
Abstract
Metastasis is one of the leading causes of cancer-related death worldwide. Fascin, a protein that bundles actin filaments to produce protrusions in cancer cells, plays a significant role in the enhancement of cell migration. This protein has been shown that the overexpression of this protein is related to the appearance of different types of cancer, such as colorectal cancer. In this study, we conducted in silico screening of the Enamine library, a compound library with a broad chemical space. Using a ligand-based virtual screening approach based on the pharmacophore model of G2, we identified the predicted inhibitors. First, these compounds were validated by physicochemical analysis. Differential scanning calorimetry (DSF) was used to study the binding between the predicted compounds and fascin protein, followed by an F-actin bundling assay to determine which compounds inhibited the bundling function of fascin. Z1362873773, which exhibited binding to fascin and inhibited F-actin bundling, was further tested in cell cultures to assess its effects on cancer cell viability and migration as well as in organoid models to evaluate potential cytotoxicity. Finally, we established a protocol that can be applied to discover anti-fascin agents from diverse compound libraries. A new molecule has been identified with considerable fascin inhibitory and migration-arresting capacity, which may lead to the development of new therapies to treat cancer.
Similar content being viewed by others

Introduction
Cancer remains one of the most formidable challenges in the medical field, presenting a myriad of complex mechanisms that evade current therapeutic strategies and demand novel approaches to treatment1. Metastasis is the primary cause of cancer2. Cancer cell migration and invasion are hallmarks of metastatic capacity3, necessitating reorganization of the actin cytoskeleton. This reorganization facilitates the creation of protrusions such as filopodia, lamellipodia, and invadopodia, which are instrumental in cancer cell movement4.
A pivotal protein in this process is fascin, which bundles actin filaments (F-actin), and is integral to the development of protrusive structures. Unlike most normal epithelial tissues, where fascin is minimally expressed or not at all, fascin is notably increased in various human cancers. This elevation is associated with enhanced tumor growth, invasion, and metastasis5,6. Fascin4,5 has been identified as a promising therapeutic target and a marker for aggressive cancer.
Previous studies have shown that the gene encoding fascin (FSCN1) is overexpressed in various types of cancer, including serrated adenocarcinoma (SAC), a subtype of colorectal carcinoma recognized by the World Health Organization (WHO) of its poor prognosis and aggressive behavior. This aggressive nature is demonstrated by increased tumor budding, loss of E-cadherin, and a higher frequency of KRAS or BRAF mutations than in conventional colorectal cancers6,7,8.
Migrastatin and its derivatives effectively inhibit fascin and have shown potential in reducing tumor cell movement, invasion, and subsequent metastasis2. However, the synthesis of these compounds is complicated because of their complex chemical structure, prompting the exploration of other anti-fascin agents, including those derived from indazol-furan-carboxamides9. Other molecules, such as G2 and its derivatives, have also demonstrated strong inhibition of fascin function9. In addition, clear progress has been made in drug repurposing, which has led to the identification of some FDA-approved compounds such as imipramine and raltegravir10,11 as promising fascin inhibitors.
However, in the search for novel fascin inhibitors, FDA-approved compounds only cover a minimal portion of the available chemical space that is yet to be explored. In addition, repurposing compounds is difficult to patent and unlikely to produce benefits in the short term. Thus, chemical compounds from large and underexplored chemical synthesis libraries could be of great value and interest to biotech and pharmaceutical companies.
In this study, we meticulously conducted a ligand-based virtual screening of the Enamine HTS (High-throughput Screening) chemical library (1 368 754 molecules)12, followed by in vitro studies for the best potential hits. This comprehensive approach allowed us to assess the effectiveness of the compounds as inhibitors of cancer cell migration and invasion, thereby providing a solid validation of our findings.
Materials and methods
Library generation
First, we selected the HTS compound library from Enamine to perform ligand-based virtual screening calculations. The library contains 1.368.049 compounds covering a broad chemical space. The compound set was split into 217 files to allow parallelization and optimization of the corresponding calculations. From the command line, we launched the command idbgen (available under the LigandScout license) to generate corresponding 217 LDB format files.
Pharmacophore model generation
Next, we generated a pharmacophore model for G2, which is an effective fascin inhibitor10. We obtained an SDF file containing the chemical structure of the inhibitor from PubChem13. The SDF file was uploaded to LigandScout GUI14, and the corresponding pharmacophore model was generated. Redundant pharmacophore features were filtered out. Additionally, interactions described in previous publications were prioritized over those without influence based on these studies15,16 The model was saved in the PMZ format to screen it against the Enamine library previously generated.
Ligand-based virtual screening by ligand scout
A massive virtual screening calculation was performed using the in-house software Metascreener developed by our research group (https://github.com/bio-hpc/metascreener). The pharmacophore model was screened against all 217 subsets of the previously generated Enamine libraries, and the maximum number of features to be omitted (-a,-allow_omit) for this calculation was 0. Our aim was to obtain compounds whose pharmacophore features matched those of the G2 model. We applied a post-filtering step to obtain only those molecules with a pharmacophore similarity value greater than 0.97, selecting 50 compounds with the highest similarity. Finally, from this set we selected the 12 available compounds in stock at Enamine from this subset to carry out the experimental validation of their inhibitory activity.
Biophysical characterization
The selected compounds were tested using two independent assays based on various principles. First, a primary assay was conducted using Differential Scanning Fluorimetry (Thermofluor), which assesses the ability of a compound to interact with a protein and induce changes in its denaturation temperature. The ability of different compounds to inhibit the catalytic activity of fascin was validated using an F-actin-bundling assay.
Differential scanning fluorimetry (DSF)/thermofluor
Fascin was assayed at a concentration of 1 µM against 79 compounds in a 384-well format, using a final assay volume of 10 µL. Using an Echo® 650 Acoustic Liquid Handler (Labcyte, Cat# LP-000061), 400 nL of each compound from an initial stock of 10 mM in 100% DMSO was dispensed to a final concentration of 400 µM compound and 4% DMSO. Four replicates for each assay point were used to obtain statistically robust measurements.
Two controls were used for the assay: a negative control containing only native fascin at 1 µM in 4% DMSO, and a positive control for inhibitory activity consisting of Fascin 1 µM in the presence of 400 µM BDP13176 (MedChemExpress, Cat# HY-119739) making a final 4% DMSO. Two complete plate columns were used for 32 replicates of positive and negative controls.
The thermal stability profile of fascin was measured using a CFX384 Touch Real-Time PCR Detection System (BioRad, Cat# 1855485) between 25 °C and 95 °C, with a temperature gradient of 1 °C/min. The Tm values and Tm shift induced by the presence of the compounds were obtained using HTSDSF Explorer17, fitting the fluorescence data to a Boltzmann sigmoidal curve.
The Z’-Factor was calculated as a quality control parameter, reflecting the robustness and reproducibility of the assay. For this Thermofluor assay, the Z’-Factor was higher than 0.75, indicating high assay quality.
