
Abstract
NiO–Mn3O4 ribbons were synthesized through electrospinning and subsequently compared with NiO nanoparticles and Mn3O4 octahedral particles to evaluate their optical and electrochemical properties. XRD analysis confirmed the presence of cubic and tetragonal phases of NiO and Mn3O4, respectively, within the nanocomposite. UV–Vis diffuse reflectance spectroscopy (DRS) revealed a bandgap of 3.53 eV for the nanocomposite, while photoluminescence emission quenching indicated an enhancement in surface defects. The ribbons exhibited superior electrochemical performance, achieving a specific capacitance of 372 F g−1 at a current density of 1 A g−1, along with 94% capacitance retention after 3000 cycles at 7 A g−1. Furthermore, the assembled NiO–Mn3O4//AC asymmetric supercapacitor device exhibited a maximum energy density of 40 Wh kg−1 at a power density of 2400 W kg−1. These findings suggest that NiO–Mn3O4 ribbons hold significant promise for high-performance energy storage devices.
Similar content being viewed by others
Introduction
During the last decade, with the rapid reduction of fossil fuels, global warming, and the worsening of environmental pollution, much research has been conducted on developing various types of sustainable and renewable energy storage devices. Presently, batteries and electrochemical capacitors are extensively used technologies that allow for the possibility of converting and storing energy in various applications1,2,3. Supercapacitors are categorized based on how they store charge into three groups: electrical double-layer capacitors, pseudocapacitors, and battery-type supercapacitors. Battery-type supercapacitors combine the advantages of electrical double-layer capacitors and pseudocapacitors, providing high charge storage and maintaining cyclic stability, which results in both high energy and power4,5,6,7. Supercapacitors are made of different materials for their application and capacity. The constituent materials of the electrodes in all types of supercapacitors are different based on the application and generally contain conductive polymers, metal oxides, and carbon materials. The material used as an electrochemical supercapacitor electrode must have characteristics such as high surface area, capacitive behavior, electrochemical stability, and high conductivity8,9,10.
Transition metal oxides (TMOs) are recognized for their ability to store energy through redox reactions. When these oxides are combined to create nanocomposites, their properties are significantly enhanced, depending on the specific ratios of each oxide involved, making them highly valuable for various applications, including batteries, fuel cells, and supercapacitors. Nickel oxide (NiO) and manganese oxide (Mn3O4) are among the TMOs that exhibit outstanding properties for different applications. NiO and Mn3O4 are notable transition metal oxides with wide-ranging applications. NiO is a p-type semiconductor with a bandgap of 3.6–4 eV, recognized for its structural integrity, environmental friendliness, and affordability. It is utilized in various domains, including gas sensors, battery cathodes, and catalysts, and shows promise in supercapacitors and solar cells due to its high capacity and stability11,12,13. Enhancements like nickel vacancies and oxygen interstitials can overcome its conductivity limitations. Wei et al. synthesized flower-like porous NiO utilizing a hydrothermal method, which delivered a specific capacitance of 546.53 F g−1 at 2 A g−1. The assembled asymmetric supercapacitor achieved a high energy density of 29.7 Wh kg−1, maintaining 83.2% of the initial capacity over 3000 cycles at 3 A g−114. Mn3O4, known for its unique valence state and wide bandgap, is thermodynamically stable and conductive. Its spinel structure, featuring Mn2+ and Mn3+ ions, contributes to its versatility15,16,17. However, it faces challenges such as low ion diffusion, cycling stability, and conductivity, which restricts its broader application. Hydrothermally synthesized spinel Mn3O4 exhibited a high specific capacitance of 352 F g−1 at a current density of 0.5 A g−118. Metal oxide composites can enhance the supercapacitive performance of their pure counterparts. Recent studies have successfully electrodeposited a composite electrode made of MnO2–NiO using the chrono-potentiometry method, which shows improved specific capacitance compared to MnO2 alone. The highest specific capacitance recorded is 435 F g−1 in a 0.5 M KOH electrolyte19. Kumar et al. synthesized a ternary hybrid composite by functionalizing reduced graphene oxide sheets with NiO/MnO2 nanoparticles using microwave irradiation for uniform distribution. The high porosity of the reduced graphene oxide enhanced diffusion rates and energy storage capacity. The composite achieved a peak specific capacity of 165.7 mA h g−1 and maintained about 83.2% cyclic stability after 2000 cycles20. He et al. developed a NiO@Mn3O4 composite supported by reduced graphene oxide (rGO) through a one-step hydrothermal method. This NiO@Mn3O4/rGO composite demonstrates impressive electrochemical performance, achieving a specific capacitance of 533.97 F g−1 at 1 A g−1. After 2000 charge–discharge cycles, it retains 75% of its capacitance even at a higher current density of 10 A g−14.
Nanofibers produced through electrospinning have established themselves as compelling materials for energy storage applications, particularly in supercapacitors. This technique allows for the fabrication of ultra-fine fibers with high surface area and tunable porosity, which are critical for enhancing charge storage capacity and improving electrochemical performance. The unique morphology of electrospun nanofibers facilitates rapid ion transport and efficient charge transfer, making them ideal candidates for active materials in supercapacitor electrodes. Recent advancements in the functionalization of nanofibers, including doping and hybridization strategies, have led to substantial improvements in energy density, power density, and cycling stability21,22,23.
This study focuses on synthesizing a novel NiO–Mn3O4 nanocomposite with a ribbon-like morphology using sol–gel electrospinning followed by calcination. We thoroughly investigated the structural, morphological, optical, and electrochemical characteristics of the composites using diverse analytical methods. The results revealed that the NiO–Mn3O4 nanocomposite, when used as a supercapacitor electrode material, exhibited high specific capacitance and sustained cycling stability throughout extended charge/discharge cycles.
Experimental
Materials
All reagents and chemicals including nickel(II) nitrate hexahydrate (Ni(No3)2·6H2O, Merck), manganese nitrate tetrahydrate (Mn(NO3)2·4H2O, Merck), polyvinylpyrrolidone (PVP, MW = 1,300,000, Sigma Aldrich), N, N-dimethylformamide (DMF, 99.8%, Merck) and ethanol (C2H5OH, 99.9%, Samchun), carbon black (Super P, Merck), polyvinylidene difluoride (PVDF, Kynar HSV900, Arkema), N-methyl-2-pyrrolidone (NMP, > 99%, Merck), and potassium hydroxide (KOH, > 85%, chem-lab) were used without further purification.
Synthesis of NiO, Mn3O4 and NiO–Mn3O4 nanocomposite
Nickel oxide (NiO), manganese oxide (Mn3O4), and nickel oxide-manganese oxide (NiO–Mn3O4) nanocomposites were synthesized using the sol–gel electrospinning technique. The preparation of these materials involves several key steps.
