
Abstract
Research indicates that minimal change disease (MCD) and focal segmental glomerulosclerosis (FSGS) may reflect varying severities of the same underlying condition, with inflammation potentially facilitating the progression from MCD to FSGS. The aim of this study was to determine the whether systemic inflammation accelerated the progression from MCD to FSGS in a mouse model of Adriamycin-induced nephrosis. In this model, systemic inflammation induced transient proteinuria without significant serum biochemical alterations or significant renal histological changes in control mice that did not develop nephrotic syndrome. In contrast, both mice with Adriamycin-induced nephrosis mice and mice with Adriamycin-induced nephrosis with systemic inflammation showed histological features of MCD at week 4 and progressive exacerbation of FSGS. However, the glomerular lesions of mice with Adriamycin-induced nephrosis in a state of systemic inflammation were more obvious than those of mice with Adriamycin-induced nephrosis. These findings suggest that systemic inflammation may hasten histological development from MCD to FSGS in this mouse model.
Similar content being viewed by others


Introduction
Idiopathic nephrotic syndrome (INS) is characterized by nephrotic-range proteinuria, hypoalbuminemia, hyperlipidemia, and edema. In pediatric INS, focal segmental glomerulosclerosis (FSGS) and minimal change disease (MCD) are the predominant histopathological patterns observed1,2,3. Although both conditions manifest as nephrotic syndrome, they exhibit significant differences in pathogenesis, pathological features, treatment response, and prognosis. MCD is more prevalent in younger children and typically responds well to corticosteroid therapy, with a favorable long-term prognosis. However, clinical observations have revealed that in some pediatric INS cases initially diagnosed as MCD, particularly in steroid-dependent or steroid-resistant scenarios, follow-up renal biopsies indicate that up to 60% of cases progress to FSGS. This pathological development significantly increases the risk of end-stage renal disease3. From a pathological mechanism perspective, both MCD and FSGS involve podocyte abnormalities3, but they exhibit significant differences in immunopathology and the levels of inflammatory markers4. In MCD, nephrotic syndrome is usually not accompanied by significant glomerular structural abnormalities. Light microscopy often reveals a normal glomerular structure, with only mild mesangial hyperplasia and fatty degeneration of proximal tubular epithelial cells. Electron microscopy reveals extensive effacement of the podocyte foot processes without electron-dense deposits. In contrast, FSGS is characterized by segmental glomerular scarring, pronounced mesangial cell proliferation, focal collapse and degradation of glomerular capillaries, accompanied by increased inflammatory cell infiltration and immune complex deposition4,5,6.
In recent years, accumulating evidence has highlighted the pivotal role of inflammatory responses in the development of idiopathic nephrotic syndrome (INS). Although MCD and FSGS exhibit distinct pathogenic pathways, both types may relapse following infection, potentially due to systemic inflammatory reactions1,5,7. Study findings have suggested that systemic inflammation may lead to elevated levels of circulating inflammatory factors, which reach the kidneys via the bloodstream. The persistent inflammatory microenvironment may exacerbate podocyte injury and promote glomerulosclerosis by activating immune responses4,6. Furthermore, research has shown that steroid-resistant patients often exhibit more pronounced systemic inflammatory responses, characterized by higher plasma levels of inflammatory factors and more significant immune cell activation1,2,3,4,5,6. Previous studies have demonstrated that the levels of proinflammatory cytokines in the kidneys of FSGS patients are significantly greater than those in MCD patients8. Case reports from the past decade have also indicated that anti-inflammatory and anti-lymphocyte therapies may be beneficial for refractory INS cases9,10.
If systemic inflammation is confirmed to be a critical factor in the onset and development of nephrotic syndrome, studying systemic inflammation will have significant theoretical and clinical implications. First, it will contribute to a deeper understanding of the mechanisms underlying the association between infections and the recurrence of nephrotic syndrome, providing a theoretical basis for preventing relapses. Second, investigating systemic inflammation may reveal the underlying pathogenesis of nephrotic syndrome, particularly in steroid-dependent or steroid-resistant cases, which are often associated with more pronounced inflammatory responses. Additionally, research on systemic inflammation may help predict patients’ responses to corticosteroid therapy, thereby offering a foundation for the development of individualized treatment plans. On the basis of these findings, approaches that target inflammatory pathways (anti-inflammatory drugs and cytokine inhibitors, e.g. could emerge as novel therapeutic strategies, providing new possibilities for improving the outcomes of steroid-resistant or steroid-dependent patients. Moreover, systemic inflammation-related biomarkers (e.g., C-reactive protein and cytokine levels) may serve as noninvasive indicators for assessing disease activity, predicting relapse or development, and guiding clinical monitoring and treatment adjustments. Therefore, the aim of this study was to determine whether experimentally induced inflammation promotes this accelerated development in a mouse model.