F-actin bundling assay
We implemented an image-based assay to visualize F-actin bundling mediated by fascin cross-linking following the methodology reported by Huang et al. (2014). Labeling was performed with phalloidin conjugated to a commercial fluorescent probe (Alexa Fluor 488-Phalloidin, Thermo Fisher, Cat# A12379), which targets the positive charges of F-actin filaments and bundles, allowing for visualization via fluorescence microscopy. The Operetta CLS High Content Analysis System (Revvity, Cat# 8900) was used to capture images, which were subsequently processed and analyzed using Harmony™ (Revvity).
A total of 79 compounds were assayed in this study. Each compound (1 µL), dissolved in 100% DMSO from an initial stock of 10 mM, was dispensed using Echo® 550 Acoustic Liquid Handling (Beckman Coulter, Cat# 100027). Following this, 15 µL of pure fascin at a concentration of 0.5 µM in buffer (20 mM HEPES, 100 mM NaCl, pH 7.45) was dispensed onto a 384-well plate (Corning, Cat# 3677) using an automatic dispenser (Multidrop Combi, Thermo Fisher, Cat# 5840300) and allowed to incubate for 30 min. Next, 15 µL of polymerized actin was added at a concentration of 0.5 µM (in 100 mM KCl, 20 mM Tris-HCl, pH 7.5, 2 mM MgCl2, 1 mM DTT, and 1 mM ATP; Cytoskeleton Inc., Cat# BK037) at a final concentration of 100 µM for each compound. After a further 30 min of incubation, 10 µL of Alexa Fluor-488 Phalloidin (diluted 50-fold from 100% methanol stocks) was added to stain F-actin, and the mixture was incubated in the dark for 1 h. Subsequently, 25 µL of the final solution was transferred to a 384-well plate coated with poly-D-lysine (Cat #354663; Corning) and incubated for 20 min before imaging.
Each assay included 16 wells as the negative control, comprising F-actin in the absence of fascin, to verify that actin bundling depends on the presence of fascin, and a positive control, BDP-13,176 (Cat# 10009022), a known fascin inhibitor used at the same concentration as the tested compounds. The Z’-Factor was calculated as a quality control parameter based on image-defined thickness and texture parameters from the Harmony software, yielding a value higher than 0.5.
Cell culture
Two human colorectal adenocarcinoma cell lines (DLD-1 (CLS Cat# 300220/p23208_DLD-1, RRID: CVCL_0248) and HCT-116 (CLS Cat# 300195/p19841_HCT116.html, RRID: CVCL_0291)) were obtained from the American Type Culture Collection (ATCC, Rockville, Maryland, USA). The cell lines were cultured at 37 °C in high-glucose Dulbecco’s modified Eagle’s medium (DMEM) containing 10% heat-inactivated fetal bovine serum (FBS) and 50 µg/mL gentamicin (Cat#15750037, Thermo Fisher) in an atmosphere of 5% CO2 and 95% humidified air. Subcultures were performed when the cells reached 90% confluence. According to the American National Standards Institute, human cell line identification by short tandem repeat profile testing has shown an appropriate match between the HCT-116 and DLD-1 cell lines.
Cell viability assay
Exponentially growing cells were plated in quintuplicate in flat-bottomed 96-well plates (Nunc, Roskilde, Denmark) at 1500 cells/well and grown in a humidified 5% CO2 incubator at 37 °C. On the day after plating, the drugs were serially diluted from 0.1 to 100 µM. The control wells contained medium without the drug plus 0.1% dimethyl sulfoxide (DMSO) (drug carrier). After 72 h, cell viability was evaluated by adding 50 µL of activated XTT solution (Biotium, VWR, catalog number: 30007) to each well, and the plates were incubated at 37 °C for 4 h18. Finally, the absorbance at 450 nm was measured by background subtraction at 630 nm using a Spectramax ID3 plate reader (Molecular Devices, VWR).
Organoids
Patient-derived colorectal cancer organoids were established and cultured in our Pathology Department, Hospital General Universitario Santa Lucía, using IntestiCult™ Organoid Growth Medium (Human) following the manufacturer’s protocol (StemCell Technologies)19. The histopathological characteristics and fascin expression of these organoids can be found in the supplementary table (Table S1). The medium from each well intended for passage was removed without disturbing the domes; each well was then filled with 500 µl of ice-cold D-PBS, the solution was pipetted up and down to facilitate matrix breakage, and the suspensions were transferred to a 15 ml conical tube, which was repeated at least once to ensure complete retrieval of all organoids, with well checks conducted under an inverted microscope. Subsequently, the tubes were centrifuged at 290 g for 5 min at a temperature between 2 and 8 °C, the supernatant was discarded, without disturbing the organoid pellet, and then treated with 1 ml of TrypLE™ Express Solution and incubated for 5 min at 37º C. After incubation, 100 µL of FBS was mixed with the suspension, the tubes were centrifuged at 250 × g for 5 min at 2–8 ºC, and the supernatant was discarded. To the obtained pellet, 1000 µl of DMEM/F-12 + 1% BSA was added, and the number of organoids in the suspension was counted using a Countess™ 3 Automated Cell Counter with Countess Reusable Slides (Invitrogen, Thermo Fisher Scientific). Considering that the domes of the 96-well plate corresponded to 5 µL and each dome had 5000 cells, the correct amount of suspension to be used for the assay was calculated, always considering the 1:1 ratio of cell suspension and Matrigel®, and that each treatment/condition was performed in quadruplicate. Pre-wetting the pipette tips with cold DMEM/F-12 + 1% BSA before manipulating the cell suspension was used to prevent sticking to the wall of the pipette tip and to place the 96-well plate in the incubator for at least 30 min before starting the assay. After plating, the plates were placed in an incubator at 37 °C and 5% CO2 for 15 min to allow the solidification of the domes. Then, approximately 100 µL of D-PBS was added to the wells at the borders to avoid evaporation of the cell culture medium, and 50 µL of complete IntestiCult™ Organoid Growth Medium at room temperature (without Y-27632 and gentamicin) was added. After 48 h, the control with the drug carrier (0.1% DMSO) and the respective drugs in serial dilutions from 0.1 to 100 µM were added to the corresponding wells (in quadruplicate) in a total volume of 100 µL per well. Subsequently, the plates were left in the incubator for 5 days, and photos of the wells were taken every day using an inverted microscope to register the growth and treatment effect on the organoids. On the fifth day, cell viability was determined using the CellTiter-Glo® Luminescent Cell Viability Assay (Promega), following the manufacturer’s guidelines. Luminescence was measured using a Spectramax ID3 plate reader (Molecular Devices)20,21,22,23,24.
Molecular modeling
We conducted molecular modeling of fascin structure to determine the interaction between the predicted compounds and fascin. First, we used the PDB code 6B0T, which corresponds to the complexation between the crystallographic structure of fascin and NP-G2-029, a derivative of G2.