To produce the nanofibers, 2 mmol of Ni(NO3)2·6H2O (or Mn(NO3)2·4H2O) is first dissolved in 5 mL of ethanol and stirred at 100 rpm for 20 min. Following this, 5 mL of Dimethylformamide (DMF) is added, and the mixture is stirred for an additional hour. Next, 1.5 g of Polyvinylpyrrolidone (PVP) is gradually incorporated into the solution while stirring continuously for 20 h to achieve a uniform and viscous sol. This prepared sol is then subjected to electrospinning under the following conditions: a voltage of 17 kV, a distance of 10 cm from the needle tip to the collector plate, and a flow rate of 0.4 mL/h. For the fabrication of the NiO–Mn3O4 nanocomposite, the same procedure is followed, using two separate sols containing 1 mmol of each metal nitrate. These sols are combined and stirred for an additional hour to ensure thorough mixing. Finally, the resulting nanofibers are dried at 100 °C for 12 h to produce PVP/Ni(NO3)2, PVP/Mn(NO3)2, and PVP/Ni(NO3)2-Mn(NO3)2 polymeric nanofibers. These nanofibers are then calcined at 500 °C with a heating rate of 2 °C/min for 1 h to eliminate any residual polymer. A schematic representation of the preparation process for NiO–Mn3O4 nanofibers is illustrated in Fig. 1.

Schematic representation of NiO–Mn3O4 nanocomposite synthesized via electrospinning method.
Characterization
The structural properties of the samples were studied by X-ray diffraction (XRD, PANalytical X’Pert Pro) analysis using Cu-Kα radiation with a wavelength of 0.15406 nm. Nitrogen adsorption–desorption tests at 77 K were performed on the prepared samples to analyze their specific surface areas by the Brunauer–Emmett–Teller method (BET, Belsorp-mini). The Fourier-transform infrared (FTIR) spectra of the samples were investigated using the Alpha Bruker spectrometer in the wavelength range of 400–4000 cm−1. The samples were analyzed for their morphology using a field emission scanning electron microscope (FESEM, MIRA3, TESCAN-XMU). Additionally, the microscope was equipped with energy dispersive spectroscopy (EDS) for further analysis. Diffuse reflectance spectra (DRS) were performed with a spectrophotometer Scinco 4100 in the wavelength range of 200–800 nm. Photoluminescence spectra of the samples were measured (Perkin Elmer, LS55).
Electrochemical measurement
Working electrodes for electrochemical testing were prepared by mixing an appropriate volume of N-methyl pyrrolidinone (NMP) with carbon black (10 wt%), polyvinylidene fluoride (10 wt%) and 80 wt% electroactive materials (NiO, Mn3O4, NiO–Mn3O4 nanocomposite). Then, they were uniformly coated on the surface of nickel foam (NF, 1 × 1 cm2) and dried in an oven at 80 °C for 12 h to remove the remaining solvent. The active mass on the surface of the fabricated working electrodes (NiO/NF, Mn3O4/NF, NiO–Mn3O4 nanocomposite/NF) is about 1 mg. The electrochemical tests were performed using a device (Potentiostat–Galvanostat, Zahner–Zennium). Cyclic voltammetry (CV) and galvanostatic charge/discharge (GCD) measurements were evaluated in a three-electrode cell system, using Ag/AgCl as the reference electrode, Pt wire as the counter electrode, and electrodes (NiO/NF, Mn3O4/NF, NiO–Mn3O4 nanocomposite/NF) as the working electrode in 3 M KOH aqueous electrolyte at room temperature. Electrochemical impedance spectroscopy (EIS) was measured in the frequency range of 0.01–100 kHz, using a 5 mV amplitude.
Results and discussion
XRD analysis
Figure 2a shows the XRD pattern for the prepared samples of NiO, Mn3O4, and NiO–Mn3O4 nanocomposite. For pure NiO, the diffraction peaks located at 2θ values of 37.44, 43.47, 57.57, 62.98, and 79.56° are associated with the crystal planes (111), (200), (220), (311), and (222), respectively. The obtained results correspond to the cubic structure of NiO (JCPDS Card No. 01-075-0269) and the Fm-3 m space group24. In addition, the peaks marked with the symbol (●) belong to Ni metal (JCPDS Card No. 04-0850)25. The diffraction pattern of pure Mn3O4 sample with peaks at 2θ values of 18.04, 28.94, 31.06, 32.36, 36.16, 38.03, 44.50, 45.37, 49.87, 50.75, 53.94, 56.07, 58.60, 59.98, 64.73, 65.83, 67.67, 69.74, 74.22, 76.66, and 77.55°, corresponds to the planes (101), (112), (200), (103), (211), (004), (220), (213), (204), (105), (312), (303), (321), (224), (400), (323), (411), (305), (413), (422), and (404), respectively. This pattern aligns with the tetragonal phase of Mn3O4 (JCPDS Card No. 24-0734) and the I41/amd space group26,27. In the NiO–Mn3O4 nanocomposite diffraction pattern, the peaks marked with the symbols (♦) and (♣) correspond to the NiO and Mn3O4 phases, respectively. The average crystallite size (D) and the lattice strain (ɛ) of the samples were estimated using the Williamson-Hall equation16:

(a) XRD patterns and (b) Williamson-Hall diagram for NiO, Mn3O4, and NiO–Mn3O4 nanocomposite.
The variables β, k, λ, and θ stand for the full-width at half-maximum (FWHM), a constant equal to 0.9, incident X-ray wavelength measuring 1.5406 Å and Bragg diffraction angle, respectively. The plot of βcosθ versus 4sinθ is shown in Fig. 2b. The average crystal sizes for NiO, Mn3O4, and NiO–Mn3O4 nanocomposite samples were 35.1, 60.5, and 30.2 nm, respectively, while the lattice strains were 0.0012, 0.00129, 0.00204, respectively.
BET analysis
To thoroughly examine the porous architectures of the samples for enhanced insights, their specific surface areas and pore dimensions were quantitatively assessed employing N2 adsorption–desorption analyses. The nitrogen adsorption–desorption isotherm corresponding to samples is graphically represented in Fig. 3. This isotherm distinctly illustrates an IV-type curve, characterized by a prominent hysteresis loop that spans a spectrum of relative pressures from P/P0 0 to 1. The specific surface area values obtained for the samples are 22.32 m2 g−1 for NiO, 44.66 m2 g−1 for Mn3O4, and 70.81 m2 g−1 for the NiO–Mn3O4 nanocomposite, respectively. The relative BJH pore size distribution diagrams of the NiO–Mn3O4 nanocomposite are depicted in the inset in Fig. 3. The NiO–Mn3O4 composite exhibited a concentration of pore sizes at 16.97 nm. This finding elucidated that synergistic integration of Mn3O4 and NiO within the composite resulted in an enhancement of porosity. The BET analysis confirmed the mesoporous characteristics, indicating that the porosity of the electrode materials provides the beneficial pathways for the intercalation–deintercalation processes of electrolyte ions at the interface between the electrode and the electrolyte.

Nitrogen adsorption–desorption isotherm for samples (the inset shows a plot of the pore size distribution for NiO–Mn3O4 nanocomposite).
