
Abstract
Loquat has both a delicious taste and natural medicinal effects. Fruit yield is the foundation of loquat production. However, as loquat fruits ripen, the time course changes in endogenous hormones and the transcriptome are still unclear, and the mining of loquat fruit development-related genes is still limited. Pulps from four stages of fruit development, including 70 days after full-blooming (DAF) (Z01), 90 DAF (Z02), 110 DAF (Z03), and 130 DAF (Z04), were collected to detect the contents of endogenous hormones and conduct transcriptome analysis. The results revealed that the pulps thickened continuously at a steady speed, whereas the rapid fruit expansion before the Z02 stage could be attributed to a sharp expansion of seeds. In addition, the endogenous hormone levels of the pulps tended to differ among these four stages. A total of 39,051 genes were obtained, and each sample was compared. Z01-VS-Z02, Z01-VS-Z03, and Z01-VS-Z04 presented 2611, 12,594, and 19,214 differentially expressed genes (DEGs), respectively. We found that there were 259, 49, 47, 70, 169, 47, 22 and 33 DEGs related to Aux, GA, CTK, ABA, Eth, BR, SA and JA, respectively. GO and KEGG enrichment analyses revealed that the most obviously enriched terms were photosynthesis-antenna proteins, starch and sucrose metabolism, and photosynthesis. Cluster analysis of the expression trends of the DEGs revealed that 25 clusters were obtained, 16 of which were highly correlated with 1 to 5 hormones. A total of 25 and 28 core genes were systematically identified from clusters 21 and 25, respectively. Heterologous expression revealed that EjHsc70, a selected core gene, promoted Arabidopsis growth and could be involved in the development process of loquat fruit. This study provides new insights into fruit development in Eriobotrya japonica.
Similar content being viewed by others


Introduction
Loquat (Eriobotrya japonica Lindl.) is a subtropical fruit that originated in China and belongs to the Maloideae subfamily of the Rosaceae1,2. Loquat fruits have a good flavour and contain a variety of nutrients, such as vitamins, organic acids, and terpenoids. In addition, fruits also have the effects of moistening the lungs, relieving cough, exerting anti-inflammatory, and so on3,4,5. Loquat fruits mature in late spring and early summer, are listed as off-season fruits, and have high economic benefits. Owing to the development of the logistics industry, the sales of loquat fruits are no longer limited to the production area and surrounding areas, and the market demand for loquat fruits and their commerciality have further increased6,7. The quality of loquat fruits is one of the important standards for the commerciality and economy of loquat8.
At the cytological level, the growth of plant organs can be divided into two coordinated processes, namely, cell proliferation and expansion, which are both regulated by endogenous hormones9. The fruit development process includes fruit set, growth, and ripening8. Early apple fruit is related to the cells produced by mitosis, and the cells expand in the middle and late stages to achieve fruit development and enlargement10. Tomato fruit size is the result of the combined action of cell number and cell size, and cell division and cell expansion together determine fruit size11. The cell division and expansion of loquat fruits are subject to complex regulation. A weak cell proliferation capacity results in a smaller cell layer, which leads to a thin cortex 12.
Plant endogenous hormones are metabolites that are involved in the process of plant growth and development. They can affect the processes of germination, rooting, flowering, fruiting, sex determination, dormancy and abscission by affecting cell division, elongation and differentiation13,14. Liu et al. reported that the differences in fruit size and shape among different cucumber inbred lines were caused mainly by differences in fruit cell number and endogenous IAA content15. The difference in the fruit size of Korla fragrant pear (Pyrus sinkiangensis Yu.) and its bud mutation Zaomeixiang pear primarily results from variations in both the cell volume and total cell count within their mesocarp. Furthermore, the distinct fruit size of these two cultivars appear to correlate with differing contents of glucose, sorbitol, zeatin riboside (ZR) and ABA, as well as the proportions of endogenous hormones16. CTK play a dual role in kiwifruit development: they not only stimulate cell division but also enhance cellular expansion within the fruit. However, prolonged carbohydrates deficiency suppresses the expression of two critical genes: CYP735a and expansin. This coordinated down-regulated, leads to reduced CTK levels, untimatedly inhibiting fruit growth17. During the fruit ripening process of strawberry and cucumber, ABA accumulates rapidly. ABA peaks in late-ripening sweet orange mutants appear later than those in wild-type fruits do18,19,20.
In tomato, fruit thickness is related to the number of cells. The overexpression of the CK-inactivating enzyme gene: Arabidopsis cytokinin oxidase/dehydrogenase 2 (AtCKX2) in tomato fruit tissues can reduce the levels of endogenous active CTKs and inhibit the expression of Aux signalling genes and GA biosynthesis genes, thereby decreasing tomato pericarp thickness and fruit size21. Self-pollinated apple fruits are smaller and more asymmetrical than cross-pollinated fruits but can grow normally after exogenous GA treatment22. Previous studies have shown that Aux, Eth and JA signals are involved in the regulation of cell growth 23,24. JA can promote BPEp (BIGPETALp) expression of the encoded bHLH transcription factor and inhibit cell swelling and petal size25. A study of the cell growth and molecular regulatory mechanism of loquat fruit growth revealed that AUX/IAA, GH3, SAUR, ARR, DELLA, PIF4 and TCH4 in the Aux, CTK, GA, and BR signal transduction pathways may be directly involved in the regulation of cell division or cell enlargement. ACS1 and ACO1, which are ethylene synthesis genes, have been shown to be associated with apple fruit firmness26,27. EjCYP90A is an activator of fruit growth in loquat. EjBZR1 (Eriobotrya japonica BRASSINAZOLE-RESISTANT 1), a BR-related transcription factor, could negatively regulate the expression of EjCYP90A gene, thereby affecting BR biosynthesis and subsequently fruit cell expansion and size28.
In recent years, with the development of high-throughput sequencing technology, transcriptome analysis has become a fast and effective method to analyse the transcription level of samples under specific conditions and has been applied in many fields, such as microbiology, zoology, and botany. It has been widely used in the production and utilization of horticultural plants, such as the resistance of Masson pine to diseases and insect pests, the influence of production areas on Angelica Dahurica, the response of maize to high-temperature stress at the flowering stage, the molecular mechanism of storage and transportation of pepper, and the resistance of chitosan to Botrytis cinerea in postharvest grapes29,30,31,32. In loquat, transcriptome analysis has been carried out to study tissue development, such as fruit setting33, flower development34, and flower bud transition35.
However, the time course changes in endogenous hormones and the transcriptome during loquat fruit development are still unclear, and the mining of loquat fruit development-related genes is still limited. In this study, the fruits of the ‘Zaozhong No. 6’ variety were used as research materials to detect the time course changes in endogenous hormones and the transcriptome of the fruit pulps from the expansion to ripening periods. Integrated endogenous hormone and transcriptome analyses were performed to screen genes related to endogenous hormone signal transduction and fruit development. Finally, a candidate fruit development-related gene from loquat was selected for heterologous expression in Arabidopsis to preliminarily confirm its function. These results could provide new insights into fruit development in Eriobotrya japonica.
Materials and methods
Experimental material and treatment
The fruits were prepared and sampled according to our previous method with some modifications36. Three uniform mature loquat trees of the ‘Zaozhong No. 6’ loquat cultivar living in an outdoor nursery at 25°45’ N and 118°55’ E (Changtai town, Putian city, Fujian Province, China) were selected and managed with common cultivation under natural conditions. By both thinning flowers and fruits, the fruits are guaranteed to be in the same developmental period. Four fruits per panicle were retained and bagged. T he same size and uniform color of healthy fruits were picked at four developmental stages, including 70 days after full-blooming (DAF), 90 DAF, 110 DAF, and 130 DAF. Three biological replicates were collected from three mature loquat trees. Each replicate was mixed with 5 individual fruits and annotated as Z01A, Z01B, and Z01C for 70 DAF; Z02A, Z02B, and Z02C for 90 DAF; Z03A, Z03B, and Z03C for 110 DAF; and Z04A, Z04B, and Z04C for 130 DAF. The fruits were then immediately transported back to the laboratory. The fruit pulps were frozen in liquid nitrogen, mechanically ground and stored at − 80 °C.
Detection of endogenous hormone contents
The contents of Aux, GA, CTK, ABA, Eth, BR, SA and JA in the fruits were detected by corresponding ELISA kits (Kmaels, China), and the test method was carried out according to the instructions of the kits. Three biological replicates were performed for each treatment. Data analysis was performed with DPS, and SigmaPlot software was used for graphing.