The structure was pre-processed using MAESTRO’s Schrödinger software25. Chain A was extracted and the corresponding hydrogens and charges were added to the structure. The new structure was saved in the mol2 format. A similar protocol was used with AutoDockTools26 software, which performed the same tasks and applied the AD type to the atoms. In this case, the structure was saved in the pdbqt format.
The predicted ligands were converted to mol2 and pdbqt formats using ChemAxon (MolConvert tools)27 and AutoDockTools, respectively. In each case, the corresponding charges were added.
Blind docking
Once the protein and candidate ligand files had been processed, two blind docking calculations were performed using the NP-G2-029 Fascin (PDB:6B0T)15 and free protein (PDB:3P53) structures to explore the conformational space and possible binding sites of the predicted compound. For each blind docking simulation, we used two docking software packages, Lead Finder28 and AutoDock Vina29. A consensus between the two methods was reached to determine the average pose.
Targeted docking
Targeted docking was performed in actin-binding site 2 coordinates, where NP-G2-044 binds to fascin in the 6B0T PDB structure15. The ligand positions for the top clusters identified in the previous blind docking were determined. Lead Finder and AutoDock Vina were used, with the respective consensus between both. In the next step, we chose the pose obtained by the Lead Finder for the molecular dynamics (MD) simulation.
MD simulations
To validate both the overall stability of the complex in an aqueous system and whether its binding to actin-binding site 2 was maintained over time, we performed an MD simulation of the fascin-Z1362873773 complex in the binding area with the pose obtained by the Lead Finder virtual screening calculations, which obtained the top one in both blind docking calculations. We also ran an MD simulation for crystallized fascin in complex with NP-G2-029 (6B0T), which is a ligand that binds to actin-binding site 2 and has been more extensively studied.
We performed 100 ns molecular dynamics simulations on the fascin structure 6b0t using the Z1362873773 pose from blind docking (BD) calculations with the top-ranked result found in the known actin-binding site 215. This pose was generated using LeadFinder for the 6b0t structure. This was generated by LeadFinder for the 6b0t structure. In addition, we performed an MD simulation for the NP-G2-029 and fascin complex using the crystallographic structure (PDB: 6B0T) to compare the results with a complex with a ligand bound at the same site15. For this, we first generated the topology of the ligand for each pose using an automatic script that utilizes ACPYPE30,31.
We followed the subsequent steps of the molecular dynamic simulations using GROMACS 2022.332. These simulations were launched on the Picasso server (https://www.scbi.uma.es/web/es/inicio/) using a GPU (NVIDIA A100-SXM4-40GB) and 4 GB of RAM. We first created protein topology using the gmx pdb2gmx command, specifying AMBER99SB as the force field. The simulation box was defined by the solvated and added ions. The next step was an energy minimization stage of 2000 ps. Subsequently, a single NvT equilibration stage of 50,000 ps and five NpT equilibration stages of 50,000 ps each were carried out. Finally, the dynamics were run at 100 ns, and the final trajectory generated was extracted in different frames to compare the ligand stability and movement with respect to the binding pose.
Trajectories analysis
The results obtained by the MD simulation were analyzed using ASGARD (https://github.com/bio-hpc/ASGARD), an in-house tool developed by our group33. We calculated the non-bonded interactions and hydrogen bonds between the final ligand and fascin structure for all simulation frames. In addition, we studied the stability of the protein-ligand complex and the flexibility and dynamics of fascin produced by its interaction with the binding site. The required input files were prepared for the tool and plots and raw data were generated.
Drug-like property predictions
Drug-like properties were calculated using the MAESTRO Schrödinger tool QikProp34. The properties were obtained for the best-known fascin inhibitors, imipramine, migrastatin, raltegravir, NP-G2-044, G2, BDP-13,176, and the predicted compound, Z1362873773. The values are saved in a CSV file. A comparative table was generated using the data. Solubility prediction calculations using the ALOGPS 2.1 program were also performed with Compound 1 and Compound 2 to make the compound solubility comparison more robust.
A ligand-based screening for two models created from Z1362873773 against the DrugBank compound database was performed to study possible alternative targets where the hit molecule could act. The first model was created by calculating all possible features of the molecule, whereas the second model only had the Z1362873773 features that matched the G2 pharmacophore model. For these calculations, the maximum number of omitted features was four for the first model and one for the second.
Results
Ligand-based in silico screening
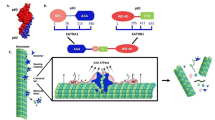
We obtained 212 compounds with features matching the pharmacophore model for G2 generated by LigandScout (Figs. 1, S1, S2). Those molecules with a Relative-Pharmacophore score higher than 0.97 were chosen for subsequent steps. Finally, the 12 compounds selected (Z1362873773, Z335377818, Z29860525, Z335377814, Z735522228, Z62748384, Z107323688, Z44912418, Z1229770825, Z79431411, Z1797081400 and Z237374404) were tested in vitro in the study’s next step. Figure 1c shows a graphical comparison of the pharmacophore model for G2 with Z1362873773, the one with the best in vitro results. The chemical structures of both compounds and the pharmacophoric features extracted from each compound using LigandScout are shown in Fig. 1.

(a) Chemical structure of G2 with the pharmacophore features assigned using LigandScout. (b) Chemical structure of Z1362873773 with the pharmacophore features assigned using LigandScout. (c) Comparison between the tridimensional chemical structures of G2 (cyan) and the Enamine compound obtained by virtual screening, Z1362873773 (orange). Color legend: pale yellow sphere—hydrophobic interactions, red sphere—hydrogen bond acceptor, green sphere—hydrogen bond donor. The arrows indicate the direction of the hydrogen bond.
Each of the 12 selected compounds underwent a biophysical characterization. Z1362873773 was the only compound that showed activity in the bundling activity, which was used to validate the activity. The rest of the compounds showed significant signals in the assays performed.
Z1362873773 reduces the fascin bundling activity
The binding of Z1362873773 to Fascin was initially assessed by DSF. These results are available in the corresponding section in the Supplementary Data (Figure S3). The effect of Z1362873773 on fascin bundling activity was assessed using an imaging-based F-actin bundling assay. Fascin cross-links the F-actin filaments into straight, compact, and rigid bundles bearing negative charges captured by the positively charged amino acid poly-D-lysine polymer that coats the well surface of the plates. By labeling F-actin with phalloidin conjugated with a fluorescent dye, we visualized the F-actin bundle structures using a high-content imaging system. When an inhibitor of fascin is present in the well, the F-actin filaments are unstructured and not visible in the assay. At a 400 µM concentration, Z1362873773 produced total inhibition of the formation of F-actin bundles (Fig. 2), confirming the results obtained in the Thermofluor assay.

