression, characterization and mechanism of ALAS2 gain-of-function mutants. Mol. Med. 25, 4 (2019).
Ducamp, S. et al. Sideroblastic anemia: Molecular analysis of the ALAS2 gene in a series of 29 probands and functional studies of 10 missense mutations. Hum. Mutat. 32, 590–597 (2011).
Bottomley, S. S., Wise, P. D., Wasson, E. G. & Carpenter, N. J. X-linked sideroblastic anemia in ten female probands due to ALAS2 mutations and skewed X chromosome inactivation. Am. J. Hum. Genet. 63(Suppl), A352 (1998).
Cazzola, M. et al. Familial-skewed X-chromosome inactivation as a predisposing factor for late-onset X-linked sideroblastic anemia in carrier females. Blood 96, 4363–4365 (2000).
Brancaleoni, V. et al. X-chromosomal inactivation directly influences the phenotypic manifestation of X-linked protoporphyria. Clin. Genet. 89, 20–26 (2016).
Fratz, E. J. et al. Human erythroid 5-aminolevulinate synthase mutations associated with X-linked protoporphyria disrupt the conformational equilibrium and enhance product release. Biochemistry 54, 5617–5631 (2015).
Balwani, M. et al. Loss-of-function ferrochelatase and gain-of-function erythroid-specific 5-aminolevulinate synthase mutations causing erythropoietic protoporphyria and X-linked protoporphyria in North American patients reveal novel mutations and a high prevalence of X-Lin. Mol. Med. 19, 26–29 (2013).
Whatley, S. D. et al. C-terminal deletions in the ALAS2 gene lead to gain of function and cause X-linked dominant protoporphyria without anemia or iron overload. Am. J. Hum. Genet. 83, 408–414 (2008).
Ducamp, S. et al. A mutation in the iron-responsive element of ALAS2 is a modifier of disease severity in a patient suffering from CLPX associated erythropoietic protoporphyria. Haematologica 106, 2030–2033 (2021).
Astner, I. et al. Crystal structure of 5-aminolevulinate synthase, the first enzyme of heme biosynthesis, and its link to XLSA in humans. EMBO J. 24, 3166–3177 (2005).
Beaumont, C. et al. Mutation in the iron responsive element of the L ferritin mRNA in a family with dominant hyperferritinaemia and cataract. Nat. Genet. 11, 444–446 (1995).
Girelli, D. et al. Molecular basis for the recently described hereditary hyperferritinemia- cataract syndrome: a mutation in the iron-responsive element of ferritin L-subunit gene (the ‘Verona mutation’) [see comments]. Blood 86, 4050–4053 (1995).
Bezerra, G. A. et al. PanDDA analysis group deposition: Crystal Structure of human ALAS2A in complex with Z2856434857. https://doi.org/10.2210/pdb5qqq/pdb (2019).
Šali, A. & Blundell, T. L. Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol. 234, 779–815 (1993).
Bekker, H. et al. GROMACS: A Parallel computer for molecular dynamics simulations. in Physics Computing ’92 (eds. DeGroot, R. & Nadrchal, J.) 252–256 (World Scientific Publishing, 1993).
Schrödinger, L. & DeLano, W. The PyMOL molecular graphics system. at http://www.pymol.org/pymol.
Kent, W. J. et al. The human genome browser at UCSC. Genome Res. 12, 996–1006 (2002).
Schubach, M., Maass, T., Nazaretyan, L., Röner, S. & Kircher, M. CADD v1.7: Using protein language models, regulatory CNNs and other nucleotide-level scores to improve genome-wide variant predictions. Nucleic Acids Res. 52, D1143–D1154 (2024).
Kopanos, C. et al. VarSome: The human genomic variant search engine. Bioinformatics 35, 1978–1980 (2019).
Adzhubei, I. A. et al. A method and server for predicting damaging missense mutations. Nat. Methods 7, 248–249 (2010).
Choi, Y. & Chan, A. P. PROVEAN web server: A tool to predict the functional effect of amino acid substitutions and indels. Bioinformatics 31, 2745–2747 (2015).
Ng, P. C. & Henikoff, S. Predicting deleterious amino acid substitutions. Genome Res. 11, 863–874 (2001).
Acknowledgements
The authors would like to express their gratitude to the patients and their families for their participation in this study. They also thank Alejandro Negro Lacort for his initial technical work and support, Joan Marc Martínez and Eva Quandt for their assistance with human protein overexpression in bacteria, and Pep Clotet’s group for generously providing the anti-GST antibody and pGEX-6P-1 plasmid. Special thanks go to Sarah Ducamp for her invaluable guidance, technical support, protocol sharing, and troubleshooting in the enzymatic activity assays. Finally, the authors appreciate Dr. Blanca Xicoy for her assistance in following up on sample collection and Pau Tomàs for receptioning and processing blood samples at BloodGenetics SL.
Funding
This work was supported by ARETHA grant PID2021122436OB-I00 funded by MCIN/AEI/https://doi.org/10.13039/501100011033 to M.S, by grant PLEC2021-007727/AEI /https://doi.org/10.13039/501100011033/Union Europea NextGenerationEU/PRTR from MCIN to M.S. and by Deutsche Josep Carreras Leukämie-Stiftung grant DJCLS R 14/04 to N.G. and M.S. D.J-S. hold a UIC predoctoral fellowship. X.F.-C. was partially supported by funds provided by the grant PID2021122436OB-I00 funded by MCIN/AEI/https://doi.org/10.13039/501100011033. V.V. was supported by funds provided by APU and ADISCON patient associations, UIC postdoctoral scholarship and by funds provided by RETOS COLABORACION grant RTC2019-007074-1 from MCIN/AEI/https://doi.org/10.13039/501100011033 from Spanish Ministry of Science and Innovation (MICINN).
Author information
Authors and Affiliations
Contributions
D.J.S. designed and performed research, analyzed and plotted the data, made figures and wrote the manuscript. M.R. performed mutagenesis and cloning of all ALAS2 variants. X.F.-C. and V.V. performed initial research and supported experimental design. M.O. performed protein modelling and wrote the manuscript. N.G. and U.G. provided patient samples and revised the manuscript. M.M. and I.H.R. provided patient clinical data, background and samples, participated in patient care, contacted patients and revised the manuscript. S.P.-M. and C.T. provided funding. M.S. designed research, wrote manuscript, provided reagents, lab facilities and funds. All authors have read and agreed to the published version of the manuscript.
Corresponding author
Ethics declarations
Competing interests
Authors declare no conflict of interest.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Jové-Solavera, D., Rámila, M., Ferrer-Cortés, X. et al. The role of genetic testing in accurate diagnosis of X-linked sideroblastic anemia: novel ALAS2 mutations and the impact of X-chromosome inactivation. Sci Rep 15, 11843 (2025). https://doi.org/10.1038/s41598-025-95590-x
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-95590-x