Total RNA extraction, library construction, sequencing and gene quantification
The samples were sequenced via high-throughput sequencing technology by Gene Denovo Biotechnology Co. (Guangzhou, China). Total RNA was extracted from 0.1 g of fruit pulp via the Omega Plant RNA Kit, with three biological replicates for each treatment. The extracted RNA samples were detected via an Agilent 2100 Bioanalyzer (Agilent, Santa Clara, CA, USA) with an RNA integrity number (RIN) > 6.3. Qualified RNA samples were used to construct a cDNA library, which was subsequently sequenced on the Illumina platform.
High-quality clean reads were obtained by removing reads containing adapters, reads with a ratio of N greater than 10%, and low-quality reads from the raw reads. Trinity software37 was used to perform de nova transcriptome assembly. The High-quality clean reads were aligned with rRNA to identify the remaining rRNA reads, and the removed rRNA reads were used for further analysis. rRNA-removed high-quality clean reads were compared to unigenes via Bowtie2 software38 and calculated the mapping ratio with the default parameter method. The gene expression level was calculated via unigene expression and normalized via the RPKM method. The evaluation of the experimental results was carried out by calculating the correlation coefficient between each two parallel experiments. Principal component analysis (PCA) was performed via the R language (http://www.r-project.org/)39.
Unigene functional annotation and enrichment analysis
The edgeR package (http://www.r-project.org/) was used with between-group differentially expressed genes (DEGs), and the differential gene screening thresholds were a q value < 0.05 and a |fold change|≥ 2. The unigene sequences were aligned to Nr, SwissProt, KEGG and COG/KOG (Clusters of Orthologous Groups of proteins) (evalue < 0.00001) sequences via BLASTx for protein function annotation. In accordance with the annotation results of the NR database, explanatory note information for the unigenes was obtained via blast2go software, which was subsequently used to classify all the unigenes according to their molecular function, cell component and biological process. Pathway enrichment analysis of the unigenes was further performed according to KEGG annotation information40,41,42. The GOplot R package was used for the visualization of the GO enrichment results.
Time series expression analysis and target gene selection
A series of cluster analysis tests were performed to determine different gene clusters via Mfuzz software. The center trends of different gene clusters were evaluated for their correlation with the content trends of different hormones to confirm hormone-related gene clusters. An absolute value of correlation > 0.8 was considered a high correlation. Gene clusters with no genes that had a membership value > 0.7 were excluded. The core genes in hormone-related gene clusters with membership value > 0.7 were screened. Finally, target genes were selected by gene annotation and previous reports of core genes43.
Heterologous function verification of selected target genes
The target gene was amplified via PCR (using the following primers: F: GGGGTACCATGGCGGGAAAGGGGGAAGG; R: CGGGATCCTTAATCGACTTCTTCAATCTTG). Then, it was inserted into the pCAMBIA1301 vector through KpnI and BamHI digestion and ligation. After that, the constructed vector was transformed into Agrobacterium strain GV3101, thereby applying the floral dip method to obtain transgenic Arabidopsis. Transgenic Arabidopsis plants were seeded on MS media supplemented with hygromycin B for selection. qRT‒PCR was used to confirm positive transgenic lines (target gene primers: Fq:TGCCAAGATGTACCAAGGTG; Rq: GGGATCCATCGACTTCTTCA; reference gene primers: At-actin-Fq:CGGCGAGTCATCGTCATCAATGT; At-actin-R1: GTTCGGGATTACGGTGGTGGGA). Transgenic Arabidopsis lines were germinated and grown in Petri dishes containing MS media and 0.8% agar. The Petri dishes were placed in a plant incubator at 22 °C with a 16 h/8 h light/dark cycle. The phenotypes of wild-type and T2 transgenic line Arabidopsis seedlings were observed after 7 d44. The hypocotyl length was determined via ImageJ software.
Results
Changes in phenotypes and endogenous hormones in loquat fruits at different developmental stages
Phenotypic observations revealed that during Z01–Z02, the fruit expanded rapidly, with larger seeds and thicker pulps. In Z03, the fruit color changed from green to yellow, and the pulp continued to thicken. However, the degree of fruit swelling during this period was not as obvious as that of Z02 because the seed volume did not change significantly. When the fruit developed to Z04, the thickness of the pulp clearly increased again, and the color changed from yellow to orange), indicating that the fruits were mature (Fig. 1). The results revealed that the pulps thickened continuously at a steady speed, whereas the rapid fruit expansion before the Z02 stage could be attributed to the sharp expansion of seeds.

Phenotypic differences in loquat fruits at different developmental stages. Z01: 70 DAF; Z02: 90 DAF; Z03: 110 DAF; Z04: 130 DAF (the same as below).
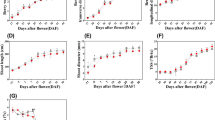
In addition, the endogenous hormone levels tended to differ across these four periods. The Aux content significantly and continuously decreased from Z01 to Z04. The GA content continued to increase significantly from Z01 to Z04. The CTK content decreased significantly from Z01 to Z03, whereas there was no significant difference between Z03 and Z04. The content change of Eth showed a trend of "rising-falling-rising", with two peaks at Z02 and Z04. The ABA content was relatively stable in Z01 and Z02, decreased significantly in Z03, then increased and reached a maximum in Z04. The content of BR increased significantly at Z01 to Z03 and stabilized between Z03 and Z04. The contents of SA and JA did not change significantly during the whole process of the four fruit periods (Fig. 2).

Endogenous hormone contents of loquat fruits at different developmental stages. Different letters on the bars indicate significant differences at P < 0.05.
Transcriptome sequencing and assembly analysis
The results of the correlation analysis revealed that the relationship among biological repeatabilities was good to conduct subsequent analysis (Fig. S1). The original number of reads in the four groups ranged from 27,647,092–37,465,976, and the number of clean reads ranged from 27,225,902–37,024,274, accounting for 98.71–99.09% of the original reads. The content of GCs was 46.96% ~ 47.63%, and the number of reads containing joint sequences ranged from 7762 to 29,568, accounting for 0.02–0.08% of the total reads. The proportion of low-quality reads was 0.79–1.4%. The number of filtered reads ranged from 27,225,902–37,024,274, and the number of bases ranged from 4,231,456,723–5,532,482,746 bp. The base number of Q30 was 3,774,227,925–5,183,979,732 bp, which accounted for 92.69–94.46% of the total base. Compared with those of the ribosomes, the number of rRNA reads aligned in all the samples ranged from 193,326–1,026,458, accounting for 0.63–3.26% of the filtered reads. The number of rRNA-removed high-quality reads ranged from 26,966,444–36,298,750, and the mapping ratio against reference unigenes accounting for 82.18–83.88%. The de novo assembly generated 58, 394 reference unigenes, among which 57,846 unigenes (99.06% of total) were exhibited detectable expression levels. The number of expressed genes in Z01, Z02, Z03 and Z04 groups were 56,152, 55,965, 53,505 and 51,809, respectively, accounting for 96.16%, 95.84%, 91.63% and 88.72% of the total number of reference genes, respectively.
Unigene annotation and expression analysis
The sequencing results were annotated via KEGG, KOG, Nr and SwissProt data, which revealed that the numbers of unigenes associated with the above four datasets were 14,572, 21,455, 38,570 and 25,723, respectively. A total of 39,051 genes were annotated, 19,345 were not annotated, and the number of annotated genes in all four databases was 10,489 (Fig. 3a).


Unigene annotation, PCA cluster analysis and DEGs statistics: (a) Venn analysis of unigenes annotated to the KEGG, NR, KOG and SwissProt databases; (b) PCA of 12 samples; (c) statistics of DEGs in Z01-vs-Z02, Z01-vs-Z03 and Z01-vs-Z04. Z01 was acted as control(the same as bellow).
According to the results of PCA, clustering of differential gene expression patterns between groups and sample clustering analysis revealed that there were differences among the four groups (Fig. 3b). There were more differentially expressed genes (DEGs) between Z01-VS-Z03 and Z01-VS-Z04 than between Z01-VS-Z02; there were more DEGs in Z03-VS-Z04 than in Z01-VS-Z02. The number of DEGs identified in the Z01-vs-Z02, Z01-vs-Z03 and Z01-vs-Z04 comparisons were 2611, 12,594 and 19,214, respectively. We identified 656 up-regulated and 1955 down-regulated, 2456 up-regulated and 10,138 down-regulated, 2909 up-regulated and 16,305 down-regulated genes in Z01-vs-Z02, Z01-vs-Z03 and Z01-vs-Z04 (Fig. 3c).