Representative fluorescence images from a high-content assay assessing the effect of Z1362873773 on Fascin that cross-links Actin filaments (F-actin) into bundles. Actin filaments are labeled with Alexa 488-phalloidin, allowing visualization of bundles by a bioimaging system. The top row shows the images of four independent wells containing 15 µM Fascin and 15 µM Actin (Fascin/Actin), where Fascin promotes the formation of thick, bundled Actin fibers. To confirm that fiber formation is Fascin-dependent, the second row displays the images of four wells containing Actin alone, where no bundled fibers are observed. Similarly, in the third row, the presence of a known Fascin inhibitor (400 µM BDP-13176) prevents fiber formation, reinforcing the requirement of Fascin for Actin bundling. The bottom-left images show two independent replicates of 400 µM Z1362873773 compound tested against Fascin/Actin, demonstrating that Z1362873773 inhibits Fascin-mediated bundling, as no fiber formation is detected. Finally, the bottom-right images serve as a control for non-specific interactions, where Z1362873773 is tested against Actin alone, confirming the absence of unwanted or nonspecific binding.
Z1362873773 affects cancer cell viability
Viability assays were performed on DLD-1 and HCT-116 cells to determine the working concentrations of the drugs. HCT-116 was more sensitive than DLD-1, and the working concentrations were set for subsequent in vitro studies at 12.5 and 20.4 µM Z1362873773 (Fig. 3).

Viability assays for HCT-116 and DLD-1 cells were performed in three independent experiments. Each line represents a normalized curve fit for an independent experiment. Each point was determined in quintuplicate (mean ± standard error of the mean).
Z1362873773 inhibits the cancer cells’ migration
To investigate the impact of Z1362873773 on cell migration, cells treated with Z1362873773 were analyzed for motility using a wound-healing assay at 24 and 48 h. As shown in Figs. 4 and 20 µM Z1362873773 significantly reduced the migration of all the tested cell lines (p < 0.05). The inhibitory effect of Z1362873773 was similar to that of G2, at the same concentration. Figure 4-A shows a detailed decrease in the migration of HCT-116 cells at 24 and 48 h, respectively. Figure 4-B shows a detailed decrease in the migration of DLD-1 cells at 24 and 48 h, respectively.

Wound healing assays for HCT-116 (A) and DLD-1 (B) cells at 24 and 48 h using G2 and Z1362873773.
Z1362873773 shows a notably cytotoxic effect in organoids
Viability curves in the presence of G2 or Z1362873773 for primary (107T and 114T) and metastatic (06 M) colorectal cancer organoids. Compound Z1362873773 showed 2–3 times significantly higher cytotoxic effect in our ex vivo model compared to G2 (Fig. 5).

Viability curves in the presence of G2 or Z1362873773 for primary (107T and 114T) and metastatic (06 M) colorectal cancer organoids.
Blind docking finds a top pose in the actin-binding site 2
We performed docking experiments using two different fascin structures. 6B0T corresponds to the structure obtained when NP-G2-029 (G2 analog) binds to actin-binding site 2 (Fig. 6). Blind docking using AutoDock (Fig. 6a) showed the pose inside the NP-G2-029 binding site as the first cluster − 8.9 kcal/mol. Using the LeadFinder method (Fig. 6b), blind docking calculations were also performed in the free protein structure (PDB:3p53) to explore the structural flexibility of fascin. In the AutoDock Vina results, the positions for the top three and four clusters were found to be close to the position of actin-binding site 2, and the third cluster pose interacted with Phe216, one of the key residues for actin-binding site 2. In the results obtained from LeadFinder (Fig. 5b), the first and second clusters were located at actin-binding site 2, exhibiting binding energies of -8.54 and − 7.39 kcal/mol, respectively. Notably, Cluster 1 interacted with Phe216 through a hydrogen bond.

Poses obtained by BD calculations for fascin-free proteins (PDB ID:3P53). (a) Poses obtained using AutoDock Vina. The poses obtained represent the top three and four clusters, respectively. (b) Poses obtained using LeadFinder. The obtained poses represent the top one and two clusters.
Regarding the blind docking calculations performed for the 6b0t structure, using AutoDock Vina (Fig. 7a) and LeadFinder (Fig. 7b) methods, actin-binding site 2 was detected in the top 1 cluster. The binding energy values were − 8.92 and − 8.72 kcal/mol for AutoDock Vina and Lead Finder, respectively. Furthermore, key residues at the binding site were identified in both the cases. We found hydrogen bond interactions between the compound and Phe216, and other hydrophobic interactions with crucial residues such as Ile93, Trp101, and Phe1415.

Poses obtained by BD calculations for fascin-NP-G2-029 (PDB ID:6B0T). (a) Pose obtained using AutoDock Vina. The obtained poses represent the top 1 cluster. (b) Pose obtained using LeadFinder. The obtained poses represented the top 1 cluster.
Target docking shows several interactions with actin-binding site 2 key residues
Blind docking calculations demonstrated that actin-binding site 2 was a potential binding site for the compound. Next, we carried out target docking at this position in the 6B0T structure (corresponding to the NP-G2-029 fascin complex). The AutoDock Vina and LeadFinder techniques obtained docking scores of -8.35 and − 8.27 kcal/mol, respectively. Subsequently, we determined the specific interactions observed by each method (Table S2). The interactions between the compound and the specific fascin residues in the binding site 1 found by AutoDock Vina calculations were the following: Phe14 (3.53 Å), Leu48 (3.80 Å), Ile93 (3.66 Å, 3.30 Å) and Glu215 (3.48 Å) for hydrophobic interactions; Trp101 (2.81 Å) and Arg217 (3.94 Å) for hydrogen bonds; and Trp101 (4.76 Å) for π-Stacking. Regarding the interactions obtained by LeadFinder software, we found hydrophobic interactions between the ligand and the residues Phe14 (3.82 Å), Leu16 (3.37 Å), Ile93 (3.33 Å, 3.27 Å), Trp101 (3.15 Å, 3.39 Å), Val134 (3.67 Å, 3.58 Å) y Phe216 (3.06 Å). LeadFinder also detects hydrogen bonds interaction with Trp101 (4.06 Å), Phe216 (3.00 Å, 3.04 Å, 2.81 Å) y Arg217 (3.41 Å).
Figure 8, a PyMOL session, visually represents the results and displays both poses obtained using AutoDock Vina and Lead Finder in the same fascin structure, allowing for a clear comparison. The residues obtained in each case matched some of the key residues at actin-binding site 215, further confirming the accuracy of our findings.

Pymol session showing the interactions between Z1362873773 and the crystallographic structure for fascin (PDB:6B0T) obtained by docking calculation using AutoDock Vina (green) and LeadFinder (purple). These residues match the known critical residues in interactions with the fascin structure. Pink interactions show hydrophobic interactions, red-colored interactions indicate hydrogen bonds, green-colored interactions indicate π-stacking, and cyan-blue-colored interactions correspond to π-cation interactions.s.
