
Abstract
Benzalkonium chloride (BAC) is the most commonly used preservative in eye drops. Unfortunately, it is potentially toxic and considered a leading cause of iatrogenic dry eye disease (DED) associated with local damage to the corneal epithelium. Corneal epithelium can be reconstituted thanks to the ability of limbal epithelial stem cells (LESCs) to self-renew, migrate, and differentiate, and can potentially be damaged by BAC. The aim of this study was to characterize the phenotype of human limbal stem cells (LSCs) isolated from the whole corneoscleral rims, and treated with BAC in vitro. The BAC dose was determined based on LSC viability assessment (MTT assay). The 48-h incubation period of LSCs with BAC was chosen to simulate long-term exposure of cells to preservative-containing eye drops. The cells were characterized by specific marker immunofluorescence staining; expression of genes related to proliferation, apoptosis, and inflammation (RT-qPCR); colony-forming ability and wound healing (scratch assay). Cell cycle stages were identified by flow cytometry. A BAC concentration of 0.0002% in the culture medium was chosen as an effective dose to inhibit LSC proliferation and migration and stimulate the expression of genes related to cell cycle, apoptosis, and inflammation. LSCs lose their clonogenic potential under the influence of BAC. It was concluded that benzalkonium chloride can develop toxic activity against limbal stem cells, limiting their regenerative potential.
Similar content being viewed by others

Introduction
The normal structure of the cornea is maintained by a population of stem cells, the so-called limbal epithelial stem cells (LESCs), located in the limbal region of the corneal epithelium. LESCs reside in the palisades of Vogt at the basal layer of the epithelium covering the anterior surface of the eye1. The limbal region (limbus) is recognized as a transition zone where the conjunctival and corneal epithelia form a superficial junction between the transparent cornea and opaque sclera of the outer coating layer of the eye (fibrous tunic)2,3.
Patients suffering from sight-damaging diseases, such as glaucoma and Sjögren’s syndrome, as well as contact lens wearers are forced to regularly use eye drops several times a day4,5,6. Regular and long-term administration of eye drops increases the risk of microbial infection. Medications such as eye drops contain preservatives whose role is to maintain their sterility and prevent microbial growth. One of the preservatives used in eye drops is benzalkonium chloride (BAC). Long-term exposure of the ocular surface to BAC is potentially toxic and leads to cellular dysfunction. A known toxic effect of BAC is the degradation of the conjunctival epithelium and apoptosis of goblet cells, resulting in a significant reduction of their total number7. This disrupts the production of tear film components, such as mucin, and subsequently damages to the corneal epithelium2 and promotes an inflammatory cascade4. Finally, long-term use of eye drops causes dry eye disease (DED), which affects approximately 20–50% of patients worldwide. DED is a growing problem defined as a multifactorial disease with tear film instability, hyperosmolarity, conjunctival cell damage, corneal cell damage, and inflammation. These processes interact with each other and constitute a vicious circle of DED8,9,10. The toxicity of BAC depends on the exposure time and concentration of the preservative. The typical concentration range of BAC used in ophthalmic medications is 0.005–0.02%11. For example, according to the product characteristics, Combigan (Allergan, Ireland) drops contain 0.005% BAC, while the concentration of BAC in Xalatan (Pfizer, USA) drops is 0.02%.
Corneal integrity depends on the self-renewing properties of its epithelium. Under physiological conditions, this ability depends on the activity of LESCs, which can divide asymmetrically, migrate from the limbus, differentiate, and gradually populate the corneal epithelium12. The corneal epithelium is also repaired after damage. The lack of corneal regeneration due to probable LESC dysfunction may negatively affect corneal homeostasis and result in abnormal vision as well as disintegration of superficial tissue providing protection from possible entry of pathogens. Corneal damage can lead to infection, ulceration, or perforation, which increases the severity of the wound, ultimately resulting in blindness13.
To date, there is a lack of studies investigating the effect of BAC on homogeneous human LESC population in vitro. Several studies have confirmed that BAC affects the conjunctival corneal epithelium, but these studies were conducted on rodent models in vivo or did not investigate its effects on a homogeneous stem cell population2,14,15,16. This may be due to, among other things, the known tendency of epithelial cells to be replaced by mesenchymal cells during cell culture17,18. As a result, limbal stem cells (LSC) may be a more appropriate term to describe the final cultured cell population.
This study aimed to investigate the BAC effect on LSCs isolated from human tissue. Development of a research model of the toxic influence of BAC on human LSCs in vitro will provide an opportunity to study methods of alleviating its side effects and will be useful in research on new drugs.