FESEM analysis
Figure 4 shows the FESEM image of NiO, Mn3O4, and NiO–Mn3O4 nanocomposite. Figure 4a–c depicts the morphology of as-spun samples. According to the images, the surface of the nanofibers is entirely smooth and notably thick prior to calcination. Figure 4d,g shows that the post-calcination of PVP/Ni(NO3)2 composite at various magnifications, exhibiting a cauliflower-like morphology of the NiO sample. The transformation of PVP/Mn(NO3)2 nanofibers into polyhedral Mn3O4 particles following calcination can be ascribed to various factors associated with the heat treatment process, as demonstrated in Fig. 4e,h. During sintering, particles may coalesce and densify, forming larger polyhedral particles. The elevated temperatures may induce surface tension-driven reshaping of fibers into configurations that lower surface energy, leading to polyhedral shapes. It’s also possible that the initial nanofiber configuration was not fully stable, and calcination simply reveals a more stable polyhedral particle structure. The transformation from nanofibers to octahedral particles in the Mn3O4 sample post-calcination can be elucidated by the synergistic roles of PVP, which serves as a reducing, stabilizing, and shape-directing agent28. PVP’s affinity for specific crystal facets enables it to act as a shape-directing agent. During electrospinning, PVP imparts viscosity to the solution, aiding in the formation of the initial nanofibers. Upon calcination, the decomposition of PVP may lead to the reorganization of manganese ions. These ions tend to align along crystallographic planes with lower surface energies, culminating in the emergence of octahedral structures. Concurrently, the manganese ions may engage in self-assembly, a process steered by the reduction of the system’s free energy, favoring the creation of thermodynamically stable, well-defined octahedral shapes. Similar morphologies have been reported for manganese oxide samples synthesized with the assistance of polymers, indicating a common influence of such organic compounds on the crystal growth and final shape of the nanoparticles29,30. Figure 4f,i demonstrates the ribbon-like morphology of the NiO–Mn3O4 nanocomposite, composed of pyramidal particles. The synergistic integration of Mn3O4 and NiO within the composite enhances the thermal stability of the nanofibers. This effect provides structural reinforcement and prevents the fibers from collapsing or converting into particles. In such composites, the overall surface energy is typically lower than that of the individual components, which reduces the driving force for morphological changes from fibers to particles.

FESEM images of as-spun (a) PVP/Ni(NO3)2, (b) PVP/Mn(NO3)2, and (c) PVP/Ni(NO3)2-Mn(NO3)2 nanocomposite, (d–i) after calcination at various magnification (Images related to each example are arranged below its as-spun counterpart), (j) EDS spectra NiO–Mn3O4 nanocomposite after calcination (inset displays the atomic percentage of elements), (K) High magnification FESEM image of NiO–Mn3O4 nanocomposite.
Figure 4j shows the EDS spectra of NiO–Mn3O4 nanocomposite. The results approve the presence of nickel, manganese, and oxygen within the configuration of the prepared sample. The table in Fig. 4j provides the atomic percentages of these elements. Figure 4k presents a high-magnification FESEM image of the NiO–Mn3O4 nanocomposite, showcasing pyramidal particles that have developed on a fibrous structure.
FTIR analysis
Figure 5a represents the FTIR spectrum of NiO, Mn3O4, and NiO–Mn3O4 nanocomposite, showing distinct absorption bands at various wavenumbers. The peaks appearing at 3430 and 1632 cm−1 are associated with the stretching vibrations of O–H and the H–O–H bending vibrations of water molecules that have been absorbed from the environment, respectively31,32. Additionally, the bands created around 1728 and 1279 cm−1 are attributed to the stretching vibrations of C=O and C–O bonds due to CO2 adsorption on the surface of the samples33,34. The absorption band at 1127 cm−1 corresponds to C–O stretching vibrations35, while the adsorption bands located at 750 and 85 cm−1 bands in NiO can be attributed to NO3− from nitrates32. Moreover, the bands at 2959, 2928, and 2863 cm−1 represent stretching vibrations of the C–H groups36. The absorption band at 1461 cm−1 indicates the presence of CH2 bending vibration37.

(a) FTIR spectra for NiO, Mn3O4, and NiO–Mn3O4 nanocomposite, (b) UV–Vis DRS and (c) Tauc plot of samples, (d) Photoluminescence spectra of NiO, Mn3O4, and NiO–Mn3O4 nanocomposite.
In the NiO sample, the distinct band at 438 cm−1 is assigned to the stretching vibrations of the Ni–O bond32. In the Mn3O4 sample, two sharp absorption bands at 613 and 505 cm−1 are linked to stretching states between the Mn–O tetrahedral and octahedral sites of the Mn3O4 nanoparticles38. The band in the specified region corresponds to the vibration of metal oxide. In NiO–Mn3O4 nanocomposite, three characteristic absorption bands at 605, 511, and 458 cm−1 can be attributed to the Ni–O bond or O–Mn–O bond39.
DRS analysis
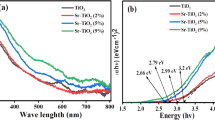
Figure 5b shows the UV–Vis diffuse reflectance spectra of NiO, Mn3O4, and NiO–Mn3O4 nanocomposite across the wavelength range of 300–800 nm. The optical bandgap energy (Eg) of the specimens was determined using the Tauc equation40:
where photon energy, absorption coefficient, and constant are the hν, α, and A, respectively, and n is equal 2 for a direct band transition. The Kubelka–Munk function F(R) is proportional to the sample absorption coefficient and is estimated using the following equation41:
where R is the diffuse reflectance of samples. According to Fig. 5c, the bandgap energy of the samples in the permissible direct transition was calculated by extrapolating the linear part of the curve (F(R) hν)2 versus hν. The estimated band gap of NiO was 3.25 eV, while Mn3O4 has higher bandgap energy of 3.60 eV. The difference in bandgap energies can be attributed to the different electronic structures and crystal field effects in these two metal oxides. For the NiO–Mn3O4 nanocomposite, the bandgap energy is 3.53 eV, which is between the bandgap energies of the individual components. This suggests that the electronic structures of NiO and Mn3O4 are interacting in such a way that the bandgap of the composite is altered42. Also, the observed energy band gap reduction in nanocomposites can be attributed to the emergence of exchange and correlation energies among the electron–electron, and hole–hole interactions during the formation of the nanocomposites. This phenomenon is indicative of strong electronic interactions that facilitate charge transfer processes within the composite material43. As such, the reduction in band gap not only signifies enhanced electronic coupling but also suggests improved charge transport properties, which are essential for applications in supercapacitor devices.
PL analysis
The photoluminescence (PL) spectrum provides information about the radiation recombination taking place in the material. The PL spectra of the NiO, Mn3O4, and NiO–Mn3O4 nanocomposite samples in the range of 200–800 nm, with an excitation wavelength of 270 nm are shown in Fig. 5d. For the NiO sample, the emission peak observed at 366 nm is linked to the near band-edge emission (NBE), stemming from exciton recombination processes occurring between the conduction and valence bands44,45. The peak at 524 nm is likely due to oxygen-related defects46. In contrast, the Mn3O4 sample exhibits an emission peak at 383 nm, which can be attributed to the recombination of free excitons through the exciton-exciton collision process at the near band-edge47. Owing to a reduced defect density or a lower density of states at the band edge, Mn3O4 demonstrates a diminished NBE emission PL intensity compared to NiO. The observed decrease in PL intensity within the NiO–Mn3O4 nanocomposite spectrum may be linked to a reduced electron–hole recombination rate48. Non-radiative recombination centers created by surface defects in the nanocomposite are probably responsible for the quenching of photoluminescence emissions. These defects may serve as trapping sites for charge carriers, causing a decrease in the radiative recombination efficiency and subsequent quenching of PL intensity. Moreover, heterojunctions formed at the NiO and Mn3O4 interface within the nanocomposite introduce alternative recombination pathways for charge carriers, potentially resulting in further PL quenching compared to the individual oxide components. Consequently, PL quenching in the NiO–Mn3O4 nanocomposite indicates an increase in surface defects, which can act as active sites for electrochemical reactions, enhancing the material’s performance in energy storage applications.