MD analysis shows the Z1362873773 stability in the actin-binding site 2 pocket
We performed an MD simulation of Z1362873773 to study the stability of the ligand in the complex over time and in an aqueous system considering fascin flexibility. In addition, we performed another MD simulation for the NP-G2-029 Fascin complex as a comparative test with a known fascin inhibitor bound to the actin-binding site 2 system. Most of the figures related to this analysis are available in the Supplementary Data.
The initial focus was on RMSD during the simulation of the protein as the ligand, a key indicator of the stability of the system in both cases. Figure S4 illustrates the stabilization of the ligands in each complex, with both NP-G2-029 and Z1362873773 RMSD values maintaining a consistent range of 0.1 to 0.2 throughout the MD (Figure S4). As expected, fascin, a known flexible protein, demonstrated significant flexibility during simulation.
Molecular Mechanics Poisson-Boltzmann Surface Area (MMPBSA) analysis estimates the binding free energy between a protein and its ligand. We performed MMPBSA calculations for both complexes to examine the stability and strength of the binding between NP-G2-029 and Z1362873773 in the actin-binding site 1 of fascin. The NP-G2-029 fascin complex stabilized the binding energy from the first 20 ns to between − 250 and − 300 kJ/mol (Figure S5-A). For Z1362873773, the binding energy of its complex with fascin started to stabilize in the first 10 ns, remaining at binding energy values of -200 and − 250 -kJ/mol2 (Figure S5-B). These analyses also revealed that van der Waals forces play a more significant role than electrostatic interactions.
Finally, we determined the main nonbonded interactions obtained by MD simulations. At this point, most of the results coincided with the docking calculation results. Two more residues were highlighted in the NP-G2-029 Fascin complex. Phe216 and Asp217 reveal high values of Total Energy, obtaining − 29.1 and 23.5 kJ/mol for Van der Waals energy and − 9.3 and − 11.1 kJ/mol for electrostatic energy, respectively (Figure S6-A). Regarding Z1362873773 (Figure S6-B), the interactions obtained followed a more even pattern. Several residues have reported total energy values between − 10 and − 20 kJ/mol. Among them, we can see some of them that match with the docking results and are known as crucial residues in the fascin union15, such as Phe14 (E_total= − 13.3 kJ/mol2), Leu48 (E_total= − 10.1 kJ/mol2), Trp101 (E_total= − 17.9 kJ/mol2), Val134 (− 11.9 kJ/mol2) and Phe216 (− 15.7 kJ/mol2). Figures S7–12 show other additional analysis performed.
Consequently, the MD simulations correlated with the experimental results, showing that Z1362873773 binds to actin-binding site 1. The consensus of the RMSD, free binding energy validation by MMPBSA, and interaction analysis presented a potential ligand with a stable and strong union with fascin, avoiding its function.
Drug-like property predictions
By meticulously predicting the ADMET (Absorption, distribution, metabolism, excretion, and toxicity) values for compound Z1362873773, we provided a comprehensive understanding of its pharmacokinetic profile. This information is crucial for its potential use in subsequent in vivo experiments and clinical trials. Our comparison with known fascin inhibitors, some of which have already been used as drugs, further confirms the significance of our findings. Table S3 presents the predicted ADMET values for each molecule, setting the acceptable ranges that deemed them favorable for a drug.
First, we discuss the predicted descriptors related to the bioavailability of Z1362873773. For this, we focused on the following properties of Table S3: the logP for aqueous solubility (logPAQS), the Apparent Caco-2 Permeability (C2P), the number of primary metabolites (NPM), the Jorgensen’s Rule of Three (Jorgensen) and the Lipinski’s Rules35. logPAQS is critical in this context because drug absorption in the body strongly depends on solubility. In this case, compound Z1362873773 showed a value of -5.831, which improved the predictions for G2 and NP-G2-044 compounds. The number of primary metabolites (4) was also low enough to not affect bioavailability because of the production of metabolites, which could affect the bioavailability of the main compound. The compound violated only one Jorgensen rule and 0 for the Lipinski rule, which is a good result for its determination as a drug. Thus, in the four cases, the values predicted by QikProp for all four descriptors were within the range considered positive for a suitable pharmacokinetic profile, even improving those calculated for some already known fascin inhibitors.
Plasma protein binding is a critical factor in determining its effectiveness. It influences the concentration of free and active drugs in the blood and their distribution and removal from the body. Strong binding to plasma proteins such as human serum albumin can decrease drug availability for therapeutic purposes, but can also prolong its effects by acting as a reservoir36. The log Khsa value provides important insights into these pharmacokinetic properties. Z1362873773 shows a value equal to 0.656. Thus, it falls within the established range to suggest that its interaction with plasma proteins is not sufficiently robust to impact the molecule’s availability and distribution35.
We also obtained favorable predicted values for human absorption. Qualitative Human Oral Absorption (HOA) and human oral absorption in the gastrointestinal tract (HOA GI) were notably high, reaching 100%. These findings suggested that Z1362873773 can be efficiently absorbed orally, indicating its potential as a drug candidate. Skin permeability is a potential route for administration of several drugs. A crucial determinant in this regard is the QP log Kp for skin permeability (log Kp) value, which indicates the ease of access of the molecule to the skin. For compound Z, QikProp predicted a QP log Kp value of − 2.157. Typically, values approaching − 2 suggest favorable permeability, indicating a probable entry route through the skin for this compound37,38. Finally, in this specific case, the drug should not cross the blood-brain barrier (BBB) because Z1362873773 is intended to act outside the central nervous system. QikProp calculates the predicted log BR/BL value, which indicates the ability of a compound to penetrate the BBB. The calculated value for Z1362873773 compound is -0.481. A negative log BR/BL value suggested a low likelihood of BBB penetration35,39.
Our predictions of ADMET properties by QikProp for Z1362873773 are within the expected range for a potentially effective drug and highlight its safety and efficacy. According to these predictions, Z1362873773 demonstrates favorable oral absorption and skin penetration. Its bioavailability is sufficient, and its plasma protein binding is weak enough not to interfere with its bioavailability significantly. Moreover, the predicted values suggest that Z1362873773 is unlikely to cross the BBB and cause adverse effects in the central nervous system. These findings, coupled with the fact that Z1362873773 displayed predicted descriptor values that were more promising than those of known inhibitors, indicate its potential as a drug candidate. For example, significant improvements were observed in the solvent-accessible surface area (SASA), hydrogen bonding, and ionization potential, which are crucial for the interaction of the drug with the target and its surrounding environment40. Furthermore, Z1362873773 showed favorable log HERG K + channel values, suggesting a reduced risk of cardiac arrhythmias as a potential side effect41.