From the sequencing results, we screened 259, 49, 47, 70, 169, 47, 22 and 33 DEGs related to Aux, GA, CTK, ABA, Eth, BR, SA and JA, respectively. The 70 d group (Z01) was used as the control group. Compared with Z01-vs-Z02, Z01-vs-Z03 and Z01-vs-Z04 presented 23, 97 and 124 Aux-related DEGs, respectively, and 16 DEGs were associated with the three groups. There were 32 GA-related DEGs, including 7, 24 and 31 in the above three comparison groups, and 7 DEGs were unique to the three groups. Twenty-three CTK-related DEGs were identified: 8, 18, 21, and 7 DEGs were common to Z01-VS-Z02, Z01-VS-Z03, and Z01-VS-Z04, respectively. There were 24 ABA-related DEGs, 3, 15, and 23 ABA-related DEGs, respectively, among the three groups. From z01-vs-z02, z01-vs-z03 and z01-vs-z04, there were 15, 63 and 96 Eth-related DEGs, respectively, and the total number of DEGs was 101, including 12 common DEGs. The 12 DEGs were divided into three major groups: GroES-like zinc-binding alcohol dehydrogenation family proteins, ethylene-responsive transcription factors and 1-aminocyclopropane-1-carboxylic acid oxidase. The number of BR-related DEGs increased to 20, with 3 codifferentially expressed genes, 6, 17 and 15, in the three comparison groups. The SA-related DEGs were an aggregate of 9 unigenes, and Z01-vs-z02, Z01-VS-Z03 and Z01-VS-Z04 accounted for 1, 5 and 7, respectively. There were 23 JA-related DEGs in all three groups, including 1, 16, and 23, which shared 1 DEG among the three groups (Table S1).
GO Enrichment analysis of differentially expressed genes
GO functional enrichment analysis classified genes according to their cellular components, molecular functions and biological processes. In Z01-vs-Z02, 12 cellular components, 11 molecular functions and 16 biological processes were annotated. In Z01-vs-Z03, 15 cellular components, 11 MFs and 17 biological processes were annotated. There were 16 cellular components and 11 molecular functions, and 17 biological processes that were annotated in Z01-vs-Z04. Among the molecular functions, binding and catalytic activity were the most differentially expressed genes; metabolic process and cellular process were the most differentially expressed genes in the biological processes category (Fig. 4).


Top 20 GO terms of Z01-VS-Z02 (a), Z01-VS-Z03 (b) and Z01-VS-Z04 (c).
The auxin response (IAA) and auxin response factor (ARF) genes were down-regulated, except for IAA4, IAA29, IAA33 and IAA11. GH3 and SAUR were 3 and 13, respectively. GH3 was down-regulated, 8 were up-regulated, and 5 were down-regulated in SAUR. There were 22 auxin-induced proteins (AAPs), 14 of which were down-regulated and 8 of which were up-regulated. There were 8 and 7 genes encoding auxin transporter protein (ATP) and auxin efflux carrier component (AECC) were down-regulated, with the exception of two AECC3s. There were 5 up-regulated and 15 down-regulated GO terms related to GA. In Z01-VS-Z02, Z01-VS-Z03, Z01-VS-Z04, 3, 6, and 6 GA-related GO terms were enriched in the molecular functions. In addition, 3, 19, and 19 GO terms were enriched in the biological processes. The number of GO terms related to CTK in Z01-VS-Z02, Z01-VS-Z03, and Z01-VS-Z04 were 5 up-regulated and 7 down-regulated, 6 up-regulated and 20 down-regulated, and 11-regulated and 21 down-regulated, respectively. In the GO terms related to ABA, Z01-VS-Z02 had 10 up-regulated and 10 down-regulated GO terms; Z01-VS-Z03 had 15 up-regulated and 29 down-regulated GO terms; and Z01-VS-Z04 had 15 up-regulated and 29 down-regulated GO terms. GO functional enrichment analysis revealed that the most enriched GO terms of biological processes among the ABA-related DEGs were GO: 0,071,554, GO: 0,009,628, etc. and reached a significant level.
In the GO terms related to Eth, 22 were up-regulated and 23 were down-regulated; the numbers of Z01-VS-Z02, Z01-VS-Z03 and Z01-VS-Z04 Eth-related GO terms were 8, 19 and 19 in the cellular component, 20, 37 and 43 in the molecular function and 89, 164 and 207 in the biological process, respectively. GO: 0,001,071, GO: 0,019,438, and GO: 1,901,362 were found to be associated with more Eth DEGs. In the GO terms relateid to BR, 7 were up-regulated and 4 were down-regulated in Z01-vs-Z02;7 were up-regulated and 6 were down-regulated in Z01-vs-Z03; 7 were up-regulated and 9 were down-regulated in Z01-vs-z04, respectively. In Z01-VS-Z02, Z01-VS-Z03, and Z01-VS-Z04, there were 5, 6, and 6 cellular component GO terms; 15, 20, 18 molecular function GO terms; and 9, 28, 28 biological process GO terms. 8 up-regulated and 18 down-regulated GO terms were related to SA. The cellular component was annotated only in Z01-VS-Z04, with 23 GO terms. The numbers of GO terms associated with SA in the molecular function of Z01-VS-Z03 and Z01-vs-z04 were 11 and 41, respectively; the numbers of biological process terms were 35 and 206, respectively. In Z01-vs-Z02, 3 molecular functions and 1 biological process GO terms were JA-related; 5 molecular functions and 32 biological processes GO terms were found in Z01-vs-Z03; and 15 cellular components, 12 molecular functions and 51 biological processes GO terms were detected in Z01-vs-Z04. (Fig. 4, Tables S1, S2 and S3).
KEGG Enrichment analysis of differentially expressed genes
KEGG functional analysis revealed that the DEGs of Z01-VS-Z02, Z01-VS-Z03, and Z01-VS-Z04 were enriched in 102, 125, and 125 metabolic pathways, respectively. Top 20 enrichment KEGG pathways were exhibited in Fig. 5. Thirteen B pathways and 58 C pathways were jointly associated with the three groups. The top 10 pathways entries with the most significant enrichment were selected for further display (Tables S4, S5). The most significant differences among the three groups were related to photosynthesis—antenna proteins—start, sucrose metabolism and photosynthesis. The paths with the greatest number of DEGs were starch and sucrose metabolism, phenylpropanoid biosynthesis, photosynthesis, and pentose and glucuronate interconversions. The above results indicate that the biosynthesis of other secondary metabolites, carbohydrate metabolism, carbohydrate metabolism, and energy metabolism continuously accumulate during fruit development (Fig. 5).


Top 20 pathways enriched in Z01-VS-Z02, Z01-VS-Z03 and Z01-VS-Z04.
GA-related genes were enriched in diterpenoid biosynthesis (ko00904) and plant hormone signal transduction (ko04075) only in Z01-VS-Z03 and Z01-VS-Z04. The expression of ent-kaurene synthase, ent-kaurenoic acid monooxygenase, gibberellin-44 dioxygenase in ko00904, gibberellin receptor (GID1), phytochrome-interacting factor, and DELLA in ko04075 were down-regulated. The KEGG analysis of the CTK-related DEGs revealed that the three comparison groups were enriched for related genes, with 1, 2, and 2 paths, respectively. The commonly enriched KEGG pathway was Zeatin biosynthesis (ko00908), and ko04075 was enriched only in Z01-VS-Z03 and in Z01-VS-Z04. The expression of cytokine synthase in ko00908 tended to decrease, whereas the expression of cytokine dehydrogenase tended to decrease. In ko04075, the cytokine metabolic pathway showed that the expression of histidine-containing phosphotransfer proteins decreased. In Z01-vs-Z02, Z01-vs-Z03 and Z01-vs-Z04, 1, 4 and 5 ABA-related DEGs were enriched in carotenoid biosynthesis (ko00906); 0, 5 and 10 genes were enriched in ko04075 (Table S4). In ko04075, the expression of PYR/PYL was downregulated, which reduced the inhibition of PP2C and upregulated its expression, further inhibiting the expression of SnRK2 and reducing the activation of ABF (Fig. 6 and Table S6).

Plant hormone signal transduction pathways (ko04075) in Z01-VS-Z02, Z01-VS-Z03 and Z01-VS-Z04. The text box in the pathway diagram represents a gene or protein, and the three consecutive text boxes above it from left to right indicate that the DEGs involved in the regulation of the gene or protein were Z01-VS-Z02 (left box), Z01-VS-Z03 (middle box) and Z01-VS-Z04 (right box). White means no DEGs, green means all DEGs were down-regulated, red means all DEGs were up-regulated, and blue means there were two kinds of DEGs that were up-regulated and down-regulated.