Materials and methods
Animals
All procedures related to animals were reviewed and approved by the Institutional Animal Care and Use Committee of Chongqing Medical University and conducted in accordance with relevant guidelines and regulations. The animal research was carried out in accordance with the ARRIVE guidelines. In this study, every effort was made to minimize the number of animals used and their suffering to the greatest extent possible, which was in line with the current animal welfare regulations.
Ninety-six six-week-old male BALB/c mice were obtained from the Center for Experimental Animals at Chongqing Medical University, China, kept in a pathogen-free environment, and randomly allocated to the control (CTL), inflammation (IFM), Adriamycin-induced nephrosis (ADR), and Adriamycin-induced nephrosis with inflammation (AWI) groups. To induce nephrotic syndrome, ADR and AWI mice received a single intravenous dose of Adriamycin (10.5 mg/kg body weight; 10% solution in 0.9% saline; Pharmacia, Italy)11. CTL mice were given an equivalent intravenous amount of 0.9% saline. IFM mice received a subcutaneous injection of 0.5 mL of 10% casein on alternate days until high-dose pentobarbital sodium injection at 4,8, or 12 weeks led to euthanasia12,13. AWI mice initially received an intravenous Adriamycin injection, followed by a subcutaneous injection of 0.5 mL of 10% casein starting one week post-Adriamycin injection and continuing every second day until t until high-dose pentobarbital sodium injection at 4,8, or 12 weeks led to euthanasia.
Specimen collection
Mice were euthanized at 4, 8, or 12 weeks post-Adriamycin administration. Urine was collected over 24 h at 2, 4, 6, 8, and 12 weeks; centrifuged at 5000 rpm for 10 min to remove particulates; and stored at -20 °C. Blood was drawn from the ophthalmic veins and spun for 20 min at 2000 rpm. The serum was obtained and preserved at -20 °C. Kidneys were excised after perfusion with 0.9% saline to pale. The upper poles of the left kidney were immersed in 4% paraformaldehyde and processed for periodic acid-Schiff (PAS), hematoxylin and eosin (H&E), and Masson trichrome staining and immunohistochemistry. A small renal cortex section from each sample was preserved in 3% glutaraldehyde for transmission electron microscopy (TEM), and the remaining kidney tissue was reserved for RNA and protein expression analysis.
Biochemical indicators
Blood urea nitrogen (BUN) and creatinine (Cr) are key indicators of renal function. While the BUN level is impacted by nonrenal factors such as a high-protein diet, gastrointestinal bleeding, dehydration, and increased catabolism, Cr levels mainly reflect glomerular filtration, making Cr a more reliable indicator of kidney function. In this study, Cr and albumin levels were examined using an automated biochemical analyzer (HITACHI 70, Japan), and 24-hour urinary protein levels were quantified using the Coomassie Brilliant Blue method. Since hyperlipidemia is a characteristic of INS, total cholesterol (TC) and low-density lipoprotein (LDL) levels were also measured with a biochemical analyzer.
Morphological analysis
Tissue samples preserved in paraformaldehyde were dehydrated using a graded ethanol series, embedded in paraffin, and sliced into 3-µm sections using a microtome. Renal tissue sections were stained with hematoxylin and eosin (H&E) for general histology, periodic acid-Schiff (PAS) for evaluation of the glomerular basement membrane and mesangial matrix, and Masson’s trichrome for the evaluation of fibrosis, following standardized protocols (Bancroft & Gamble, 2008). For TEM, the samples were rinsed three times with PBS, preserved in 1% osmium tetroxide for 1 h, dehydrated in a graded ethanol series, washed with acetone, embedded in epoxy resin (Epon812), sectioned with an ultrathin slicer (LKB-type I), and stained with uranyl acetate and lead citrate.