Regarding the solubility of Z1362873773, the values obtained by solubility prediction calculations performed by the ALOGPS 2.1 program show that Z1362873773 (logP: 3.70; logS: − 4.08) has values more according to a more soluble compound as NP-G2-044 (logP: 4.63; logS: − 4.08). Z1362873773 showed a predicted solubility value slightly better than NP-G2-044, a fascin inhibitor already tested with in vivo assays and clinical trials.
In our study of toxicity target prediction, we identified several potential targets for each of the two pharmacophoric models. Initially, we analyzed the hit molecules and their corresponding targets using LigandScout based on a model incorporating all features (Table S4). Tasquinimod, a recognized anti-angiogenic agent, has emerged as a primary therapeutic target. Although the literature references some immunomodulatory proteins, no specific target has yet been confirmed for Tasquinimod42,43. Other compounds identified using this method lacked notable targets associated with adverse effects. Overall, no targets associated with toxicity or adverse effects were identified in this model.
Our analysis using the Z1362873733 model derived from the G2 compound model features (Table S5), NP-G2-044, a derivative of the Genin compound structure, stands out as the top candidate in our calculations. Furthermore, we have come across various other molecules that serve different functions, including acting as androgen receptor modulators, antidepressants, and even treatments for other types of cancer, such as enasideb for acute myeloid leukemia (AML). Nonetheless, we did not encounter any targets or functions within these identified compounds that could be considered harmful or raised concerns regarding potential side effects.
Feasibility of the synthesis pathways for Z1362873773
Finally, to compare the advantages of Z1362873773 against G2, we analyzed the synthesis pathways for each compound (Figure S13). The first advantage of Z1362873773 is related to the quantity of compounds required for each synthetic pathway. Specifically, Z1362873773 requires a relatively small number of compounds and reagents, generally on the milligram (mg) scale, whereas G2 synthesis requires a larger gram (g) scale. The starting compounds in the synthesis of Z1362873773 (EN300-31270 and EN300-39965) are available from both enamine and other chemical vendors. Furthermore, the number of synthesis steps is a key factor for optimizing the process. Z1362873773 synthesis requires just one step, whereas G2 synthesis involves two distinct steps.
When considering the reagents required, G2 necessitates a broader range, including t-butoxide (t-BuOK), Tetrahydrofuran (THF), acetonitrile (MeCN), 1-methyl-1 H-imidazole, and methane sulfonyl chloride (MsCl), the latter of which is notably toxic, light-sensitive, corrosive, and causes irritation. In contrast, Z1362873773 synthesis relies on two primary reagents, 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide (EDC) and 1-hydroxybenzotriazole (HOBt). Both syntheses demonstrate comparable safety and feasibility regarding solvents. The synthesis of Z73 involves using water, DMF, chloroform, and methanol. In comparison, the synthesis of G2 utilizes tetrahydrofuran (THF), acetonitrile (MeCN), dichloromethane (DCM), water, and diethyl ether (Et2O).
Additionally, cost and time are distinguishing factors, as Z1362873773 is more affordable, and its synthesis is slightly faster than that of G2.
Discussion
Fascin is one of the main target proteins involved in CRC migration and propagation of colorectal cancer. This protein is overexpressed in different types of cancer, especially in those with a poor prognosis and more aggressive invasion. Several studies have identified possible molecules that can inhibit fascin function. However, the chemical space explored in this context is limited. This study explored a more extensive chemical space using the Enamine HTS library, which contains more than one million compounds, to identify potential fascin inhibitors.
We completed a workflow to filter the entire library, searching for potential inhibitors. The workflow starts with ligand-based virtual screening, where we calculate the pharmacophoric similarity between G2, a known fascin inhibitor, and all the compounds from the library. Next, we validated the interactions of these compounds with fascin using biophysical in vitro assays. The next step was to determine the effect of the inhibitors on the migration and viability of assays in colorectal cancer cell lines and organoids. Finally, the results obtained by docking and MD calculations of the complex formed between the final hit and fascin showed a high correlation with the mechanistic information about already known fascin inhibitors in terms of interaction and binding in the actin-binding sites of fascin.
Our results demonstrate that our study protocol holds promise for identifying more potential compounds. Moreover, this workflow could be applied in various contexts, including those with new potential or even larger libraries than the HTS Enamine library. This could significantly enhance the ability to identify compounds with high activity against specific backgrounds, potentially revolutionizing cancer treatment and drug development.
From our workflow, we isolated a novel compound, Z1362873773, which shares similarities with the known fascin inhibitor, G2. This synthetic molecule binds to fascin and inhibits the formation of actin bundles that are crucial for cancer cell migration. In migration and viability assays, Z1362873773 demonstrated values comparable to those of G2 in both in vitro and ex vivo models. Although the dose-response curves are based on only four different concentrations, it is feasible to estimate an IC50 value under these conditions. This is particularly relevant when working with novel compounds that are expensive and challenging to produce in large quantities. While adding more concentrations could provide a more defined ‘bottom’ for the curve, the data obtained with the four concentrations still permits a reasonable estimation of the IC50.
Fascin expression in DLD-1 is low in contrast to HCT-116. The variation between HCT-116 and DLD-1 regarding endogenous fascin expression levels was previously stated in the studies by Montoro-García et al. (2020)10 and Alburquerque-González et al. (2020)11. The differential expression of fascin could explain a superior on-target efficacy in HCT-116. These studies allow us to postulate a correlation between endogenous fascin expression levels and the efficacy of the evaluated compounds in affecting cell viability, potentially associated with fascin inhibitory activity. Despite the similar cytotoxicity observed among the organoids, all derived from conventional MSS adenocarcinoma, this could be attributed to the uniform expression of fascin and their shared histological subtype, likely establishing a common baseline in cellular response.
Furthermore, the prediction of ADME properties and target proteins involved in toxicity suggested that Z1362873773 possesses ideal characteristics for drug action, marking a significant step forward in potential cancer treatments.
Ultimately, we compared the synthesis routes for G2 and Z1362873773 to explore the significant differences and ease of synthesis of the newly discovered compound compared to the G2 compound. Several differences can position Z1362873773 as a compound with greater ease of synthesis than the original G2 compound.
This study represents a significant advancement in the field of fascin inhibitors. Even though some fascin inhibitors have been previously proposed, this work expands the chemical space by identifying a structurally distinct compound (Z1362873773) through a large-scale screening approach, in contrast to prior studies that primarily focused on known or repurposed drugs. The proposed workflow integrates ligand-based virtual screening, in vitro validation, and molecular dynamics simulations to identify novel fascin inhibitors and could be adapted to drug discovery in other contexts. Furthermore, these findings reveal a simpler synthetic alternative to existing fascin inhibitors, such as G2 or migrastatin, while maintaining comparable efficacy. As a result, this study sets a precedent for lead optimization, with the potential to enhance potency and selectivity in future preclinical investigations.