Methods
All methods were carried out in accordance with relevant guidelines and regulations. The experiments were conducted on human LSCs isolated from postoperative clinically rejected tissue from the corneoscleral rim of a deceased donor’s eye, after the extraction of the central part of the cornea for transplantation. The tissue sections were collected at the Clinical Department of Ophthalmology of the District Railway Hospital in Katowice. The study involving human cells was approved, but the need to obtain informed consent was waived by the Bioethics Committee of the Medical University of Silesia (SUM) in Katowice (decisions no. PCN/CBN/0052/KB/26a/22 and PCN/CBN/0052/KB/26b/22).
LSCs were expanded in vitro, identified, and tested for various supplements of the standard culture medium, namely hydrocortisone (H), insulin (I), and epidermal growth factor - EGF (E) for 12 days. The aim of the first stage of the experiment was to select a more appropriate composition of the culture medium compared to the standard medium (SM) containing DMEM/F-12 with L-glutamine, 10% fetal bovine serum (FBS), and 1% antibiotic-antimycotic solution (AA). The second stage of the experiment assessed the viability of cells cultured in H/I/E-supplemented medium as a control (CTRL) and treated with different doses of BAC for 48 h. The incubation time of LSCs in the BAC was 48 h, as such a period of time can simulate long-term exposure to eye drops with preservatives19. A dose of 0.0002% BAC was chosen for further experiments. The MTT assay, colony-forming (clonogenic) assay, scratch assay, RT-qPCR, and flow cytometry were used to evaluate the effect of BAC on LSC. The experimental scheme is presented in the Fig. 1.

Scheme of the experiment. SM, standard medium (DMEM/F12, 10% FBS, 1% AA); E, EGF; I, insulin; H, hydrocortisone; LPS, lipopolysaccharide.
Isolation of LSCs
Tissue portions were transported to the Department of the Cytophysiology, WNMK, SUM Katowice and stored in Eusol C (Alchimia, Austria) until the tests were performed. LSCs were isolated from the corneoscleral rim by explant culture. All superfluous tissue portions, such as outer residual corneal ring, sclera, and conjunctiva, were discarded, leaving only the corneal limbus with its basal layer. The limbal pieces were washed with PBS (Corning, USA) solution with antibiotics (2%) (Corning, USA) and diced into 2 × 2 × 2 mm portions. The obtained tissue sections were placed on culture dishes and flooded with the culture medium after adhesion.
Cell culture
LSCs were primarily maintained in DMEM/F-12 with L-glutamine (Dulbecco’s Modified Eagle Medium/Nutrient Mixture F-12, Gibco, USA), supplemented with 20% fetal bovine serum (FBS, Eurx, Poland) and 1% antibiotic-antimycotic solution (Corning, USA), and incubated at 37 °C and 5% CO2 in an incubator (Sanyo MCO-19 M, Japan). Isolated LSCs were characterized for the presence or absence of specific markers after the first passage and banked after the second passage for further evaluation. The culture medium was changed every two days, then the cells were transferred into new dishes for residual tissue removal. The LSCs used for the experiments were at the third to fifth passages. We performed the appropriate test using the Mycoalert kit (Lonza, Switzerland), which showed that the final cell population was not contaminated with mycoplasma.
To achieve the most efficient LSC outgrow, several compositions of the cell culture medium supplemented with H, I, and E or combined H/E, I/E, and H/I/E were tested. The addition of H/I/E to the culture medium was expected to enhance cell proliferation, maintain epithelial colonies, and support cell migration. After 24 h incubation, viability was assessed with MTT assay. All groups were analyzed in six replicates (n = 6).
The final selected culture medium contained DMEM/F-12 with L-glutamine, 10% FBS, 1% antibiotic-antimycotic solution (AA), 5 µg/ml insulin (Sigma-Aldrich, USA), 0.45 µg/ml hydrocortisone (Sigma-Aldrich, USA), and 20 ng/ml EGF (Sigma-Aldrich, USA). The LSCs were then treated with BAC ranging from 0.00002 to 0.02% for 48 h. The concentration of 0.0002% BAC solution for further experiments was chosen based on MTT assay. The assays were performed in eight replicates (n = 8). LSCs cultured in H/I/E medium constituted the control group, and the cells treated with BAC solution for 48 h were the study group (BAC group). Cell morphology was assessed using an Olympus IX73 microscope (Olympus, Shinjuku, Tokyo) with a camera (ORCA-spark, Hamatsu, Japan).