The path statistics of Eth-related DEGs revealed that Z01-VS-Z04 was the most common and that Z01-VS-Z02 was the least common. Cysteine and methionine metabolism (ko00270) were the common paths of the three comparison groups. ko04075 is enriched in Z01-VS-Z03 and Z01-VS-Z04. S-methyl-5-thio-d-ribolphose 1-phosphate and aminocyclopropanecarboxylate oxidase gene expression increases with fruit development, promoting ethylene accumulation. S-Methyl-5-thio-D-ribose-1-phosphate and aminocyclopropanecarboxylate oxidase gene expression increased continuously. The expression of ETR genes in ko04075 was up-regulated, and the expression of EIN3 was down-regulated. During late fruit ripening, the ERF1/2 gene was down-regulated. The results of BR-related DEGs enrichment revealed that the expression of steroid 22 s-hydroxylase, 3beta, 22alpha-dihydroxy3-dehydrogenase and PHYB activation-tagged suppressors were down-regulated with fruit ripening. In Z01-VS-Z03 and Z01-VS-Z04, the brassinosteroid resistance 1/2 of ko04075 was up-regulated. The KEGG enrichment analysis of SA revealed that only two pathways were enriched in Z01-VS-Z04: the phosphatidylinositol signalling system (ko04070) and inositol phosphate metabolism (ko00562). Both phosphatidylinositol-3,4,5-trisphosphate 3-phosphatase and dual-specificity protein phosphatase (PTEN) of ko04070 and ko00562 were down-regulated. Pathway enrichment analysis of JA-related DEGs revealed that alpha linolenic acid metabolism (ko00592) was enriched in the three groups. However, the number of DEGs increased with the development of loquat fruit, and the expression of jasmonate O-methyltransferase decreased. Alpha linolenic acid metabolism (ko00592) and ko04075 were enriched only in the middle and late periods of fruit development, and jasmonate Zim domain-containing protein (JAZ) was down-regulated. The expression of jasmonate O-methyltransferase in ko00592 was down-regulated. JA-related DEGs in ko04075 were enriched from the middle fruiting stage, and the expression of jasmonate Zim domain-containing protein (JAZ) and transcription factor (MYC2) were decreased (Fig. 6 and Table S6).
Time series expression analysis and target gene selection
To detect the gene expression patterns among different developmental stages of loquat fruits, trend analysis was performed. In total, 25 clusters with different expression trends were identified via Mfuzz software. Among them, 9 gene clusters had no genes with a membership value > 0.7 and were excluded, including clusters 3, 6, 10, 11, 12, 13, 15, 16, and 23 (Figs. 7 and 8). Cluster 17 was not highly correlated with any of the eight hormones. Clusters 2 and 9 were highly associated with BR and Eth, respectively. Clusters 7, 20 and 24 were highly correlated with 3 hormones(Cluster 7: Aux, CTK and JA; Cluster 20: GA, SA and ABA; Cluster 24: Aux, GA and BR). Cluster 4 was highly associated with Aux, CTK, JA and GA. Clusters 1, 4, 14, 19, 21, 22, and 25 were all highly correlated with Aux, CTK, JA, GA and BR. Interestingly, the gene expression trends of clusters 21 and 25 were positively correlated with GA and BR and negatively correlated with Aux, CTK and JA, whereas those of clusters 1, 4, 14, 19, and 22 were the opposite. Therefore, clusters 21 and 25 were selected for further gene mining (Fig. 8). There were 25 and 28 core genes in clusters 21 and 25, respectively. According to the gene annotation and previous reports45, a heat shock cognate 70 kDa protein gene (Unigene0026363) was selected as the target gene (Tables S7 and S8).

Trend analysis of loquat developmental RNA-seq data. The membership value is used to judge the possibility that a gene belongs to a certain cluster. If the membership value is close to 1, it means that the gene belongs to a certain cluster very clearly. If the membership value is close to 0, it means that the gene almost does not belong to the cluster. The genes with membership > 0.7 was the core gene of the gene clusters.

Correlation analysis between the center trends of different gene clusters and the content of different hormones. Gene clusters without core genes(membership value > 0.7) were not shown in this figure.
Functional validation of candidate genes
Transgenic and wild-type Arabidopsis seedlings were seeded at the same time under the same conditions. Morphological phenotypes were recorded after 7 d of growth. The EjHsc70 gene (Unigene0026363) was overexpressed in all three transgenic Arabidopsis lines (Fig. 9A.) and significantly augmented development phenotypes, especially hypocotyl length (Fig. 9B, C). These results indicated that the EjHsc70 gene promoted Arabidopsis growth and could be involved in the development process of loquat fruit.

Morphological phenotypes of the transgenic lines harboring the loquat EjHsc70 gene (Unigene0026363). A. Relative expression level of the EjHsc70 gene in the wild-type and transgenic lines; B. Hypocotyl length statistics of the wild-type (WT) and transgenic lines (OE1, OE2 and OE3); C. Morphological phenotypes of the wild-type and transgenic lines.
Discussion
Auxin plays a very important role in regulating fruit size and inhibiting fruit ripening. The auxin concentration was negatively correlated with seed maturation. Auxin is associated with delayed softening of strawberry fruit and delayed ripening of the fruit after harvest46. A decrease in the auxin concentration can be considered a signal of seed maturity47. At 80–119 DAF, the expansion period of pulp cells in loquat rapidly occurred, and the auxin content peak appeared at both the young fruit stage and the fruit expansion stage48. This conclusion was consistent with the change trend of the Aux level during the loquat expansion period in this study. Reig et al. suggested that the CTK content in the pulp cells of loquat fruits decreases during fruit growth until the fruit stops growing49 and Leng et al. reported that production of ABA increased with increasing Eth content50, which was consistent with the trends in CTK, ABA and Eth contents observed in this study. Abscisic acid is a plant hormone that regulates plant growth and development. It plays a role in coloring and promoting fruit abscission during fruit ripening. The abscisic acid content was positively correlated with the peak of fruit drop, which was consistent with the trend of the change in the endogenous hormone concentration in this study51. During the early expansion of Solanum tuberosum L., if the expression of genes related to GA degradation was upregulated, the content of GA decreased, and the swelling rate of tubers accelerated52. In addition, when paclobutrazol, a GA synthesis inhibitor, was applied to Lilium brownii, the standard of endogenous GA in the bulb decreased, and nutrients accumulated rapidly53. On the basis of these findings, it was speculated that the low GA content of loquat fruits in this study was due to the accumulation of nutrients for fruit expansion at 70–90 DAF. When the fruit matured at 130 DAF, the demand for nutrients decreased. At this time, the increase in the GA content reduced the accumulation of nutrients. BR can not only increase the fruit setting rate, vitamin C content, and anthocyanin content in peels but also increase the fruit diameter, weight, hardness, total sugar content and color54. In this study, the content of BR increased significantly during each period before maturity, which was consistent with the previous summary of the effects of BRs. Jasmine can relieve abiotic and biotic stresses, such as drought, salt, temperature and disease, in the environment. In addition, jasmonate can promote the production of anthocyanins in fruit through the action of the MYB transcription factor, which is used to improve fruit color. SA can interact with Eth to regulate fruit senescence55. However, the JA and SA contents of the loquats in this study did not change significantly, indicating that the color changes and that fruit swelling in loquat are not strongly related to JA.
In recent years, with the development of high-throughput sequencing technology, transcriptome analysis of the transcript levels of samples under specific conditions has become a fast and effective method. This method has been used in many horticultural plants such as Malus x domestica, Vitis vinifera L., Eriobotrya japonica56,57,58. In loquat, transcriptome analysis has been carried out to study tissue development and biotic stress, such as fruit setting59, flower development34, flower bud transition60, temperature stress61 etc. In this study, 70 DAF was used as the control group and compared with 90, 110, and 130 DAF materials. Through KEGG enrichment analysis, the changes in the expression trends of each gene involved in endogenous hormone signal transduction were found to be consistent with the phenotype and were obtained through screening of gene expression trends.
The increased expression of Sl ARF10 could induce down-regulation of the expression of genes related to cell enlargement, thus inhibiting leaf growth62. AUX/IAA are frequently coexpressed with ARF, and their encoded proteins interact with each other under different conditions to regulate specific growth and development processes63. SAURs can be divided into two types: growth inhibitors and growth promoters64. In this study, the auxin concentration decreased continuously during the “Zaozhong-6” fruit expansion period. Moreover, the expression of most AUX1, AUX/IAA, ARF, GH3, and SAUR genes was down-regulated in response to a decrease in the auxin content. In this way, the inhibition of fruit ripening was relieved; we speculated that these five signal transduction factors were mainly negative regulators during the loquat fruit expansion period.