Glomerulosclerosis index
The glomerulosclerosis index was determined by analyzing H&E- and PAS-stained sections under a light microscope. The evaluation process involved the following steps: (1) ten glomeruli were randomly selected per section; (2) glomerulosclerosis was categorized into four grades on the basis of the degree of tuft involvement: grade 0 for normal structure, grade 1 for < 25% involvement, grade 2 for 25–50% involvement, grade 3 for 50–75% involvement, and grade 4 for > 75% involvement. The index was calculated as [(1×number of glomeruli with grade 1) + (2×number with grade 2) + (3×number with grade 3) + (4×number with grade 4)] × 100%/total glomeruli observed. Scoring was conducted by investigators who were blinded to the treatment groups.
Immunohistochemical analysis
Immunohistochemistry was utilized to measure IL-1β and TGF-β1 protein levels. After three washes with PBS, the rehydrated sections were incubated in 10 mM citrate buffer (pH 6.0) at 100 °C for 10 min to unmask the antigens. Sections were then incubated with 0.3% H2O2 in methanol for 15 min to block endogenous peroxidase and exposed to either a rabbit anti-mouse IL-1β antibody (dilution 1:100; sc-7884, Santa Cruz Biotechnology, USA) or a rabbit anti-mouse TGF-β1 antibody (dilution 1:100; sc-146, Santa Cruz Biotechnology) overnight at 4 °C. Afterward, horseradish peroxidase-conjugated goat anti-rabbit secondary antibody (PV-6001, Zhongshan, China) was applied at 37 °C for 30 min, after which diaminobenzidine (DAB) staining and hematoxylin counterstaining were performed. Negative control sections were incubated with PBS instead of primary antibodies, whereas validated positive sections served as positive controls. The target protein levels were visualized using an Image-Pro Plus System and expressed as the integral optical density.
RNA extraction and real-time polymerase chain reaction
RNA was retrieved from frozen kidneys with RNA isolation kits (RP1201, Bioteke, China) per the manufacturer’s protocols. The RNA purity and concentration were determined spectrophotometrically. Complementary DNA (cDNA) was generated from 1 µl of RNA via a reverse transcription kit (DRR047A, Takara, China). Real-time PCR was conducted in a 10 µL system comprising 1 µL of cDNA, 0.8 µL of primer mixture, 5 µL of SYBR Green Master Mix, and 3.2 µL of ddH2O for 40 cycles of 30 s at 95 °C, 30 s at 59 °C, and 30 s at 65 °C, with an additional 5 min at 65 °C. Primers were selected using TaqMan Primer Express (Table 1), and the relative TGF-β1 and IL-1β mRNA levels were standardized to those of β-actin via the 2ΔΔCt method.
Correlation analysis
Associations between inflammatory cytokine levels and glomerulosclerosis severity in ADR- and AWI mice were assessed using Spearman’s correlation analysis.
Statistical evaluation
The data were assessed using SPSS version 18.0 and are presented as the means ± standard deviations. Differences were examined using one-way ANOVA, with post hoc analysis via the Student‒Newman‒Keuls test. A p value of < 0.05 was considered significant.
Results
Systemic inflammation increased the 24-hour urinary protein level

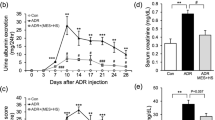
Twenty-four-hour urine protein levels in each group over 12 weeks. Data are presented as the means ± standard deviations. CTL: control; IFM: inflammation; ADR: Adriamycin-induced nephrosis; AWI: Adriamycin-induced nephrosis with inflammation. n = 8 per group; *P < 0.05 vs. the control; ▲P < 0.05 vs. the AWI group at the same time point.
AWI mice presented the highest levels of proteinuria throughout the study. Both the ADR and AWI mice developed proteinuria by the second week post-Adriamycin injection, with proteinuria levels peaking at 8 weeks and persisting for the duration of the study (Fig. 1). From week 6 onward, proteinuria was significantly greater in AWI mice than in other mice. IFM mice also displayed proteinuria beginning at week 6, which decreased over time. The decline in proteinuria may reflect compensatory tubular adaptation or a GFR reduction due to nephron loss in advanced FSGS.
Systemic inflammation increased serum lipid and cr levels

Levels of low-density lipoprotein (LDL), serum albumin (ALB), total cholesterol (TC) and creatinine (Cr) in all groups at 4, 8, and 12 weeks. Data are presented as the means ± standard deviations. CTL: control; IFM: inflammation; ADR: Adriamycin-induced nephrosis; AWI: Adriamycin-induced nephrosis with inflammation. n = 8 per group; *P< 0.05 vs. the control; ▲P < 0.05 vs. the AWI group at the same time point.