In summary, we developed an efficient ligand-based virtual screening workflow, which led to the discovery of a novel anti-fascin agent from a large-scale combinatorial library (Enamine). Unlike previous studies that used smaller libraries of FDA-approved compounds, our approach explored a more comprehensive chemical space. Starting with the pharmacophoric features of the G2 compound, we identified a new fascin inhibitor with unique chemistry that is potentially free of intellectual property. Additionally, we provided molecular insights into how these compounds interact structurally with fascin, suggesting that this protocol could be applied to discover other anti-fascin agents.
Data availability
Data is provided within the manuscript and in the supplementary information document.
References
The global challenge of cancer. Nat. Cancer 1, 1–2. https://doi.org/10.1038/s43018-019-0023-9 (2020).
Chen, L. et al. Migrastatin analogues target fascin to block tumour metastasis. Nature 464, 1062–1066. https://doi.org/10.1038/nature08978 (2010).
Hanahan, D. Hallmarks of cancer: new dimensions. Cancer Discov. 12, 31–46. https://doi.org/10.1158/2159-8290.CD-21-1059 (2022).
Machesky, L. M. & Li, A. Fascin: invasive filopodia promoting metastasis. Commun. Integr. Biol. 3, 263–270. https://doi.org/10.4161/cib.3.3.11556 (2010).
Conesa-Zamora, P. et al. Expression profiling shows differential molecular pathways and provides potential new diagnostic biomarkers for colorectal serrated adenocarcinoma. Int. J. Cancer. 132, 297–307. https://doi.org/10.1002/ijc.27674 (2013).
Tan, V. Y. et al. Association of fascin-1 with mortality, disease progression and metastasis in carcinomas: a systematic review and meta-analysis. BMC Med. 11, 52. https://doi.org/10.1186/1741-7015-11-52 (2013).
Ristic, B. et al. Emerging role of Fascin-1 in the pathogenesis, diagnosis, and treatment of the Gastrointestinal cancers. Cancers (Basel). 13, 2536. https://doi.org/10.3390/cancers13112536 (2021).
García-Solano, J. et al. Immunohistochemical expression profile of β-catenin, E-cadherin, P-cadherin, laminin-5γ2 chain, and SMAD4 in colorectal serrated adenocarcinoma. Hum. Pathol. 43, 1094–1102. https://doi.org/10.1016/j.humpath.2011.08.020 (2012).
Han, S. et al. Improving fascin inhibitors to block tumor cell migration and metastasis. Mol. Oncol. 10, 966–980. https://doi.org/10.1016/j.molonc.2016.03.006 (2016).
Montoro-García, S. et al. Novel anti-invasive properties of a Fascin1 inhibitor on colorectal cancer cells. J. Mol. Med. 98, 383–394. https://doi.org/10.1007/s00109-020-01877-z (2020).
Alburquerque-González, B. et al. The FDA-Approved antiviral raltegravir inhibits Fascin1-Dependent invasion of colorectal tumor cells in vitro and in vivo. Cancers 13, 861. https://doi.org/10.3390/cancers13040861 (2021).
HTS Collection (Enamine, 2024). https://enamine.net/compound-collections/screening-collection/hts-collection
Kim, S. et al. PubChem 2023 update. Nucleic Acids Res. 51, D1373–D1380. https://doi.org/10.1093/nar/gkac956 (2023).
Wolber, G. & Langer, T. LigandScout: 3-D pharmacophores derived from Protein-Bound ligands and their use as virtual screening filters. J. Chem. Inf. Model. 45, 160–169. https://doi.org/10.1021/ci049885e (2005).
Huang, J. et al. Structural insights into the Induced-fit Inhibition of fascin by a Small-Molecule inhibitor. J. Mol. Biol. 430, 1324–1335. https://doi.org/10.1016/j.jmb.2018.03.009 (2018).
Lin, L. et al. Potential inhibitors of fascin from A database of marine natural products: A virtual screening and molecular dynamics study. Front. Chem. 9. https://doi.org/10.3389/fchem.2021.719949 (2021).
Martin-Malpartida, P. et al. HTSDSF explorer, A novel tool to analyze High-throughput DSF screenings. J. Mol. Biol. 434, 167372. https://doi.org/10.1016/j.jmb.2021.167372 (2022).
de Jong, P. R. et al. ERK5 signalling rescues intestinal epithelial turnover and tumour cell proliferation upon ERK1/2 abrogation. Nat. Commun. 7, 11551. https://doi.org/10.1038/ncomms11551 (2016).
Bouffi, C. et al. In vivo development of immune tissue in human intestinal organoids transplanted into humanized mice. Nat. Biotechnol. 41, 824–831. https://doi.org/10.1038/s41587-022-01558-x (2023).
Itatani, J. & Olsen, C. Measure cancer cell viability using a homogeneous, stable luminescence assay. (2019).
Sato, T. et al. Long-term expansion of epithelial organoids from human colon, adenoma, adenocarcinoma, and Barrett’s epithelium. Gastroenterology 141, 1762–1772. https://doi.org/10.1053/j.gastro.2011.07.050 (2011).
Fujii, M. et al. A colorectal tumor organoid library demonstrates progressive loss of niche factor requirements during tumorigenesis. Cell. Stem Cell. 18, 827–838. https://doi.org/10.1016/j.stem.2016.04.003 (2016).
Dijkstra, K. K. et al. Generation of tumor-Reactive T cells by Co-culture of peripheral blood lymphocytes and tumor organoids. Cell 174, 1586–1598e12. https://doi.org/10.1016/j.cell.2018.07.009 (2018).
Vlachogiannis, G. et al. Patient-derived organoids model treatment response of metastatic Gastrointestinal cancers. Science 359, 920–926. https://doi.org/10.1126/science.aao2774 (2018).
Schrödinger Release 2022-1: Desmond Molecular Dynamics System. D. E. Shaw Research, 2021. Maestro-Desmond Interoperability Tools (Schrödinger, 2021).
Morris, G. M. et al. AutoDock4 and AutoDockTools4: automated Docking with selective receptor flexibility. J. Comput. Chem. 30, 2785–2791. https://doi.org/10.1002/jcc.21256 (2009).
ChemAxon. Cheminformatics platforms and desktop applications. http://www.chemaxon.com
Stroganov, O. V. et al. Lead finder: an approach to improve accuracy of Protein – Ligand docking, binding energy estimation, and virtual screening. J. Chem. Inf. Model. 48, 2371–2385. https://doi.org/10.1021/ci800166p (2008).
Trott, O. & Olson, A. J. AutoDock Vina: improving the speed and accuracy of Docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 31, 455–461. https://doi.org/10.1002/jcc.21334 (2010).
Sousa da Silva, A. W. & Vranken, W. F. ACPYPE - AnteChamber python parser interface. BMC Res. Notes. 5, 367. https://doi.org/10.1186/1756-0500-5-367 (2012).