The modification and mutual transformation of CTKs in vivo not only affects the dynamic balance of CTKs but also affects its transport, which is highly important for regulating its physiological activity. It is generally believed that bound cytokinin is a storage form, such as a glycosylated or transport form (nucleosylation), whereas the free state is the main form of biological activity65. In this study, the free cytokinin isopentenyl adenine (IP)- and trans zeatin (t-z)-related catalytic enzyme-encoding genes were down-regulated, and the bound cytokinin catalytic enzyme-encoding genes were up-regulated. It was speculated that active CTKs play a negative regulatory role in fruit expansion. Gibberellin is thought to inhibit the expansion of tubers. In the gibberellin signal transduction pathway, biologically active GA binding to the GID1 receptor promotes interaction with the DELLA protein. FSLY1/GID2E 3 ubiquitin ligands containing F-box proteins can mediate the ubiquitination of DELLA and can be degraded by the 26S proteasome. DELLA degradation can induce GA responses66. The expression of GID1 and Della was down-regulated during the expansion of Lycoris radiata bulbs52. It was inferred that the GA content in loquat pulp was lower at 70–90 d, that GID2 was upregulated and that TF downregulation could decrease the degree of GA signal transduction. At 110–130 d, the GA content increased, and the expression of GID1 began to decrease, whereas the expression of TF and DELLA further decreased to cope with the increase in GA content to promote nutrient accumulation and fruit expansion. DELLAs act not only on the GA signalling pathway but also on other plant hormone signal transduction pathways. For example, the ethylene signalling pathway regulates root growth by regulating growth inhibition by DELLAs53,67. In the absence of GA, DELLA releases jasmonic acid insensitive 1 (JAZ1/MYC2) through competitive binding to JA Zim domain 1 (JAZ1) to promote JA signalling. In the presence of GA, the combination of JAZ1 and MYC2 was enhanced by mediating the degradation of DELLAs, which can inhibit the output of the JA signal68,69,70. BR has a growth-promoting effect, and it conducts signal crosstalk and connection with GAs through the interaction between BZR1/BES1 and GAs71. BR not only induced the biosynthesis of GAs but also mediated and activated the catabolism of GA. BR activated GA catabolism with the help of the basic helix–loop–helix transcription factor CESTA (CES) and its homologues BRASSINOSTEROID-ENHANCED EXPRESSION (BEE) 1 and 3 in Arabidopsis thaliana. CES and BEEs are regulated by BR at the transcriptional level and can induce the expression of the class III GA2 oxidase GA2ox7 at the same time. BRs can increase the activity of this enzyme to regulate GA72. The changes in the concentrations of BR and GA should be consistent, similar to the results of this study.
In addition to inducing plant disease resistance, salicylic acid also acts as an ethylene antagonist to regulate plant maturation and senescence. For example, exogenous SA may delay the ripening and senescence process by inhibiting the biosynthesis of ethylene in the frui of Malus pumila Mill73,74,75,76. From 70 to 90 d of loquat fruit development, the SA content decreased slightly, and only PR-1 was down-regulated in the signalling pathway in response to changes in SA content. In this study, there was no significant difference in the SA concentration or PR-1 expression in the signal transduction pathway from 110 to 130 days; however, the number of DEGs associated with NPR1 and TGA increased. These findings indicated that in the early stage of fruit expansion, PR-1 was the main response factor in the SA signalling pathway, and in the middle and late stages of fruit expansion, NPR1, TGA and PR-1 were involved in signal regulation. At 130 d, the SA content reached a maximum, presumably to inhibit ethylene synthesis to delay fruit senescence. However, the content of JA did not change significantly during fruit development. This may be because pulp cell division mainly occurs in the early stage of development, and the latter is dominated by cell enlargement. Studies have shown that JA is involved mainly in the rapid defense response of plants and inhibits growth processes such as photosynthesis and cell division. It has been reported that the JAZ protein can repress the expression of genes involved in the jasmonic acid response at the transcriptional level and is a negative regulator of MYC2. JAI3 (a JAZ protein) can bind to MYC2, resulting in loss of MYC2 function. JAI3 protein-dependent binding to the SCF complex and degradation of the 26S proteasome led to the release of MYC2, which acted as a negative feedback regulator of the expression of jasmonic acid-responsive genes77,78. Presumably, in this study, although the expression of JAZ was down-regulated, the inhibition of JA-responsive genes was reduced. However, the expression of MYC2 was also down-regulated, which reduced the signal transmission of JA, slowed the inhibition of fruit cell division, and accelerated fruit cell proliferation and fruit enlargement. We predicted that MYC2 was the main element of JA involved in loquat fruit development.
A clustering algorithm can be used to cluster the expression trends of different genes. It can be correlated with changes in phenotype, shape, metabolites and so on to screen relevant candidate genes. Ye79 used a K-means clustering algorithm to analyse the expression trends of DEGs in tall and dwarf Brassica napus, divided them into six clusters, and found that they were related to the formation of plant structure. Sixteen candidate genes related to Cd tolerance were screened by combining qtl and RNA-Seq analysis data in the study of Cd toxicity in barley80. The cluster analysis of differential gene expression trends in this study can be divided into 25 clusters, 16 of which were highly correlated with 1 to 5 hormones. 25 and 28 core genes were screened from cluster 21 and 25, respectively. Exogenous BR can promote fruit ripening and increase soluble sugar, ascorbic acid, lycopene contents, respiration rates, and ethylene yields81. Su demonstrated that the expression of the EjBZR1 gene was negatively correlated with cell and fruit size and regulated BR biosynthesis through EJCYP90A, which in turn affected cell expansion and fruit size28. The ATCKX2 gene can effectively inhibit poplar root germination. The overexpression of ATCKX2 can decrease endogenous active CTK levels, resulting in reduced pericarp thickness, which was confirmed by paraffin sections to be due mainly to a reduction in cell number and thus reduced cell division with respect to fruit size; moreover, the thickness of the pericarp and single-fruit weight of transgenic plants decreased significantly21,82. Expansin (EXP) is a type of plant Expansin. In many plant tissues, the expression of the EXP gene is closely related to growth and maturation83. In strawberry, Faexp1, FaEXP2 and FAEXP5 have been shown to be closely related to fruit hardness84. In this study, EXP3 (UNIGENE0022981) was highly expressed during the expansion stage, and its expression level was up-regulated exponentially from 70 to 110 DAF and down-regulated until 130 DAF, indicating that EXP3 was related to the expansion and maturation of loquat fruit. PG acts on pectin molecules, which degrade pectin, disintegrates the cell wall structure and results in fruit softening. It has been shown to be involved in the ripening process of tomatoes, peaches, strawberries and other fruits85,86,87. Although the expression of PG in this study was always low, the fold difference in expression during each period was obvious, which may be related to the change in the cell wall structure during the fruit expansion process. DNA binding with one finger (DOF) transcription factor family member OBP4 (octopine synthase element binding factor binding protein) is a negative regulator of cell proliferation and cell expansion; nuclear chromatin co-immunoprecipitation experiments revealed that CyclinB1, cyclin-dependent kinase B1 and XTH (xyloglucan endotransglucosylases/hydrolases) are downstream target genes of OBP488. The auxin-responsive factor ARF8 can interact with the b HLH (basic helix-loop-helix transcription factor) transcription factor BIGPETALp to regulate cell expansion and thus affect Arabidopsis petal size89. Among the three bHLH candidate genes, bHLH147 has the highest expression, which may be closely related to pulp cell expansion. In the ethylene biosynthesis pathway, 1-aminocyclopropane-1-carboxylate synthase (ACS) and 1-aminocyclopropane-1-carboxylate oxidase (ACO) are two important rate-limiting enzymes. ACS catalyzes the synthesis of 1-aminocyclopropane-1-carboxylate (ACC) from S-adenosylmethionine (SAM), and ACO catalyzes the synthesis of ethylene from 1-aminocyclopropane-1-carboxylate90,91. The results showed that BRs could affect ethylene biosynthesis. After BR treatment, the expression of leacs2, leacs4 and leaco1 is upregulated in tomato92. These results indicated that BR was positively correlated with ACS and ACO gene expression. In this study, the BR content was consistent with the expression levels of ACS and ACO. One of the ACO genes (Unigene0026400) had a relatively high expression level, and the expression level was significantly different. It is speculated that this gene is more sensitive to changes in BR in loquat.
The cpHSC70-1 and cpHSC70-2 proteins are essential for plant vegetative growth and reproduction. In Arabidopsis, mtHSC70-1 mutations cause severe embryonic defects. Studies have shown that Arabidopsis mtHSC70-1 loss-of-function mutations lead to mitochondrial abnormalities, decreased respiratory complex IV activity and abundance, and a dwarf phenotype93,94. The EjHsc70-2 gene may be related to the development of different tissues in loquat according to its expression patterns45. We found that the cluster 21 were positively correlated with GA and BR and negatively correlated with Aux, CTK and JA. The core gene EjHsc70 was found in cluster 21 and selected to verify its function. Heterologous validation revealed that the EjHsc70 promoted the elongation of Arabidopsis hypocotyl, which illustrated that the EjHsc70 could be involved in the development process of loquat fruit. However, the function of the EjHsc70 gene needs to be further verified in loquat plants, and the molecular mechanism of the EjHsc70 gene is still unknown and needs to further study.