Serum TC, LDL, albumin, and Cr levels were comparable in CTL and IFM mice throughout the experiment (Fig. 2). In contrast, compared with those in CTL mice, serum TC and LDL levels were significantly elevated in ADR and AWI mice at 8 and 12 weeks (Fig. 2). Notably, the serum Cr level increased earlier in AWI mice (8 weeks) than in ADR mice (12 weeks).
Systemic inflammation accelerated glomerulosclerosis

Hematoxylin and eosin (H&E) staining of renal tissues. Representative images from the CTL group (a, b, c), IFM group (d, e, f), ADR group (g, h, i), and AWI group (j, k, l) at 4, 8, and 12 weeks, respectively, after Adriamycin injection. Scale bars = 50 μm.

Periodic acid-Schiff staining of renal tissues. Representative images from the CTL group (a, b, c), IFM group (d, e, f), ADR group (g, h, i) and AWI group (j, k, l) at 4, 8, and 12 weeks, respectively, after Adriamycin injection. Scale bars = 50 μm.

Masson’s trichrome staining of renal tissues. Representative images from the CTL group (a, b), IFM group (c, d), ADR group (e, f), and AWI group (g, h) at 8 and 12 weeks after Adriamycin injection. Scale bars = 50 μm.
Histopathological examinations revealed normal glomeruli in CTL mice throughout the study period. No lesions were detected in any of the groups at 4 weeks. However, by 8 weeks, IFM mice displayed inflammatory cell infiltration, whereas ADR and AWI mice exhibited mesangial cell proliferation and mild matrix expansion, progressing to FSGS at 12 weeks, with more pronounced changes in AWI mice at week 12(Figs. 3, 4 and 5).
Systemic inflammation promoted podocyte foot process fusion and mesangial cell proliferation

Morphological changes in the renal tissues under TEM. CTL group: tall and narrow foot processes (a, b, c); IFM group: glomerular basement membrane thickening at 12 weeks (e, d, f); ADR group: increase in foot process fusions from 4 weeks to 12 weeks (g, h, i); AWI group: more podocyte foot process fusions were observed than in the ADR samples at the same time point (j, k, l).
TEM examination revealed no significant pathological changes in the CTL or IFM groups, with only minimal basement membrane thickening observed in the IFM group at 12 weeks. Conversely, both the ADR group and the AWI group demonstrated pathological alterations, including podocyte foot process fusion, mesangial cell proliferation, and dense basement membrane deposits, beginning at week 4 and persisting at later time points, with the AWI group exhibiting the most severe changes (Fig. 6).
Systemic inflammation increased the glomerulosclerosis index (GSI, %)
CTL: control, ADR: Adriamycin-induced nephrosis, IFM: inflammation, AWI: Adriamycin-induced nephrosis with inflammation. Values represent the means ± standard deviations. n = 8 per group; *P < 0.05 vs. the control; ▲P < 0.05 vs. the AWI group at the same time point.
GSI values did not significantly differ between CTL and IFM mice throughout the experiment. However, the GSI values of ADR and AWI mice were significantly greater than those of CTL mice, with increases at 12 weeks in ADR mice and as early as 8 weeks in AWI mice. From 8 weeks onward, the GSI values of AWI mice remained significantly greater than those of ADR mice (Table 2).
Systemic inflammation upregulated TGF-β1 and IL-1β expression in the kidney

Renal mRNA expression of transforming growth factor-β1 (TGF-β1) and interleukin-1 beta (IL-1β) at 4, 8, and 12 weeks as determined by real-time PCR. The expression levels were normalized to those of the housekeeping gene β-actin. CTL: control, IFM: inflammation, ADR: Adriamycin-induced nephrosis, AWI: Adriamycin-induced nephrosis with inflammation. Data are presented as the means ± standard errors. n = 8 per group; *P < 0.05 vs. control; ▲P < 0.05 vs. AWI at the same time point.

Immunostaining for TGF-β1: CTL group (a, b, c), IFM group (d, e, f), ADR group (g, h, i), and AWI group at 4, 8, and 12 weeks, respectively, after Adriamycin injection. Scale bars = 50 μm.

Immunostaining for IL-1β in the CTL group (a, b, c), IFM group (d, e, f), ADR group (g, h, i), and AWI group (j, k, l) at 4, 8, and 12 weeks, respectively, after Adriamycin injection. Scale bars = 50 μm.