Electrochemical analysis
The electrochemical performance of NiO, Mn3O4, and NiO–Mn3O4 nanocomposite was examined using cyclic voltammetry and galvanostatic charge–discharge analysis. Figure 6a–c shows the cyclic voltammetry curves of electrodes made of NiO, Mn3O4, and NiO–Mn3O4 nanocomposite at different scan rates of 10, 30, 50, and 80 mV s−1 within the potential window of 0–0.5 V. The shape of the curves indicates the quasi-capacitive behavior of the electrodes. In the CV curves, two distinct redox peaks are detected due to Faradaic electrochemical reactions. The corresponding reaction equations in the KOH electrolyte are as follows4,49:

CV curves at different scan rates of (a) NiO, (b) Mn3O4, (c) NiO–Mn3O4 nanocomposite, (d) CV profiles of the electrodes recorded at a scan rate of 80 mV s−1.
As can be seen, with increasing scanning rate, the redox currents increase. Concurrently, the position of the anode peaks shifts to a more positive potential, while the cathode peaks shift to a more negative potential. This shift may be attributed to an increase in ohmic resistance and polarization during the reaction50. Despite the changes in scan rate, the symmetrical nature of the curves remains unchanged, suggesting that the reactions are reversible and that the redox processes at the surface of the sample are rapid51.
The enclosed area under the CV curves indicates the amount of charge stored, which is directly related to the capacitance. The specific capacitance for the electrodes is obtained from the CV curves by applying the following equation52:
where, Cs is the specific capacitance, \(\int idV\) integral area beneath the curve, m is the mass of the electrode’s active material, υ indicates the scan rate, and ∆V signifies the potential window.
The CV curves of the NiO, Mn3O4, and NiO–Mn3O4 nanocomposite electrodes at a scan rate of 80 mV s−1 are shown in Fig. 6d. The specific capacitance measured at scan rates of 10, 30, 50, and 80 mV s−1 was approximately 334, 298, 263, and 205 F g−1 for NiO; 402, 343, 323, and 275 F g−1 for Mn3O4; and 472, 441, 408, and 356 F g−1 for the NiO–Mn3O4 composite. The higher specific capacitance of the NiO–Mn3O4 nanocomposite, in comparison to the individual NiO and Mn3O4 samples, suggests that it can store more charge within the electrode, thereby exhibiting superior capacitive performance.
Figure 7a shows the variations in specific capacitance for NiO, Mn3O4, and NiO–Mn3O4 nanocomposite electrodes at different scan rates. This trend may be attributed to the restricted ion distribution within the electrode surface cavities during higher scan rates53. Using cyclic voltammetry, the change in current response to variations in sweep rate can offer insights into whether the redox reaction is controlled by diffusion. Established techniques are available to distinguish between diffusion-controlled and surface processes in electrode materials. The total capacitance of the electrode consists of two parts: rapid electrochemical processes, such as ion adsorption/desorption electric double layer capacitance process (EDLC), and fast Faradaic reactions of redox species. The second part is affected by ion diffusion within the electrode material. Identifying the dominant process is crucial for accurately interpreting the performance of the electrode material54. During a linear voltage scan at a constant rate, the electrode current follows a power law relationship55:

(a) Specific capacitance vs different scan rates, (b) plot of log i vs log ν for NiO–Mn3O4 nanocomposites electrode (inset: linear fitting of k1 and k2 parameters), (c) CV curve at a scan rate of 30 mV s−1 (Shaded region shows the diffusive contribution), (d) percentage of Diffusive and capacitive contribution at various sweep rates.
Here, ‘i’ stands for the current, ‘ν’ indicates the sweep rate, while ‘a’ and ‘b’ are constants; specifically, ‘b’ equals 1 for EDLC processes and 0.5 for reactions governed by diffusion. For NiO–Mn3O4 supercapacitor tested in KOH electrolyte, ‘b’ value of approximately 0.77 (0.5 < b < 1) suggests a combination of battery-type and capacitive mechanisms, which we refer to as supercapattery behavior. This relationship can be expressed as55:
where “i” is the current at a set voltage, and “k” and “k′” represent constants. By plotting i/ν1/2 against ν1/2, we can determine the slope k. By calculating the values of “k” and “k′”, we can graph the diffusion contribution (shaded region), as illustrated in Fig. 7c at a scan rate of 30 mV s−1. Diffusion-controlled and capacitive-controlled contribution percentages at sweep rates from 20 to 80 mV s−1 are depicted in Fig. 7d. At slower scan rates, the surface contribution is reduced compared to faster scan rates. As the scan rate increases, the diffusion-controlled contribution declines slightly. Nevertheless, even at high scan rates, the diffusion-controlled component remains more stable and substantial compared to the capacitance contribution. This indicates that the significant capacitance of the NiO–Mn3O4 composite electrode mainly originates from battery behavior56.
To more precisely assess the electrochemical performance and cyclic stability of NiO, Mn3O4, and NiO–Mn3O4 nanocomposite electrodes, GCD analysis was performed within a potential range of 0–0.5 V at current densities of 1, 3, 5, 7, and 10 A g−1. The electrodes’ charge–discharge curves at different current densities are presented in Fig. 8a–c. The nonlinear and relatively symmetrical forms of the charge–discharge curves underscore the battery-like nature, which is consistent with the oxidation–reduction peaks observed in CV curves50.

GCD curves of (a) NiO, (b) Mn3O4, and (c) NiO–Mn3O4 nanocomposite in various current density, (d) comparative GCD curve of three electrodes in current density 1 A g−1, (e) cycling stability of electrodes, (f) Nyquist plots of the electrodes (The inset shows the equivalent circuit and a close-up view of the high-frequency region).
The specific capacitance (Cs) and specific capacity (Q) of electrodes were calculated based on GCD curves using Eqs. (9) and (10).
Here, I, ∆t, m, and ∆V refer to the current, discharge time, and mass of active material in the electrode and potential range, respectively57,58,59.
The GCD curves of NiO, Mn3O4, and NiO–Mn3O4 nanocomposite electrodes at a current density of 1 A g−1 are displayed in Fig. 8d. The specific capacitance values obtained for the samples are 230 F g−1 for NiO, 246 F g−1 for Mn3O4, and 372 F g−1 for the NiO–Mn3O4 nanocomposite, as shown in Table 1. Notably, the NiO–Mn3O4 nanocomposite revealed a longer discharge time than the pure samples, suggesting a higher specific capacitance, which can be attributed to the combined structure and synergistic effect between NiO and Mn3O460.
The changes in specific capacitance of NiO, Mn3O4, and NiO–Mn3O4 nanocomposite electrodes according to different current densities are shown in Table 1. The specific capacitance values of the electrodes decrease with increasing current density. At lower current densities, electrolyte ions have enough time to engage in charge exchange at the electrode–electrolyte interface, allowing for a thorough distribution across all active sites of the electrode material. In contrast, higher current densities may lead to underutilization of the electrode materials due to limited ion diffusion. Additionally, the GCD curves reveal an escalation in voltage drop corresponding with the increase in current density, which contributes to the reduction in specific capacitance at elevated current densities61.