Conclusions
In this study, the relationships between the endogenous hormone content and gene expression were explored in four developmental stages of loquat fruits, and fruit development-related genes were identified. The results revealed that the endogenous hormone levels of the pulps tended to differ among these four stages, and the DEGs among the different stages and hormone-related genes were screened. Cluster analysis divided the DEGs into 25 profiles, 16 of which were highly correlated with 1 to 5 hormones. A total of 25 and 28 core genes were screened from clusters 21 and 25, respectively.. Heterologous expression revealed that EjHsc70, a selected core gene, promoted Arabidopsis growth and could be involved in the development process of loquat fruit. This study provides new insights into fruit development in Loquat.
Data availability
The raw sequence data reported in this paper have been deposited in the Genome Sequence Archive in the National Genomics Data Center, China National Center for Bioinformation/Beijing Institute of Genomics, Chinese Academy of Sciences (project accession: PRJCA009904), which are publicly accessible at https://ngdc.cncb.ac.cn/bioproject/browse/PRJCA009904.
Abbreviations
- Aux:
-
Auxin
- GA:
-
Gibberellin
- CTK:
-
Cytokinin
- ABA:
-
Abscisic acid
- Eth:
-
Ethylene
- BR:
-
Brassinosteroid
- SA:
-
Salicylic acid
- JA:
-
Jasmonic acid
References
Lin, S. Q. Verification of the international introduction history of cultivated loquats. J. Fruit Sci. 37, 272–277 (2020).
Lin, S. et al. Morphological and developmental features of stone cells in Eriobotrya fruits. Front Plant Sci 13, 823993. https://doi.org/10.3389/fpls.2022.823993 (2022).
Tian, S.; Qin, G.; Li, B. Loquat (Eriobotrya japonica L.) . Postharvest Bio. Tec. Tropical Subtropical Fruits PB. 2011, 424–444. https://doi.org/10.1533/9780857092885.424
Su, W. B. et al. Advances in terpenoid identification and triterpene acid biosynthesis in loquat. Acta Hortic. Sin. 46, 1727–1738. https://doi.org/10.16420/j.issn.0513-353x.2018-0886 (2019).
Lü, H. et al. A new HPLC–UV method for the quantification of terpenoids and antioxidant activity of commercial loquat leaf tea and preparation. J. Food meas Charact. https://doi.org/10.1007/s11694-019-00358-3 (2020).
Pu, H. H. The current situation, problems and countermeasures of Wuxing loquat industry in Duanqiao town, Guanling Buyei and Miao Autonomous County province. South China Agr. 16, 154–156 (2022).
Huang, H. H. & Guo, M. Current situation, development trend and countermeasures of Liandu White Loquat Industry. Contemporary Hortic. 5, 58. https://doi.org/10.14051/j.cnki.xdyy.2020.05.025 (2020).
Oberts, J.A. But weight, there’s more: Genetic and hormonal control of fruit size in fleshy fruits. Annual P. Reviews. 2020, 3, 215–250. https://doi.org/10.1002/9781119312994.apr0727
Chen, Q. et al. The putative role of the NAC transcription factor EjNACL47 in cell enlargement of loquat (Eriobotrya japonica L.). Hortic. 7, 323. https://doi.org/10.3390/horticulturae7090323 (2021).
Dash, M. & Malladi, A. The AINTEGUMENTA genes, MdANT1 and MdANT2, are associated with the regulation of cell production during fruit growth in apple (Malus×domesticaBorkh.). BMC Plant Biol. 12, 1–15. https://doi.org/10.1186/1471-2229-12-98 (2012).
Azzi, L. et al. Fruit growth-related genes in tomato. J. Exp. Bot. 66, 1075–1086 (2015).
Su, W. et al. The cellular physiology of loquat (Eriobotrya japonica L.) fruit with a focus on how cell division and cell expansion processes contribute to pome morphogenesis. Sci. Hortic. 224, 142–149. https://doi.org/10.1016/j.scienta.2017.06.012 (2017).
Mayorga, G. A. & Nambeesan, S. U. Temporal expression patterns of fruit-specific α-EXPANSINS during cell expansion in bell pepper (Capsicum annuum L.). BMC Plant Biol 20, 1–12. https://doi.org/10.1186/s12870-020-02452-x (2020).
Chiba, Y. et al. Identification of Arabidopsis thaliana NRT1/PTR FAMILY (NPF) proteins capable of transporting plant hormones. J. Plant Res. 128, 679–686. https://doi.org/10.1007/s10265-015-0710-2 (2015).
Liu, X. X. et al. Cucumber fruit size and shape variations explored from the aspects of morphology, histology, and endogenous hormones. Plants 9, 1–17. https://doi.org/10.3390/plants9060772 (2020).
Tian, J. et al. Effects of endogenous hormones and sugars on fruit size driven by cell division between Korla fragrant pear and its bud mutation. Hortsci. 56, 881–888 (2021).
Nardozza, S. et al. Phytohormone and transcriptomic analysis reveals endogenous Cytokinins affect kiwifruit growth under restricted carbon supply. Metabolites 10, 1–18. https://doi.org/10.3390/metabo10010023 (2020).
Jia, H. F. et al. Abscisic acid plays an important role in the regulation of strawberry fruit ripening. Plant Physiol. 157, 188–199. https://doi.org/10.1104/pp.111.177311 (2011).
Wang, Y. P. et al. The expression profiling of the CsPYL, CsPP2C and CsSnRK2 gene families during fruit development and drought stress in cucumber. J. Plant Physiol. 169, 1874–1882. https://doi.org/10.1016/j.jplph.2012.07.017 (2012).
Wu, J. et al. An integrative analysis of the transcriptome and proteome of the pulp of a spontaneous late-ripening sweet orange mutant and its wild type improves our understanding of fruit ripening in citrus. J Exp Bot. 6, 1651–1671. https://doi.org/10.1093/jxb/eru044 (2014).
Gan, L. J. et al. Cytokinins are involved in regulation of tomato pericarp thickness and fruit size. Hortic Res. 9, 1–10. https://doi.org/10.1093/hr/uhab041 (2022).
Liu, C.S.; Xiao, P.S.; Jiang, F.; Wang, S.Y.; Liu, Z.; Song, G.Z.; Li, W.; Lv, T.X.; Li, .J; Wang, D.M.; Li, Y.; Wu, C.B.; Li, T.Z. Exogenous gibberellin treatment improves fruit quality in self-pollinated apple. Article. 2022, 174, 11–21. https://doi.org/10.1016/j.plaphy.2022.01.029
Czesnick, H. & Lenhard, M. Size control in plants-lessons from leaves and flowers. Cold Spring Harb. Perspect. Biol. 7, 1–17. https://doi.org/10.1101/cshperspect.a019190 (2015).
Huang, J., Li, Z. & Zhao, D. Deregulation of the Osmi R160 target gene OsARF18 causes growth and developmental defects with an alteration of auxin signalling in rice. Sci. Rep. 6, 1–14. https://doi.org/10.1038/srep29938 (2016).
Varaud, E. et al. AUXIN RESPONSE FACTOR8 regulates arabidopsis petal growth by interacting with the bHLH transcription factor BIGPETALp. Plant Cell 23, 973–983 (2011).
Zhang, H. L., Yao, K. F., Yi, D. H. & Lin, S. Q. Changes of endogenous hormones levels in three loquat varieties during fruit development. Botanical RES. 10, 6 (2021).
Zhu, Y. & Barritt, B. H. Apple cultivar genotypes for Md-ACS1 and Md-ACO1 ethylene production genes and implications for breeding. Tree Genet Genome. 4, 555–562 (2008).
Su, W. B. et al. EjBZR1 represses fruit enlargement by binding to the EjCYP90 promoter in loquat. Hortic. Resh. 8, 1–10. https://doi.org/10.1038/s41438-021-00586-z (2021).
Liu, X., Luo, M., Li, M. & Wei, J. Transcriptomic analysis reveals lncRNAs associated with flowering of Angelica sinensis during vernalization. Curr. Issues in Mol. Biol. 44(5), 1867–1888 (2022).
Li, C. et al. Transcriptome and metabolome analysis to reveal the mechanisms responding to high temperature stress in Anthesis stage of Maize. Acta Agric. Boreali-Sin. 35, 8–21. https://doi.org/10.7668/hbnxb.20190649 (2020).
Sun, H. et al. Investigating the molecular mechanisms of pepper fruit tolerance to storage via transcriptomics and metabolomics. Hortic. 7, 242. https://doi.org/10.3390/horticulturae7080242 (2021).