Quantification of histopathological results based on the integral optical density (IOD). The IOD was calculated for each image using the following equation: positive area × average density. Results are presented as the means ± standard deviations. CTL: control; IFM: inflammation; ADR: Adriamycin-induced nephrosis; AWI: Adriamycin-induced nephrosis with inflammation. *P < 0.05 vs. CTL, ▲P < 0.05 vs. AWI at the same time point.
TGF-β1 and IL-1β levels, primarily in renal tubular cells, were increased in all experimental mice compared with those in CTL mice (P < 0.05; Figs. 8, 9 and 10), with AWI mice showing the highest levels. These findings were further validated using real-time PCR, with AWI mice showing the highest expression from week 4 onward (P < 0.05; Fig. 7).
Correlation analysis

Correlation analysis of significant associations between inflammatory markers and the GSI in the ADR and AWI groups at different experimental time points. ADR: Adriamycin-induced nephrosis, AWI: Adriamycin-induced nephrosis with inflammation.
Inflammatory cytokine expression was positively correlated with glomerulosclerosis severity in ADR and AWI mice (P < 0.05; Fig. 11).
Discussion
The method of subcutaneous injection of casein to induce systemic inflammation was first used by researchers to explore the relationships between inflammation and metabolic, kidney, and cardiovascular diseases. Owing to its simple procedure, good repeatability and stable inflammatory response, casein subcutaneous injection has gradually become one of the classic methods for establishing models of systemic inflammation. Casein, an exogenous protein, triggers systemic inflammation by triggering a local immune response and activating inflammatory signaling pathways. Experimental data have shown that casein can lead to the release of proinflammatory cytokines and induce systemic inflammation by activating inflammatory signaling pathways such as the NF-κB, MAPK and TLR4 pathways13. For example, the Lipid Research Center at Chongqing Medical University, which has provided critical reagents and technical support, frequently employs this approach to establish foundational models of systemic inflammation for experimental research12.
In this study, we administered casein subcutaneously to trigger chronic systemic inflammation in BALB/c mice regardless of Adriamycin-induced nephrosis. Our immunohistochemical staining and real-time PCR analyses revealed significant increases in the protein and mRNA levels of TGF-β1 and IL-1β in the kidney tissues of casein-injected mice (IFM group) from weeks 4 to 12 compared with those of control mice, confirming the successful induction of chronic systemic inflammation in our mouse model. Notably, casein-induced inflammation led to transient proteinuria without severe histological changes in the kidneys. While the specific underlying mechanism remains unclear, it is well documented that proteinuria secondary to systemic inflammation is often transient14,15. Interestingly, Reiser et al. reported that inflammation induced by lipopolysaccharide injection in mice resulted in transient expression of CD80 on podocytes, which was correlated with proteinuria16,17.
Adriamycin-induced nephrosis mimics MCD in mice and serves as a valuable model for studying factors involved in the progression of MCD to FSGS11. TGF-β1 and IL-1β, both proinflammatory cytokines, have been implicated in glomerulosclerosis development18,19,20. In our experimental model, we observed histological changes characteristic of MCD at week 4 in both the ADR and AWI groups and features of FSGS at week 12. This development was accompanied by increased TGF-β1 and IL-1β mRNA and protein levels and renal inflammatory cell infiltration, indicating that inflammation is a crucial factor in the progression from MCD to FSGS. The positive correlation between the levels of these proinflammatory cytokines and the glomerulosclerosis index (GSI) further supports their pathogenic roles in FSGS. Although IL-1β and TGF-β1 are detected primarily in the tubulointerstitium, their systemic release or paracrine signaling to glomerular cells (e.g., podocytes and mesangial cells) likely mediates sclerosis. This aligns with the ‘tubuloglomerular crosstalk’ hypothesis, where tubular injury drives glomerular pathology via inflammatory mediators21.
Immune system perturbation has been linked to the onset of INS. Compared with patients with MCD and healthy controls, FSGS patients exhibit a significantly greater degree of interstitial infiltration by CD3+ T cells, CD20+ B cells, and CD68+ macrophages4. Previous studies have also reported that FSGS patients have elevated serum IL-1, IL-2, IL-13, and TNF-α levels, which can induce the differentiation of T helper 1, T helper 2, and T helper 17 cells4,22,23,24. Moreover, proteinuria itself may exacerbate inflammation by promoting proinflammatory and profibrotic cytokine production in the kidney25.