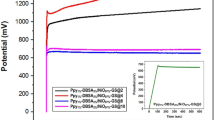
Figure 8e illustrates the cycling stability of electrodes at a current density of 7 A g−1 over 3000 cycles. The NiO–Mn3O4 nanocomposite electrode exhibits a capacitance retention of about 94%, higher than the Mn3O4 (91%) and NiO (89%) electrodes. This indicates commendable cyclic stability, likely due to the reduction of impurities and enhanced electrochemical characteristics, including better charge transfer and structural integrity, resulting in superior performance and long-term cycling stability23.
To more accurately study the charge-transfer and capacitive nature of NiO, Mn3O4, and NiO–Mn3O4 nanocomposite electrodes, electrochemical impedance spectroscopy (EIS) was conducted over a frequency range of 100 kHz to 0.01 Hz in a 3 M KOH electrolyte solution. Figure 8f shows the Nyquist plots for each of the electrode materials, with inset plots highlighting the data in the high-frequency domain. The intercept found on the x-axis is indicative of the equivalent series resistance (Rs), which comprises the cumulative effect of solution (KOH) resistance, working electrode resistance, and the contact resistance between the electrode material and current collector material. The diameter of the semicircle in the high-frequency range is directly correlated to the charge transfer resistance (Rct) of the electrode, which plays a crucial role in governing the movement of charge within the electrode–electrolyte interface. On the other hand, the Warburg impedance (Ws) associated with electrolyte diffusion is depicted by the upright line in the low-frequency range62,63,64. By employing a suitable equivalent circuit model using Z-View software (as shown in the inset of Fig. 8f), it is evident that the value of Rct is smaller for the NiO–Mn3O4 nanocomposite electrode (0.50 Ω) compared to both the Mn3O4 electrode (17.38 Ω) and the NiO electrode (23.76 Ω). This reduction in Rct observed in the NiO–Mn3O4 nanocomposite electrode can be attributed to the highly effective interaction between the electrolyte and the electrode material, thereby enhancing the accessibility of electrochemically active sites and promoting efficient charge transfer mechanisms65,66. As delineated by the equivalent circuit analysis, the NiO–Mn3O4 nanocomposite showcases the lowest Rs value (1.83 Ω), which is notably lower than the corresponding values for both the Mn3O4 (1.97 Ω) and NiO (2.48 Ω) electrodes. Taken together, all these findings unequivocally support the assertion that the NiO–Mn3O4 nanocomposite electrode demonstrates superior specific capacitance compared to all the tested samples.
Notwithstanding, given that the magnitude of capacitance plays a critical role in determining the energy density of the resultant device, the NiO–Mn3O4 electrode has been chosen to assess the electrochemical properties of the devices. The efficacy of supercapacitors is influenced not only by the composition of the positive electrode but also significantly by the composition of the negative electrode. Activated carbon (AC) is applied on the surface of nickel foam to fabricate a flexible asymmetric device. Moreover, a charge balancing procedure between the positive and negative electrodes is implemented to ensure that the device demonstrates optimal performance. The CV curves for both the positive and negative electrodes, in addition to the device, assessed at a scan rate of 30 mV s−1, are depicted in Fig. 9a. The voltage range for AC extends from − 1 to 0 V, thereby affording a considerable voltage range for the device. CV and GCD measurements at varying voltages are performed to ascertain a suitable voltage window for the device, indicating that the voltage window can achieve up to 1.6 V.

(a) CV curves of NiO–Mn3O4 and AC at 30 mV s−1 in the three-electrode system, (b) CV and (c) GCD curves of the NiO–Mn3O4//AC ASC device in the two-electrode system, (d) Cycling stability performance of the NiO–Mn3O4//AC ASC at 7 A g−1, (e) Specific capacitance and coulombic efficiency of the NiO–Mn3O4//AC ASC at different current densities, (f) Ragone plot.
Figure 9b elucidates that the CV curves of the apparatus manifest a quasi-rectangular shape at scan rates varying from 10 to 80 mV s−1, which can be ascribed to the dominant pseudocapacitive nature of the device. The specific capacitances recorded across different scan rates are 209, 183, and 114 F g−1, respectively, showcasing negligible deterioration despite a tenfold escalation in the scan rate, thus signifying the device’s outstanding rate capability. Figure 9c displays the GCD curves of the device at current densities spanning from 1 to 10 A g−1, with specific capacitances documented at 120, 115, 106, 84, and 62 F g−1 corresponding to the current densities of 1, 3, 5, 7, and 10 A g−1, respectively. In Fig. 9d, the device exhibits exceptional cycling stability, demonstrating a capacitance retention of 89.75% after 5000 cycles at a current density of 7 A g−1. The coulombic efficiency of the electrode (ηt) was assessed utilizing Eq. (11) based on charge and discharge times (tc and td)67, and the findings are illustrated in Fig. 9e. The derived coulombic efficiency is estimated to be approximately 93.1%, corroborating the device’s notable reversibility.
Taking into account the measured Cs of the device across a range of current densities, both power density and energy density are computed employing the equations outlined below67.
E (Wh kg−1) represents the energy density, whereas P (W kg−1) denotes the power density. A Ragone plot is utilized to elucidate the correlation between power density and energy density within the asymmetric storage device. The energy and power density values for the NiO–Mn3O4//AC electrode were calculated using Eqs. (12) and (13). The energy densities of 40, 38, 35, 29, and 22 Wh kg−1 were obtained at power densities of 2400, 3800, 6300, 7450, and 8800 W kg−1, respectively, and at current densities of 1, 3, 5, 7, and 10 A g−1. The corresponding Ragone plots are provided in Fig. 9f. The enhanced energy density of the NiO–Mn3O4 //AC system is ascribed to the synergistic interaction present between the NiO and Mn3O4, both of which exhibit a high specific capacitance. Even at an elevated current density of 10 A g−1, the energy density retains a value of 22 Wh kg−1 while simultaneously achieving a peak power density of 8800 W kg−1. The comparative analysis of other literature with the present study is listed in Table 2, indicating that the performance is related to the composition, morphology of the samples, as well as the electrolyte used.
Conclusion
The NiO–Mn3O4 ribbons, embedded with pyramid-shaped particles, were synthesized using the sol–gel electrospinning method. Their optical and electrochemical properties were compared to those of NiO nanoparticles and Mn3O4 octahedral particles. X-ray diffraction confirmed the presence of cubic and tetragonal phases of NiO and Mn3O4, respectively. UV–Vis diffuse reflectance spectroscopy measurements indicated that the bandgap energy of the NiO–Mn3O4 nanocomposite is 3.53 eV, positioned between the bandgap energies of its individual components. The observed quenching of PL emission in the NiO–Mn3O4 nanocomposite is likely attributed to surface defects, traps, or non-radiative recombination centers. The NiO–Mn3O4 ribbons exhibited remarkable electrochemical performance, achieving a maximum specific capacitance of 372 F g−1 at a current density of 1 A g−1. Notably, after 3000 cycles at a current density of 7 A g−1, the specific capacitance retention of NiO–Mn3O4 nanocomposite significantly improved to 94% compared to individual components. Additionally, the assembled NiO–Mn3O4//AC asymmetric supercapacitor device reached a maximum energy density of 40 Wh kg−1 at a power density of 2400 W kg−1. NiO–Mn3O4 electrode with safe and suitable electrochemical performance is promising for practical application in energy storage devices and might play an important role in renewable energy, potentially reducing pollution and decreasing the consumption of hydrocarbon fuels. We hope that this work can open up new possibilities for exploring novel binary transition metal oxides as electrode materials for application in the energy storage field.