Zhang, Z. B. et al. Integrative transcriptomics and metabolomics data exploring the effect of chitosan on postharvest grape resistance to Botrytis cinerea. Postharvest Biol. Tec. 167, 111248. https://doi.org/10.1016/j.postharvbio.2020.111248 (2020).
Jiang, S., Luo, J., Xu, F. & Zhang, X. Transcriptome analysis reveals candidate genes involved in gibberellin-induced fruit setting in triploid loquat (Eriobotrya japonica). Front. Plant Sci. 7, 1–11 (2016).
Jing, D. et al. An integrative analysis of transcriptome, proteome and hormones reveals key differentially expressed genes and metabolic pathways involved in flower development in Loquat. Int. J. Mol. Sci. 21, 1–22. https://doi.org/10.3390/ijms21145107 (2020).
Xia, Y. et al. Comparative transcriptome analysis of flower bud transition and functional characterization of EjAGL17 involved in regulating floral initiation in loquat. PLoS ONE https://doi.org/10.1371/journal.pone.0239382 (2020).
Wu, J. C. et al. Effects of exogenous nitric oxide on anti-oxidation capacities in young loquat fruits under low temperature stress. Sci. Silvae Sinicae. 9, 73–78 (2010).
Langmead, B. & Salzberg, S. L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 9(4), 357–359. https://doi.org/10.1038/nmeth.1923 (2012).
Grabherr, M. G., Haas, B. J., Levin, Y. M. & J.Z., Amit, I.,. Trinity: Reconstructing afull-length transcriptome without a genome from RNA-Seqdata. Nat. Biotechnol. 29, 644–652. https://doi.org/10.1038/nbt.1883 (2011).
Hu, Z. et al. novo assembly and characterization of the leaf, bud, and fruit transcriptome from the vulnerable tree Juglans mandshurica for the development of 20 new microsatellite markers using Illumina sequencing. Mol. Genet. Genom. 291, 849–862. https://doi.org/10.1007/s00438-015-1147-y (2016).
Huang, Z.X.; Ke, C.; H.; Chen, J. Bioinformatics procedure of large-scale GO annotation. J. Xiamen Univ. 2012, 51, 139–143.
Ogata, H. et al. KEGG: Kyoto encyclopedia of genes and genomes. Nucl. Acids Res. 27, 29–34. https://doi.org/10.1093/nar/27.1.29 (1999).
Wang, D. et al. Study on fructose and mannose metabolism pathway of Tremella fuciformis based on transcriptome. Southwest China J. Agric. Sci. 32, 1347–1352 (2019).
Tang, D. et al. SRplot: A free online platform for data visualization and graphing. PLoS ONE 18(11), e0294236 (2023).
Li, H. Q., Xu, J., Chen, L. & Li, M. R. Establishment of an efficient Agrobacterium tumefaciens-mediated leaf disc transformation of Thellungiella halophila. Plant Cell Rep. 26, 1785–1789. https://doi.org/10.1007/s00299-007-0391-y (2007).
Lin, S.K.; Wu, T.; Li, M.; Huang, X.M.; Zhang, Y.Q.; Han, L.X.; Wu, B.S.; Chen, Y.; Lin, S.Y.; Lin, D.H.; Wu, M.L.; Wu, J.C*. Cloning, in silico characterization, subcellular localization, and expression of a heat shock cognate 70 kDa protein/gene (EjHsc70–2) from Eriobotrya japonica. Acta. Physiol. Plant. 2019, 41, 1–13. https://doi.org/10.1007/s11738-019-2908-8
González, F. A., Muñoz, V. M., Valenzuela, R. F., Parra, P. C. & Morales, Q. L. Effect of exogenous auxin treatment on cell wall polymers of strawberry fruit. Int. J. Mol. Sci. 22, 1–14. https://doi.org/10.3390/ijms22126294 (2021).
Devoghalaere, F. et al. A genomics approach to understanding the role of auxin in apple (Malus x domestica) fruit size control. BMC Plant Biol. 12, 1–5. https://doi.org/10.1186/1471-2229-12-7 (2012).
Cai, J. X., Liu, G. Q. & Chen, W. Changes in endogenous polyamine and hormone contents during different fruit developing stages in loquat. Chin. Agric. Sci. Bull. 23, 317–322. https://doi.org/10.3969/j.issn.1000-6850.2007.09.068 (2007).
Reig, C. et al. Soil temperature regulates fruit color change in “algerie” loquat: Nutritional and hormonal control. J. Plant Growth Regul. 35(4), 1108–1117. https://doi.org/10.1007/s00344-016-9608-z (2016).
Leng, P., Yuan, B., Guo, Y. & Chen, P. The role of abscisic acid in fruit ripening and responses to abiotic stress. J. Exp. Bot. 65, 4577–4588. https://doi.org/10.1093/jxb/eru204 (2014).
Ayub, R. A. et al. Abscisic acid involvement on expression of related gene and phytochemicals during ripening in strawberry fruit Fragaria×ananassav. Camino Real. Sci. Hortic. 203, 178–184. https://doi.org/10.1016/j.scienta.2016.03.026 (2016).
Roumeliotis, E., Visser, R. G. F. & Bachem, C. W. B. A crosstalk of auxin and GA during tuber development. Plant Signall. Behav. 7, 1360–1363. https://doi.org/10.4161/psb.21515 (2012).
Patrick, A., Wim, H. V., Dominique, V. D. S. & Nicholas, P. H. Ethylene regulates Arabidopsis development via the modulation of DELLA protein growth repressor function. Plant Cell 15, 2816–2825. https://doi.org/10.1105/tpc.015685 (2003).
Attia, S. M. & Adss, I. A. A. Effect of preharvest applied brassinosteroid on “Anna” apple fruit retention, coloration and quality. Bio Res. 18, 1416–1425. https://doi.org/10.1007/978-94-007-0189-2_9 (2021).
Kondo, S. Usage and action mechanism of oxylipins including jasmonic acid on physiological aspects of fruit production. Sci. Hortic. 295, 1–10. https://doi.org/10.1016/j.scienta.2022.110893 (2022).
El-Sharkawy, I., Liang, D. & Xu, K. Transcriptome analysis of an apple (Malus x domestica) yellow fruit somatic mutation identifies a gene network module highly associated with anthocyanin and epigenetic regulation. J. Exp. Bot. 66(22), 7359–7376. https://doi.org/10.1093/jxb/erv433 (2015).
Sweetman, C., Wong, D. C., Ford, C. M. & Drew, D. P. Transcriptome analysis at four developmental stages of grape berry (Vitis vinifera cv Shiraz) provides insights into regulated and coordinated gene expression. BMC Genomics 13(1), 691–691. https://doi.org/10.1186/1471-2164-13-691 (2012).
Lin, S. et al. De novo analysis reveals transcriptomic responses in Eriobotrya japonica fruits during postharvest cold storage. Genes 9(12), 639. https://doi.org/10.3390/genes9120639 (2018).
Jiang, S., An, H., Xu, F. & Zhang, X. Proteome analysis provides new insight into major proteins involved in gibberellin-induced fruit setting in triploid loquat (Eriobotrya japonica). Genes Genomics. 42, 383–392. https://doi.org/10.1007/s13258-019-00912-9 (2020).
Xia, Y. et al. Comparative transcriptome analysis of flower bud transition and functional characterization of EjAGL17 involved in regulating floral initiation in loquat. PLoS ONE 15, 1–18. https://doi.org/10.1371/journal.pone.0239382 (2020).
Ma, S. et al. De novo analysis reveals transcriptomic responses to heat stress in loquat leaves. Agron. J. 114(4), 1902–1914. https://doi.org/10.1002/agj2.20898] (2022).
Hendelman, A., Buxdorf, K., Stav, R., Kravchik, M. & Arazi, T. Inhibition of lamina outgrowth following Solanum lycopersicum AUXIN RESPONSE FACTOR 10 (Sl ARF10) derepression. Plant Mol. Biol. 78, 561–576. https://doi.org/10.1007/s11103-012-9883-4 (2012).
Piya, S.; Shrestha, S.K.; Binder, B.; Stewart, C.; Neal; Hewezi, T. Protein‒protein interaction and gene co-expression maps of ARFs and Aux/IAAs in Arabidopsis. Front. Plant SCI. 2014, 5, 744. https://doi.org/10.3389/fpls.2014.00744
Maryam, M. & Marina, P´., Andrea, C., Sumaira, F., Sergi, M.B.,. Leaf size modulation by cytokinins in sesame plants. Plant Physiol Bioch. 2021(167), 763–770. https://doi.org/10.1016/j.plaphy.2021.09.013 (2021).
Xue, H., Gao, X., He, P. & Xiao, G. H. Origin, evolution, and molecular function of DELLA proteins in plants. Crop J. 10, 287–299. https://doi.org/10.1016/j.cj.2021.06.005 (2021).