Our study provides evidence that the systemic inflammation induced by casein may hasten the development of MCD into FSGS in mice with Adriamycin-induced nephrosis. This hastened development is evidenced by the earlier appearance of the histological features of FSGS in AWI mice than in ADR mice. Glomerular and tubular cells release inflammatory mediators after injury, forming a local inflammatory environment in the kidney. Through inflammatory cell infiltration, inflammatory mediator release and excessive deposition of the extracellular matrix, the glomerular structure and function are destroyed26. Systemic inflammation affects the kidney through circulating inflammatory mediators such as cytokines and chemokines, which may exacerbate the inflammatory response in the kidney, thereby promoting glomerular damage and sclerosis27. Moreover, systemic inflammation is often accompanied by metabolic disorders (such as high blood pressure and high blood glucose levels) and oxidative stress, which may indirectly accelerate the process of glomerulosclerosis development. Intrarenal inflammation interacts with systemic inflammation, which may lead to a vicious cycle that further accelerates the development of FSGS.
While the exact mechanisms remain to be elucidated, previous investigations have revealed that proinflammatory cytokines are essential for apoptosis and sclerosis in podocytes2,28,29,30,31. Additionally, TGF-β1, known for inducing various critical podocyte changes, is recognized as a key mediator of podocyte injury32,33. IL-1β promotes podocyte foot process effacement and apoptosis while stimulating fibronectin and collagen secretion by mesangial cells, potentially accelerating glomerulosclerosis35. Our research, along with findings from other scholars, indicates that inflammatory stress magnifies lipid-modulated renal impairment in patients with INS, potentially leading to glomerulosclerosis and tubulointerstitial fibrosis8,34,35,36.
In summary, our study revealed that systemic inflammation can lead to transient proteinuria in healthy mice; however, in the context of Adriamycin-induced nephrosis, systemic inflammation may accelerate the development of MCD into FSGS, with TGF-β1 and IL-1β playing pivotal roles in this process. The beneficial effects of anti-inflammatory agents on glomerulosclerosis development in ADR mice may inform future therapeutic strategies for managing FSGS clinically36.
Data availability
The datasets used and analyzed during the current study are available from the corresponding author upon reasonable request.
References
Vivarelli, M., Massella, L., Ruggiero, B. & Emma, F. Minimal change disease[J]. Clin. J. Am. Soc. Nephrol. 12 (2), 332–345 (2017). https://pubmed.ncbi.nlm.nih.gov/27940460/
Fogo, A. B. Causes and pathogenesis of focal segmental glomerulosclerosis[J]. Nat. Rev. Nephrol. 11 (2), 76–87 (2015). https://pubmed.ncbi.nlm.nih.gov/25447132/
Maas, R. J., Deegens, J. K., Smeets, B., Moeller, M. J. & Wetzels, J. F. Minimal change disease and idiopathic FSGS: manifestations of the same disease[J]. Nat. Rev. Nephrol. 12 (12), 768–776 (2016). https://pubmed.ncbi.nlm.nih.gov/27748392/
Campbell, R. E. & Thurman, J. M. The immune system and idiopathic nephrotic syndrome[J]. Clin. J. Am. Soc. Nephrol. 17 (12), 1823–1834 (2022). https://pubmed.ncbi.nlm.nih.gov/36198505/
Hackl, A. et al. The role of the immune system in idiopathic nephrotic syndrome[J]. Mol. Cell. Pediatr. 8 (1), 18 (2021). https://pubmed.ncbi.nlm.nih.gov/34792685/
Ahmadian, E. et al. The role of cytokines in nephrotic syndrome[J]. Mediators of Inflammation, 2022: 6499668. (2022). https://pubmed.ncbi.nlm.nih.gov/35185384/
Roca, N. et al. Activation of the acute inflammatory phase response in idiopathic nephrotic syndrome: association with clinicopathological phenotypes and with response to corticosteroids[J]. Clin. Kidney J. 14 (4), 1207–1215 (2021). https://pubmed.ncbi.nlm.nih.gov/33841866/