Data availability
All data generated or analyzed during this study are included in this published article, and the datasets used and analyzed during the current study are available from the corresponding author upon reasonable request.
References
Senthilkumar, V. et al. NiO nanoarrays of a few atoms thickness on 3D nickel network for enhanced pseudocapacitive electrode applications. J. Power Sources. 303, 363–371. https://doi.org/10.1016/j.jpowsour.2015.11.034 (2016).
Feng, X. et al. Construction of carnations-like Mn3O4@NiCo2O4@NiO hierarchical nanostructures for high-performance supercapacitors. Electrochim. Acta. 308, 142–149. https://doi.org/10.1016/j.electacta.2019.04.048 (2019).
Bejjanki, D. & Puttapati, S. K. Easy synthesis of NiO-Mn2O3@reduced graphene oxide ternary composite as electrode material for supercapacitor application. J. Electron. Mater. 52, 4729–4737. https://doi.org/10.1007/s11664-023-10436-4 (2023).
He, X., Cheng, Y., Qi, H. & Zhang, Y. One-step hydrothermal synthesis of bimetallic oxides (NiO@ Mn3O4) supported on rGO: A highly efficient electrode material for supercapacitors. Electrochim. Acta. 388, 138609. https://doi.org/10.1016/j.electacta.2021.138609 (2021).
Yuan, Y. F. et al. Freestanding hierarchical NiO/MnO2 core/shell nanocomposite arrays for high-performance electrochemical energy storage. Electrochim. Acta. 227, 303–309. https://doi.org/10.1016/j.electacta.2017.01.002 (2017).
Chen, J. et al. Synthesis of NiO@MnO2 core/shell nanocomposites for supercapacitor application. Appl. Surf. Sci. 360 534–539. https://doi.org/10.1016/j.apsusc.2015.10.187 (2016).
Yaseen, S. et al. Bimetallic NiO/Mn2O3 nano-pyramids as battery-type electrode material for high-performance supercapacitor application. Electrochim. Acta. 470, 143340. https://doi.org/10.1016/j.electacta.2023.143340 (2023).
Sh. Ashok, C. & Thomas, N. Facile synthesis of hierarchical NiCo2O4/NiO nanorods for high-performance supercapacitor. Chem. Phys. Lett. 826, 140651. https://doi.org/10.1016/j.cplett.2023.140651 (2023).
Ariyamparambil, V. J. & Kandasubramanian, B. A mini-review on the recent advancement of electrospun MOF-derived nanofibers for energy storage. Chem. Eng. J. Adv. 11, 100355. https://doi.org/10.1016/j.ceja.2022.100355 (2022).
Karuppaiah, M. et al. Solvent dependent morphological modification of micro-nano assembled Mn2O3/NiO composites for high performance supercapacitor applications. Ceram. Int. 45, 4298–4307. https://doi.org/10.1016/j.ceramint.2018.11.104 (2019).
Sumantha, H. S., Rajagopal, S., Nagaraju, G., Shashank, M. & Suresha, B. L. Facile and eco-friendly combustion synthesis of NiO particles for photodegradation studies. Chem. Phys. Lett. 779, 138837. https://doi.org/10.1016/j.cplett.2021.138837 (2021).
Duraisamy, N., Numan, A., Fatin, S. O., Ramesh, K. & Ramesh, S. Facile sonochemical synthesis of nanostructured NiO with different particle sizes and its electrochemical properties for supercapacitor application. J. Colloid Interface Sci. 471, 136–144. https://doi.org/10.1016/j.jcis.2016.03.013 (2016).
Anitha, S. et al. Synthesis and characterization of NiO-CdO composite materials towards photoconductive and antibacterial applications. Mater. Chem. Phys. 211, 88–96. https://doi.org/10.1016/j.matchemphys.2018.01.048 (2018).
Wei, Y. et al. Simple controllable fabrication of novel Flower-Like hierarchical porous NiO: Formation mechanism, shape evolution and their application into asymmetric supercapacitors. ChemElectroChem 16, e202200462. https://doi.org/10.1002/celc.202200462 (2022).
Zhao, Y., Ma, C. & Li, Y. One-step microwave preparation of a Mn3O4 nanoparticles/exfoliated graphite composite as superior anode materials for Li-ion batteries. Chem. Phys. Lett. 673, 19–23. https://doi.org/10.1016/j.cplett.2017.02.002 (2017).
Sunaina, P., Chand, A., Joshi, S., Lal, V. & Singh Effect of hydrothermal temperature on structural, optical and electrochemical properties of α-MnO2 nanostructures for supercapacitor application. Chem. Phys. Lett. 77, 138742. https://doi.org/10.1016/j.cplett.2021.138742 (2021).
Wang, B. et al. Preparation of Mn3O4 microspheres via glow discharge electrolysis plasma as a high-capacitance supercapacitor electrode material. J. Alloys Compd. 926, 166775. https://doi.org/10.1016/j.jallcom.2022.166775 (2022).
Shunmugapriya, B. & Vijayakumar, T. Comparative study on the electrochemical property of Mn3O4 electrode material synthesised using chemical techniques. Electrochim. Acta. 512, 145492. https://doi.org/10.1016/j.electacta.2024.145492 (2025).
Saha, P. et al. Recent progress of high-energy density supercapacitors based on nanostructured nickel oxides. Electrochim. Acta. https://doi.org/10.1016/j.electacta.2024.1448 (2024).
Kumar, R. et al. Homogeneous reduced graphene oxide supported NiO-MnO2 ternary hybrids for electrode material with improved capacitive performance. Electrochim. Acta. 303, 246–256. https://doi.org/10.1016/j.electacta.2019.02.084 (2019).
Setayesh, M., Mazloom, J. & Safari, M. Highly efficient asymmetric supercapacitor based on magnetic CeO2@γ-Fe2O3 composite and reduced graphene oxide electrodes. Ceram. Int. 50, 18185–18194. https://doi.org/10.1016/j.ceramint.2024.02.302 (2024).
Safari, M., Mazloom, J., Boustani, K. & Monemdjou, A. Hierarchical Fe2O3 hexagonal nanoplatelets anchored on SnO2 nanofibers for high-performance asymmetric supercapacitor device. Sci. Rep. 12, 14919. https://doi.org/10.1038/s41598-022-18840-2 (2022).
Shirzad Choubari, M., Rahmani, S. & Mazloom, J. Boosted electrochemical performance of magnetic caterpillar-like Mg0.5Ni0.5Fe2O4 nanospinels as a novel pseudocapacitive electrode material. Sci. Rep. 13, 7822. https://doi.org/10.1038/s41598-023-35014-w (2023).
Venkatalakshmi, N., Kini, H. J. & Naik, H. B. Green-synthesized nickel oxide nanoparticles: Magnetic and biomedical applications. Inorg. Chem. Commun. 151, 110490. https://doi.org/10.1016/j.inoche.2023.110490 (2023).
Wang, P., Zhang, X., Wei, Y. & Yang, P. Ni/NiO nanoparticles embedded inporous graphite nanofibers towards enhanced electrocatalytic performance. Int. J. Hydrogen Energy. 44, 19792–19804. https://doi.org/10.1016/j.ijhydene.2019.05.121 (2019).
Lett, J. A. et al. Hydrothermal synthesis and photocatalytic activity of Mn3O4 nanoparticles. Top. Catal. 66, 126–138. https://doi.org/10.1007/s11244-022-01710-6 (2023).