Bernardi, A. et al. ACPYPE update for nonuniform 1–4 scale factors: conversion of the GLYCAM06 force field from AMBER to GROMACS. SoftwareX 10, 100241. https://doi.org/10.1016/j.softx.2019.100241 (2019).
Abraham, M. J. et al. GROMACS: high performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 1–2, 19–25. https://doi.org/10.1016/j.softx.2015.06.001 (2015).
Rodríguez-Martínez, A. et al. Enhancing MD simulations: ASGARD’s automated analysis for GROMACS. J. Biomol. Struct. Dynamics. 1–13. https://doi.org/10.1080/07391102.2024.2349527 (2024).
Schrödinger, L. L. C. QikProp, version 2024-1. Maestro, version 2024-1. (Schrödinger, 2024). https://www.schrodinger.com/products/qikprop.
Ntie-Kang, F. et al. Assessing the Pharmacokinetic profile of the CamMedNP natural products database: an in Silico approach. Org. Med. Chem. Lett. 3, 10. https://doi.org/10.1186/2191-2858-3-10 (2013).
Yuan, Y. et al. A novel strategy for prediction of human plasma protein binding using machine learning techniques. Chemometr. Intell. Lab. Syst. 199, 103962. https://doi.org/10.1016/j.chemolab.2020.103962 (2020).
Ntie-Kang, F. An in Silico evaluation of the ADMET profile of the streptomedb database. SpringerPlus 2, 353. https://doi.org/10.1186/2193-1801-2-353 (2013).
Daina, A., Michielin, O. & Zoete, V. SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 7, 42717. https://doi.org/10.1038/srep42717 (2017).
Carpenter, T. S. et al. A method to predict Blood-Brain barrier permeability of Drug-Like compounds using molecular dynamics simulations. Biophys. J. 107, 630–641. https://doi.org/10.1016/j.bpj.2014.06.024 (2014).
Anandan, S. et al. Integrated approach for studying bioactive compounds from cladosporium spp. Against Estrogen receptor alpha as breast cancer drug target. Sci. Rep. 12, 22446. https://doi.org/10.1038/s41598-022-22038-x (2022).
Garrido, A. et al. hERG toxicity assessment: useful guidelines for drug design. Eur. J. Med. Chem. 195, 112290. https://doi.org/10.1016/j.ejmech.2020.112290 (2020).
Armstrong, A. J. et al. Long-term survival and biomarker correlates of Tasquinimod efficacy in a multicenter randomized study of men with minimally symptomatic metastatic Castration-Resistant prostate cancer. Clin. Cancer Res. 19, 6891–6901. https://doi.org/10.1158/1078-0432.CCR-13-1581 (2013).
Raymond, E. et al. Mechanisms of action of Tasquinimod on the tumour microenvironment. Cancer Chemother. Pharmacol. 73, 1. https://doi.org/10.1007/s00280-013-2321-8 (2014).
Acknowledgements
This study was supported by a grant from the Scientific Foundation of the Spanish Association against Cancer (Predoctoral Fellowship, Grant number: PRDMU246084RODR). This research was funded by JUNTA ANDALUCIA GRANT, the Andalusian Regional Government, through Grant Proyectos de Excelencia (P18-RT-1193). This work was funded by FEDER/Junta de Andalucía-Consejería de Transformación Económica, Industria, Conocimiento y Universidades (Grant numbers PY20_00678 and B-BIO-18-UGR20). This work was supported by Ministerio de Ciencia e Innovation through the grant Ramón and Cajal program (Grant RYC2022-037702-I, funded by MCIU/AEI/10.13039/501100011033 and by the ESF+). Project support for this research was provided in part by the European Union Horizon 2020 Grant (REVERT project GA848098) and Instituto de Salud Carlos III (PI23/00601). This work was partially supported by the Fundación Séneca del Centro de Coordinación de la Investigación de la Región de Murcia under Project 22616/PI/24. This study formed part of the AGROALNEXT programme and was supported by MCIN with funding from European Union NextGenerationEU (PRTR-C17.I1) and by Fundación Séneca with funding from Comunidad Autónoma Región de Murcia (CARM). The authors acknowledge the computing resources and technical support provided by the Plataforma Andaluza de Bioinformática at the University of Málaga. Powered@NLHPC research was partially supported by the super-computing infrastructure of NLHPC (ECM-02). The author(s) thankfully acknowledges RES resources provided by Barcelona Supercomputing Center in MareNostrum5 to BCV-2025-1-0004. The authors declare that this work has been included in a patent application (application number: EP24382860.5)
Author information
Authors and Affiliations
Contributions
(A) R.M. contributed to Conceptualization, Methodology, Software, Validation, Formal analysis, Investigation, Data Curation, and Writing (Original Draft and Review & Editing). L G.R. contributed to Validation, Investigation, Writing (Original Draft and Review & Editing) and Visualization. M C.R. contributed to Investigation, Resources, Writing (Original Draft and Review & Editing), Supervision, Project administration, and Funding acquisition. I. L. contributed to Methodology, Formal analysis, Investigation, Resources, Writing (Original Draft and Review & Editing), Supervision, Project administration, and Funding acquisition. D. R. contributed to Validation, Investigation, and Writing (Original Draft and Review & Editing). F P.C. contributed to Validation, Investigation, and Writing (Original Draft and Review & Editing). (B) A.G. contributed to Investigation, and Writing (Original Draft and Review & Editing). S M.G. contributed to Methodology, Investigation, Resources, and Writing (Original Draft and Review & Editing)g. A.B. A.R. contributed to Validation, Investigation, and Writing (Original Draft and Review & Editing). P. C.Z. contributed to Methodology, Formal analysis, Investigation, Resources, Writing (Original Draft and Review & Editing), Supervision, Project administration, and Funding acquisition. A. M.H. contributed to Validation, Investigation, and Writing (Original Draft and Review & Editing). G. L.G. contributed to Methodology, Formal analysis, Investigation, Resources, Writing (Original Draft and Review & Editing), Visualization, Supervision, Project administration, and Funding acquisition. H. P.S. contributed to Conceptualization, Software, Formal analysis, Investigation, Resources, Writing (Original Draft and Review & Editing), Supervision, Project administration, and Funding acquisition.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethical approval
The study was conducted in accordance with the Declaration of Helsinki, and approved by the Ethics Committee of the Santa Lucía University General Hospital (protocol code: REVERT, date of approval: 06/02/2021). All procedures performed in this study involving patients and human subjects were in accordance with the ethical standards of the institutional research committee and the Declaration of Helsinki. For human-derived organoids, written informed consent was obtained in previous studies, and patients authorized the use of their samples in other research studies.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Rodríguez-Martínez, A., Giraldo-Ruiz, L., Ramos, M.C. et al. Discovery of Z1362873773: a novel fascin inhibitor from a large chemical library for colorectal cancer. Sci Rep 15, 14906 (2025). https://doi.org/10.1038/s41598-025-96457-x
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-96457-x