Zhang, G. et al. The effects of inflammation on lipid accumulation in the kidneys of children with primary nephrotic syndrome[J]. Inflammation 34 (6), 645–652 (2011). https://pubmed.ncbi.nlm.nih.gov/21103916/
Gauckler, P. et al. Rituximab in adult minimal change disease and focal segmental glomerulosclerosis: what is known and what is still unknown?[J]. Autoimmun. Rev. 19 (11), 102671 (2020). https://pubmed.ncbi.nlm.nih.gov/32942039/
Li, J., Zhao, M., Xiang, X., He, Q. & Gui, R. A novel biomimetic nanomedicine system with anti-inflammatory and anti-osteoporosis effects improves the therapy efficacy of steroid-resistant nephrotic syndrome[J]. J. Nanobiotechnol. 19 (1), 417 (2021). https://pubmed.ncbi.nlm.nih.gov/34903236/
Simic, I., Tabatabaeifar, M. & Schaefer, F. Animal models of nephrotic syndrome[J]. Pediatr. Nephrol. 28 (11), 2079–2088 (2013). https://pubmed.ncbi.nlm.nih.gov/23250714/
Ma, K. L. et al. Inflammatory stress exacerbates lipid accumulation in hepatic cells and fatty livers of Apolipoprotein E knockout mice[J]. Hepatology 48 (3), 770–781 (2008). https://pubmed.ncbi.nlm.nih.gov/18752326/
Wojtecka-Lukasik, E. et al. Histamine chloramine modifies casein-induced inflammation[J]. Inflamm. Res. 58 (1), 20–21 (2009). https://pubmed.ncbi.nlm.nih.gov/19271131/
Nasr, S. H. et al. Kidney biopsy findings in patients with COVID-19, kidney injury, and proteinuria[J]. Am. J. Kidney Dis. 77 (3), 465–468 (2021). https://pubmed.ncbi.nlm.nih.gov/33217501/
Gurevich, E., Israel, E., Segev, Y. & Landau, D. Febrile proteinuria in hospitalized children: characterization of urinary proteins[J]. Front. Pead. 6, 202 (2018). https://pubmed.ncbi.nlm.nih.gov/30175088/
Reiser, J. & Mundel, P. Danger signaling by glomerular podocytes defines a novel function of inducible B7-1 in the pathogenesis of nephrotic syndrome[J]. J. Am. Soc. Nephrol. 15 (9), 2246–2248 (2004). https://pubmed.ncbi.nlm.nih.gov/15339973/
Reiser, J. et al. Induction of B7-1 in podocytes is associated with nephrotic syndrome[J]. J. Clin. Invest. 113 (10), 1390–1397 (2004). https://pubmed.ncbi.nlm.nih.gov/15146236/
Galozzi, P., Bindoli, S., Doria, A. & Sfriso, P. The revisited role of interleukin-1 alpha and beta in autoimmune and inflammatory disorders and in comorbidities[J]. Autoimmun. Rev. 20 (4), 102785 (2021). https://pubmed.ncbi.nlm.nih.gov/33621698/
Yan, J. et al. Interleukin-17A participates in podocyte injury by inducing IL-1β secretion through ROS-NLRP3 inflammasome-caspase-1 pathway[J]. Scand. J. Immunol. 87 (4), e12645 (2018). https://pubmed.ncbi.nlm.nih.gov/29446486/
Qiu, Y. et al. Rhodojaponin II attenuates kidney injury by regulating TGF-β1/Smad pathway in mice with adriamycin nephropathy[J]. J. Ethnopharmacol. 243, 112078 (2019). https://pubmed.ncbi.nlm.nih.gov/31301369/
Kriz, W. & LeHir, M. Pathways to nephron loss starting from glomerular diseases-insights from animal models[J]. Kidney Int. 67 (2), 404–419 (2005). https://pubmed.ncbi.nlm.nih.gov/15673288/
Choi, M. E., Ding, Y. & Kim, S. I. TGF-β signaling via TAK1 pathway: role in kidney fibrosis[J]. Semin. Nephrol. 32 (3), 244–252 (2012). https://pubmed.ncbi.nlm.nih.gov/22835455/
Shao, X. S. et al. The prevalence of Th17 cells and FOXP3-regulate T cells (Treg) in children with primary nephrotic syndrome[J]. Pediatr. Nephrol. 24 (9), 1683–1690 (2009). https://pubmed.ncbi.nlm.nih.gov/19499249/
Yap, H. K. et al. Th1 and Th2 cytokine mRNA profiles in childhood nephrotic syndrome: evidence for increased IL-13 mRNA expression in relapse[J]. J. Am. Soc. Nephrol. 10 (3), 529–537 (1999). https://pubmed.ncbi.nlm.nih.gov/10073603/
Camici, M. The nephrotic syndrome is an Immunoinflammatory disorder[J]. Med. Hypotheses. 68 (4), 900–905 (2007). https://pubmed.ncbi.nlm.nih.gov/17184932/