Akhtar, M. S. et al. Impact of porous Mn3O4 nanostructures on the performance of rechargeable lithium ion battery: Excellent capacity and cyclability. Solid State Ion. 336, 31–38. https://doi.org/10.1016/j.ssi.2019.03.010 (2019).
Koczkur, K. M., Mourdikoudis, S., Polavarapu, L. & Skrabalak, S. E. Polyvinylpyrrolidone (PVP) in nanoparticle synthesis. Dalton Trans. 44, 17883–17905. https://doi.org/10.1039/C5DT02964C (2015).
Li, Y. et al. Well shaped Mn3O4 nano-octahedra with anomalous magnetic behavior and enhanced photodecomposition properties. Small 7, 475–483. https://doi.org/10.1002/smll.201001403 (2011).
Kong, Y. et al. Study on the synthesis of Mn3O4 nanooctahedrons and their performance for lithium ion batteries. Nanomater10, 367. https://doi.org/10.3390/nano10020367 (2020).
Jokandan, Z. A., Mazloom, J., Ghaziani, M. M. & Ghaziani Optimized electrospun MnFe2O4 nanofibers as promising electrode materials for supercapacitor applications: Physical and electrochemical properties. J. Mater. Sci. Mater. Electron. 35, 113. https://doi.org/10.1007/s10854-023-11918-1 (2024).
Anand, G. T., Nithiyavathi, R., Ramesh, R., Sundaram, S. J. & Kaviyarasu, K. Structural and optical properties of nickel oxide nanoparticles: Investigation of antimicrobial applications. Surf. Interfaces. 18, 100460. https://doi.org/10.1016/j.surfin.2020.100460 (2020).
Olajire, A. A., Mohammed, A. A. & Mohammed Green synthesis of nickel oxide nanoparticles and studies of their photocatalytic activity in degradation of polyethylene films. Adv. Powder Technol. 31, 211–218. https://doi.org/10.1016/j.apt.2019.10.012 (2020).
Foroushani, M. S. et al. A theranostic system based on nanocomposites of manganese oxide nanoparticles and a pH sensitive polymer: Preparation, and physicochemical characterization. Bioelectrochemistry 130, 107347. https://doi.org/10.1016/j.bioelechem.2019.107347 (2019).
Sundaresan, N. & Ravichandran, S. Biosynthesis of nickel oxide nanoparticles using evolvulus alsinoides extract and their potential photocatalytic and invitro anticancer activity. Inorg. Chem. Commun. 150, 110489. https://doi.org/10.1016/j.inoche.2023.110489 (2023).
Ramesh, R., Catherine, G., Sundaram, S. J., Khan, F. L. A. & Kaviyarasu, K. Synthesis of Mn3O4 nano complex using aqueous extract of Helianthus annuus seed cake and its effect on biological growth of Vigna radiata. Mater. Today Proc. 36, 184–191 https://doi.org/10.1016/j.matpr.2020.02.883 (2021).
Bhagwan, J., Sahoo, A., Yadav, K. L., Sharma, Y. & Sharma Porous, one dimensional and high aspect ratio Mn3O4 nanofibers: Fabrication and optimization for enhanced supercapacitive properties. Electrochim. Acta. 174, 992–1001. https://doi.org/10.1016/j.electacta.2015.06.073 (2015).
Hu, Z. et al. Retracted Article: Ffacile synthesis of hierarchical Mn3O4 superstructures and efficient catalytic performance. Phys. Chem. Chem. Phys. 18, 26602–26608. https://doi.org/10.1039/C6CP05004B (2016).
Majeed, K. et al. Effective removal of methylene blue by Mn3O4/NiO nanocomposite under visible light. Sep. 10 200. https://doi.org/10.3390/separations10030200 (2023).
Silambarasan, M., Ramesh, P. S., Geetha, D. & Venkatachalam, V. A report on 1D MgCo2O4 with enhanced structural, morphological and electrochemical properties. J. Mater. Sci. Mater. Electron. 28, 6880–6888. https://doi.org/10.1007/s10854-017-6388-6 (2017).
Dhiwahar, A. T., Sundararajan, M., Sakthivel, P., Dash, C. S. & Yuvaraj, S. Microwave-assisted combustion synthesis of pure and zinc-doped copper ferrite nanoparticles: Structural, morphological, optical, vibrational, and magnetic behavior. J. Phys. Chem. Solid. 138, 109257–109291. https://doi.org/10.1016/j.jpcs.2019.109257 (2019).
Mohamed, M. M. & El-Farsy, H. Rapid reduction of nitroarenes photocatalyzed by an innovative Mn3O4/α-Ag2WO4 nanoparticles. Sci. Rep. 10, 21495. https://doi.org/10.1038/s41598-020-78542-5 (2020).
Rahaman, H., Barman, K., Jasimuddin, S., Ghosh, S. K. & Ghosh Hybrid Mn3O4–NiO nanocomposites as efficient photoelectrocatalysts towards water splitting under neutral pH conditions. RSC Adv. 114, 113694–113702 https://doi.org/10.1039/C6RA22499G (2016).
Goel, R., Jha, R. & Ravikant, C. Investigating the structural, electrochemical, and optical properties of p-type spherical nickel oxide (NiO) nanoparticles. J. Phys. Chem. Solids. 144, 109488. https://doi.org/10.1016/j.jpcs.2020.109488 (2020).
Loukil, A., Boukhachem, A., Amor, M. B., Ghamnia, M. & Raouadi, K. Effects of potassium incorporation on the structural, optical, vibrational and electrical properties of NiO sprayed thin films for p-type optical windows. Ceram. Int. 42, 8274–8289. https://doi.org/10.1016/j.ceramint.2016.02.040 (2016).
Raha, S., Mohanta, D. & Ahmaruzzaman, M. Novel CuO/Mn3O4/ZnO nanocomposite with superior photocatalytic activity for removal of Rabeprazole from water. Sci. Rep. 11, 15187. https://doi.org/10.1038/s41598-021-94066-y (2021).
Diallo, A. et al. Green synthesis of single phase hausmannite Mn3O4 nanoparticles via aspalathus linearis natural extract. SN Appl. Sci. 3, 1–11. https://doi.org/10.1007/s42452-021-04550-3 (2021).
Mannaa, M. A. et al. Role of NiO nanoparticles in enhancing structure properties of TiO2 and its applications in photodegradation and hydrogen evolution. ACS Omega. 6, 30386–30400. https://doi.org/10.1021/acsomega.1c03693 (2021).
Yi, T. F. et al. Construction of spherical NiO@ MnO2 with core-shell structure obtained by depositing MnO nanoparticles on NiO nanosheets for high-performance supercapacitor. Ceram. Int. 46, 421–429. https://doi.org/10.1016/j.ceramint.2019.08.278 (2020).
Ghaziani, M. M., Mazloom, J., Ghodsi, F. E. & Ghodsi Electrospun MgCo2O4 nanofibers as an efficient electrode material for pseudocapacitor applications: Effect of calcination temperature on electrochemical performance. J. Phys. Chem. Solids. 152, 109981. https://doi.org/10.1016/j.jpcs.2021.109981 (2021).
Wang, Y. et al. Facile solvothermal synthesis of novel MgCo2O4 twinned-hemispheres for high-performance asymmetric supercapacitors. J. Alloys Compd. 818, 152905–152914. https://doi.org/10.1016/j.jallcom.2019.152905 (2019).