Xu, J. X. et al. Differential expression of genes related to endogenous hormone during bulb development in Lycoris radiata. Acta Hortic. Sin. 47, 1126–1140 (2020).
Hou, X. L., Ding, L. H. & Yu, H. Crosstalk between GA and JA signalling mediates plant growth and defense. Plant Cell Rep. 32, 1067–1074. https://doi.org/10.1007/s00299-013-1423-4 (2013).
Navarro, L. et al. DELLAs control plant immune responses by modulating the balance of jasmonic acid and salicylic acid signalling. Curr. Biol. 18, 650–655. https://doi.org/10.1016/j.cub.2008.03.060 (2008).
Hong, G. J., Xue, X. Y., Mao, Y. B., Wang, L. J. & Chen, X. Y. Arabidopsis MYC2 interacts with DELLA proteins in regulating sesquiterpene synthase gene expression. Plant Cell 24, 2635–2648. https://doi.org/10.1105/tpc.112.098749 (2012).
Abbas, H. M. K. et al. Mechanism associated with brassinosteroids crosstalk with gibberellic acid in plants. Brassinosteroids Signall. https://doi.org/10.1007/978-981-16-5743-6_6 (2022).
Pablo, A. et al. Brassinosteroid-regulated bHLH transcription factor CESTA induces the gibberellin 2-oxidase GA2ox7. Plant Physiol. 188, 2012–2025. https://doi.org/10.1093/plphys/kiac008 (2022).
Yang, W.; Xu, X.N.; Li, Y.; Wang, Y.Z.; Li, M.; Wang, Y.; Ding, X.H.; Chu, ZH.H; Lam, H.M. Rutin-mediated priming of plant resistance to three bacterial pathogens initiating the early SA signal pathway. PLOS ONE. 2016, 11, 0146910. https://doi.org/10.1371/journal.pone.0146910
Arif, Y., Sami, F., Siddiqui, H., Bajguz, A. & Hayat, S. Salicylic acid in relation to other phytohormones in plant: A study towards physiology and signal transduction under challenging environment. Environ. Exp. Bot. 175, 104040. https://doi.org/10.1016/j.envexpbot.2020.104040 (2020).
Gu, X. Y., Liu, Y. & Liu, L. J. Progress on the biosynthesis and signal transduction of phytohormone salicylic acid. Hereditas 42, 858–869 (2020).
Yu, X. D., Xu, Y. & Yan, S. H. P. Salicylic acid and ethylene coordinately promote leaf senescence. JIPB. 63, 823–827 (2021).
Fernandez-Calvo, P. et al. The arabidopsis bHLH transcription factors MYC3 and MYC4 Are targets of JAZ repressors and act additively with MYC2 in the activation of Jasmonate responses. Plant Cell 23, 70–715. https://doi.org/10.1105/tpc.110.080788 (2011).
Andrea, C., Sandra, F., Jose, M. C., Patriciam, F. C. & Roberto, S. The ZIM domain mediates homo- and heteromeric interactions between Arabidopsis JAZ proteins. Plant J. 59, 77–87. https://doi.org/10.1111/j.1365-313X.2009.03852.x (2009).
Ye, S. et al. Combined BSA-Seq based mapping and RNA-Seq profiling reveal candidate genes associated with plant architecture in Brassica napus. Int. J. Mol. Sci. 23, 1–15. https://doi.org/10.3390/ijms23052472 (2022).
Derakhshani, B. et al. Combined QTL mapping and RNA-Seq profiling reveals candidate genes associated with cadmium tolerance in barley. PLoS ONE 15, 1–19. https://doi.org/10.1371/journal.pone.0230820 (2020).
Zhu, T. et al. Effects of brassinosteroids on quality attributes and ethylene synthesis in postharvest tomato fruit. Postharest. Biol. Tec. 100, 196–204. https://doi.org/10.1016/j.postharvbio.2014.09.016 (2015).
Li, W.; Zhai, L.M.; Steven, H.S.; Huseyin, Y.; Emily, M.; Chen, j.m.; Wang, X.M.; Zhuang, W.B.; Fang, C.; Chen, Y.N.; Richard, M.A.; Han, Z.H.; Li, Y. Transgenic reduction of cytokinin levels in roots inhibits root-sprouting in populus. Plant Physiol. 2019, 180, 1788–1792. https://doi.org/10.1104/pp.19.00217
Sampedro, J. & Cosgrove, D. J. The expansin superfamily. Genome Biol. 6, 1–11. https://doi.org/10.1186/gb-2005-6-12-242 (2005).
Dotto, M. C., Martínez, G. A. & Civello, P. M. Expression of expansin genes in strawberry varieties with contrasting fruit firmness. Plant Physiol. Biochem. 44, 301–307. https://doi.org/10.1016/j.plaphy.2006.06.008 (2006).
Jiang, F. et al. Disassembly of the fruit cell wall by the ripening-associated polygalacturonase and expansin influences tomato cracking. Hortic. Res. 6, 1–15. https://doi.org/10.1038/s41438-018-0105-3 (2019).
Chen, X.; Li, W.; Wang, W.T.; Yang, D.L. Cloning and sequencing of cDNA of polygalacturonase in peach. Biotec. Bulletin. 2009, 96–99.
Hao, Q. N., Ma, C. & Ma, B. G. Construction and expres-sion identification of RNAi vector of polygalacturonase gene in strawberry. J Shi-hezi Univ. Nat. Sci. 27, 5. https://doi.org/10.3969/j.issn.1007-7383.2009.04.007 (2009).
Xu, P. P., Chen, H. Y., Ying, L. & Cai, W. M. At DOF5.4/OBP4, a DOF transcription factor gene that negatively regulates cell cycle progression and cell expansion in Arabidopsis thaliana. Sci. Rep. 6, 1–13 (2016).
Varaud, E. et al. Auxin response factor8 regulates arabidopsis petal growth by interacting with the b HLH transcription factor BIGPETALp. Plant Cell 23, 973–983. https://doi.org/10.1105/tpc.110.081653 (2011).
Paulo, F. J. & Ramos, P. S. B. Fast and furious: Ethylene-triggered changes in the metabolism of papaya fruit during ripening. Front. Plant Sci. 10, 1–11. https://doi.org/10.3389/fpls.2019.00535 (2019).
Atkinson, R. G. et al. Dissecting the role of climacteric ethylene in kiwifruit (Actinidia chinensis) ripening using a 1-aminocyclopropane-1-carboxylic acid oxidase knockdown line. J. Exp. Bot. 62, 3821–3835. https://doi.org/10.1093/jxb/err063 (2011).
Zhu, T. et al. Effects of brassinosteroids on quality attributes and ethylene synthesis in postharvest tomato fruit. Postharvest Biol. Tec. 100, 196–204. https://doi.org/10.1016/j.postharvbio.2014.09.016 (2015).
Su, P. H. & Li, H. Arabidopsis stromal 70-kD heat shock proteins are essential for plant development and important for thermotol-erance of germinating seeds. Plant Physiol. 146, 1231–1241. https://doi.org/10.1104/pp.107.114496 (2008).
Li, G. C. et al. Mitochondrial heat-shock cognate protein 70 contributes to auxin-mediated embryo development. Plant Physiol. https://doi.org/10.1093/plphys/kiab138 (2021).
Wei, S. S. et al. Arabidopsis mtHSC70–1 plays important roles in the establishment of COX-dependent respiration and redox homeostasis. J. Exp. Botany. 70, 5575–5590. https://doi.org/10.1093/jxb/erz357 (2019).
Acknowledgements
We appreciate that Guangzhou Gene Denovo Biotechnology Co., Ltd offered assistance in the sequencing and basic data analysis. The English language of this manuscript was edited by American Journal Experts (AJE).
Funding
This research was funded by the Fujian Provincial Science and Technology Project (2021N5014, 2022N5006, 2024N0062, 2021J011106, 2022J011162, 2023J011006), the Science and Technology Plan Project of Putian (2023GJGZ001, 2021ZP08, 2021ZP10, 2021ZP11), and the Research Projects of Putian University (2024177).
Author information
Authors and Affiliations
Contributions
S.L. and J.W. supervised this study and revised the manuscript. S.K.L., B.W. and Y.X. carried out the bioinformatics analysis, data mining, and functional validation and completed the manuscript. L.H. and D.L. performed the measurement of endogenous hormones. J.L. checked and revised the manuscript. All the authors have read and agreed to the published version of the manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
All methods were carried out in accordance with relevant guidelines and regulations.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Lin, S., Wu, B., Xiong, Y. et al. Integrated endogenous hormones and transcriptome analysis contribute to fruit development related gene mining in Eriobotrya japonica. Sci Rep 15, 14794 (2025). https://doi.org/10.1038/s41598-025-96870-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-025-96870-2