Anders, H. J. & Schaefer L.Beyond tissue injury—damage-associated molecular patterns,toll-like receptors,and inflammasomes also drive regeneration and fibrosis[J]. J. Am. Soc. Nephrol. 25 (7), 1387–1400 (2014). https://pubmed.ncbi.nlm.nih.gov/24762401/
Ruiz-Ortega, M. et al. Targeting the progression of chronic kidney disease[J]. Nat. Rev. Nephrol. 16 (5), 269–288 (2020). https://pubmed.ncbi.nlm.nih.gov/32060481/
Brähler, S. et al. Intrinsic Proinflammatory signaling in podocytes contributes to podocyte damage and prolonged proteinuria[J]. Am. J. Physiology-Renal Physiol. 303 (12), F1473–F1485 (2012). https://pubmed.ncbi.nlm.nih.gov/22975019/
Suleiman, H. Y. et al. Injury-induced actin cytoskeleton reorganization in podocytes revealed by super-resolution microscopy[J]. JCI Insight. 2 (16), e94137 (2017). https://pubmed.ncbi.nlm.nih.gov/28814668/
Faa, G. et al. Morphogenesis and molecular mechanisms involved in human kidney development[J]. J. Cell. Physiol. 227 (3), 1257–1268 (2012). https://pubmed.ncbi.nlm.nih.gov/21830217/
Lee, H. S. Mechanisms and consequences of TGF-β overexpression by podocytes in progressive podocyte disease[J]. Cell Tissue Res. 347 (1), 129–140 (2012). https://pubmed.ncbi.nlm.nih.gov/21541658/
Kim, J. H., Kim, B. K., Moon, K. C., Hong, H. K. & Lee, H. S. Activation of the TGF-β/Smad signaling pathway in focal segmental glomerulosclerosis[J]. Kidney Int. 64 (5), 1715–1721 (2003). https://pubmed.ncbi.nlm.nih.gov/14531804/
Kim, T. S., Kim, J. Y., Hong, H. K. & Lee, H. S. mRNA expression of glomerular basement membrane proteins and TGF-β1 in human membranous nephropathy[J]. J. Pathol. 189 (4), 425–430 (1999). https://pubmed.ncbi.nlm.nih.gov/10547606/
Ruan, X. Z., Varghese, Z. & Moorhead, J. F. Inflammation modifies lipid-mediated renal injury[J]. Nephrol. Dialysis Transplantation. 18 (1), 27–32 (2003). https://pubmed.ncbi.nlm.nih.gov/12480956/
Zhang, G., Li, Q., Li & Wang et a1. Interleukin-1b enhances the intracellular accumulation of cholesterol by up-regulating the expression of low-density lipoprotein receptor and 3-hydroxy-3-methylglutaryl coenzyme A reductase in podocytes[J]. Mol Cell Biochem, Ja;346(1–2):197–204. (2011). https://pubmed.ncbi.nlm.nih.gov/20936497/
Zhang, W., Li, Q., Wang, L. & Yang, X. Simvastatin ameliorates glomerulosclerosis in Adriamycin-induced-nephropathy rats[J]. Pediatr. Nephrol. 23 (12), 2185–2194 (2008). https://pubmed.ncbi.nlm.nih.gov/18791746/
Acknowledgements
We thank Dr. Xiong Z Ruan (Renal Research Unit, Department of Nephrology, Royal Free Hospital School of Medicine, London UK) and Yaxi Chen (Lipids Research Center, Chongqing Medical University, Chongqing, China) for their generous provision of key reagents and technical assistance.
Funding
This research was sponsored by the Natural Science Foundation Project of the Chongqing Science and Technology Commission in China (cstc2020jcyj-msxmX0273).
Author information
Authors and Affiliations
Contributions
L.L was in charge of gathering data, analyzing statistics, and drafting the manuscript. Q.L handled data interpretation and literature search and reviewed and edited the manuscript. G.f. Z designed the study, interpreted the data, secured funding, and reviewed and edited the manuscript.All the authors have reviewed the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethical approval
All the animal investigations were authorized by the Committee of Chongqing Medical University Institutional Animal Care and Use Committee.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Liu, L., Li, Q. & Zhang, G. Systemic inflammation accelerates the development of focal segmental glomerulosclerosis in a mouse model of adriamycin induced nephrosis. Sci Rep 15, 14304 (2025). https://doi.org/10.1038/s41598-025-96125-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-025-96125-0