Vijayan, S., Kirubasankar, B., Pazhamalai, P. & Angaiah, S. Electrospun Nd3+ doped LiMn2O4 nanofibers as high-performance cathode material for Li-Ion capacitors. ChemElectroChem 4 2059–2067 https://doi.org/10.1002/celc.201700161 (2017).
Zhao, J., Liu, H. & Zhang, Q. Preparation of NiO nanoflakes under different calcination temperatures and their supercapacitive and optical properties. Appl. Surf. Sci. 392, 1097–1106. https://doi.org/10.1016/j.apsusc.2016.09.128 (2017).
Safartoobi, A., Mazloom, J., Ghodsi, F. E. & Ghodsi Novel electrospun bead-like Ag2MoO4 nanofibers coated on Ni foam for visible light-driven heterogeneous photocatalysis and high-performance supercapacitor electrodes. Phys. Chem. Chem. Phys. 26, 430–444. https://doi.org/10.1039/d3cp04751b (2024).
Wang, J., Polleux, J., Lim, J. & Dunn, B. Pseudocapacitive contributions to electrochemical energy storage in TiO2 (anatase) nanoparticles. J. Phys. Chem. C. 111, 14925–14931. https://doi.org/10.1021/jp074464w (2007).
Wang, Y. et al. Controllable preparation of nickel cobalt manganese ternary metal-organic frameworks for high-performance supercapacitor. J. Energy Storage. 58, 106395. https://doi.org/10.1016/j.est.2022.106395 (2023).
Mona, Y. et al. Reduced graphene oxide anchored MnCo2S4 for solid state asymmetric capacitor device. Results Eng. 25, 103879. https://doi.org/10.1016/j.rineng.2024.103879 (2025).
Veerasamy, U. S. et al. Investigation on electrochemical behavior of rGO nanosheet supported Cobalt doped vanadium pentoxide nanorods as a battery type asymmetric capacitor device. J. Energy Storage. 84, 110956. https://doi.org/10.1016/j.est.2024.110956 (2024).
Veerasamy, U. S. et al. Multidimensional woodchips-like Mn-metal-organic framework for asymmetric supercapacitor devices. Appl. Surf. Sci. 24, 100650. https://doi.org/10.1016/j.apsadv.2024.100650 (2024).
Govindan, R. et al. Ceria and ceria-based hybrid nanostructures as potential electrode materials for high-performance electrochemical supercapacitors. Mater. Today Chem. 34, 101779. https://doi.org/10.1016/j.mtchem.2023.101779 (2023).
Liu, X., Wang, J. & Yang, G. Amorphous nickel oxide and crystalline manganese oxide nanocomposite electrode for transparent and flexible supercapacitor. J. Chem. Eng. 347, 101–110. https://doi.org/10.1016/j.cej.2018.04.070 (2018).
Dhas, S. D. et al. Fabrication of efficient electrochemical capacitors rooted in sol-gel derived NiMn2O4 nanoparticles. J. Electroanal. Chem. 897, 115548. https://doi.org/10.1016/j.jelechem.2021.115548 (2021).
Tian, D. et al. Fabrication of two-dimensional metal-organic frameworks on electrospun nanofibers and their derived metal doped carbon nanofibers for an advanced asymmetric supercapacitor with a high energy density. J. Power Sources. 413, 50–58. https://doi.org/10.1016/j.jpowsour.2018.12.014 (2019).
Han, X. G. et al. Addition of dissimilar metal nodes to improve the electrochemical performance of MOF as a supercapacitor. Inorgan. Chim. Acta. 536, 120916. https://doi.org/10.1016/j.ica.2022.120916 (2022).
Shaik, D. P., Pitcheri, R., Qiu, Y. & Hussain, O. M. Hydrothermally synthesized porous Mn3O4 nanoparticles with enhanced electrochemical performance for supercapacitors. Ceram. Int. 45, 2226–2233. https://doi.org/10.1016/j.ceramint.2018.10.135 (2019).
Feng, X. et al. Controllable synthesis of porous NiCo nanoflowers asymmetric all-solid-state supercapacitors. J. Chem. Eng. 368, 51–60. https://doi.org/10.1016/j.cej.2019.02.191 (2019).
Ansari Jokandan, Z., Mazloom, J. & Safari, M. Electrospun Mn1 – xCoxFe2O4 (x = 0–0.7) nanofibers for supercapacitors and oxygen evolution reaction. Mat. Chem. Phys. 28, 129948. https://doi.org/10.1016/j.matchemphys.2024.129948 (2024).
Sheokand, S., Kumar, P., Samra, Samra, K. S. Optimization of CuS@NiO nanocomposite for enhanced charge storage in supercapacitors. J. Appl. Electrochem. 1–14. https://doi.org/10.1007/s10800-025-02267-4 (2025).
Xu, J. et al. NiO-rGO composite for supercapacitor electrode. Surf. Interfaces. 18, 100420. https://doi.org/10.1016/j.surfin.2019.100420 (2020).
Lee, J. W., Hall, A. S., Kim, J. D. & Mallouk, T. E. A facile and template-free hydrothermal synthesis of Mn3O4 nanorods on graphene sheets for supercapacitor electrodes with long cycle stability. Chem. Mater. 24, 1158–1164. https://doi.org/10.1021/cm203697w (2012).
KT, M. S., Kumar, J. P. J. & Kumar, S. NiO/Co3O4 nanocomposite electrode for high performance supercapacitor and oxygen evolution reaction applications, ionics. 1–11. (2025). https://doi.org/10.1007/s11581-025-06137-z
Maheshwarana, G. et al. Exploration of Cr2O3-NiO nanocomposite as a superior electrode material for supercapacitor applications. Mater. Lett. 300, 130191. https://doi.org/10.1016/j.matlet.2021.130191 (2021).
Liu, E. H. et al. Preparation and characterization of nanostructured NiO/MnO2 composite electrode for electrochemical supercapacitors. Mater. Res. Bull. 44, 1122–1126. https://doi.org/10.1016/j.materresbull.2008.10.003 (2009).
Xi, S. et al. Facile synthesis of free-standing NiO/MnO2 core-shell nanoflakes on carbon cloth for flexible supercapacitors. Nanoscale Res. Lett. 12, 171. https://doi.org/10.1186/s11671-017-1939-6 (2017).
Acknowledgements
The authors gratefully acknowledge the University of Guilan Research Council for the support of this research.
Author information
Authors and Affiliations
Contributions
Z.S.D.: Investigation, Formal analysis, Writing original draft. J.M.: Conceptualization, Methodology, Writing-review & editing, Validation, Supervision. M.M.G.: Writing-review, Visualization, Formal analysis, Methodology.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Doroudkhani, Z.S., Mazloom, J. & Ghaziani, M.M. Optical and electrochemical performance of electrospun NiO–Mn3O4 nanocomposites for energy storage applications. Sci Rep 15, 11436 (2025). https://doi.org/10.1038/s41598-025-96008-4
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-96008-4
Keywords
This article is cited by
-
Correlation of structural, electrical and electrochemical response of Mn-doped nickel ferrite, via auto-combustion method as a pseudocapacitive electrode
Journal of Materials Science: Materials in Electronics (2026)
-
Transformation of recovered manganese metal from spent lithium batteries to Mn3O4 catalyst - applied for supercapacitor application
Journal of Material Cycles and Waste Management (2025)