Cell phenotyping
Gene expression - RT-qPCR
RT-qPCR was preceded by total RNA extraction from cell pellets using a RNA Extracol (EURx, Poland) according to the manufacturer’s protocol. The nucleic acid concentration was determined using Nanodrop 2000 (Thermo Fisher Scientific, USA). Experiments were carried out in three biological and two technical replicates (n = 6). The sequences of specific primers used in the experiments are presented in Supplementary Information; Table S1.
The qualitative detection of ABCG2 and TP63 characteristic for LESC/LSCs, as well as KRT3 and KRT12 negative marker gene mRNAs was carried out for the first time after 1 passage using RT-qPCR with SYBR Green chemistry (GoTaq 1-Step RT-qPCR System, Promega, USA) and a LightCycler 96 Instrument (Roche, USA). The ACTβ gene served as a reference control. The amplicons were identified by 2% agarose gel electrophoresis. For this purpose, 5 µl of samples was mixed with 1 µl of 6x Loading Buffer TryDye (EURx, Poland) and placed in the gel. Electrophoresis was carried out at a constant voltage of 120 V for 25 min. The Perfect 100–1000 bp DNA Ladder (EURx, Poland) was used to assess the fragment lengths of the obtained products. Bands were detected using Gel Doc EZ instrument (Bio Rad, USA).
Cells were cultured and cell pellets were collected for RNA isolation also after passages 3, 4, and 5 to assess TP63 and ABCG2 gene expression to determine the stability of the LSC phenotype. RT-qPCR was followed by electrophoresis according to the previously established protocol.
The expression of genes related to the cell cycle (CDKN2C, CDKN2A, CCND1, CCND2, CCNE1, CCNE2, RB1, E2F2), apoptosis (BAX, BCL2, CASP3, CASP7, TP53), and inflammation (IL1β, IL6, IL4, IL10, NLRP3, IL1β, IL18, CASP1, MMP3, MMP9, TIMP1), including NLRP3 pathway and MMP activation, was quantified. The LPS, as a well-known pro-inflammatory agent, was used at a dose of 2 µg/ml as a positive control to evaluate the expression of inflammation-related genes. The dose of LPS was chosen based on previous studies20.
Marker identification - immunofluorescent staining and flow cytometry
LSCs after first passage were cultured on cover slips, fixed with 4% formaldehyde, and permeabilized with 0.5% Triton X-100 solution (Sigma-Aldrich, USA) and Tween 20 (Sigma-Aldrich, USA). After rinsing with PBS, the cells were incubated with blocking buffer for 1 h, rinsed again, and labelled with rabbit anti-ABCG2 polyclonal (1:50; 27286-AP; Proteintech, USA), rabbit anti-p63 polyclonal (1:50; 12143-1-AP; Proteintech, USA), mouse anti-CK3 monoclonal (1:50; ab68260; Abcam, UK), and rabbit anti-CK12 monoclonal (1:50; ab185627; Abcam, UK) primary antibodies at 4 °C overnight. The expression of proteins specific for LSCs: ABCG2 and p63, as well as negative controls (CK3 and CK12) were correlated with the expression of corresponding genes. The samples were then rinsed three times for 5 min with PBS and incubated for 1 h with Alexa Fluor 488 fluorochrome-conjugated secondary antibodies (1:1000; ab150077 for p63, ABCG2, CK12, and ab150113 for CK3; Abcam, UK). Cell nuclei were stained with DAPI (VECTASHIELD Vibrance Antifade Mounting Medium, H-1700; Vector, USA). Stained sections were compared with the isotype control (DA1E for p63, ABCG2, CK12; Cell Signaling, USA and ab184437 for CK3, Abcam, UK).
In addition, LSCs were cultured, passaged and characterized by flow cytometry (CytoFlex; Beckman Coulter, Brea, CA, USA) after passages 3 and 5 for the presence of mesenchymal markers CD73, CD90 and CD105 using the Human Mesenchymal Stem Cell Verification Flow Kit (FMC020, R&D Systems, USA). Cells were digested from the surface of culture vessels, suspended in a labeling solution containing specific antibodies conjugated with fluorochromes and (in separate tubes) appropriate isotype controls. After incubation, the cell suspension was washed and centrifuged. In the last step, cells were suspended in a labeling solution and analyzed in a flow cytometer. The information on the antibodies used for immunocytochemistry and flow cytometry is provided in Supplementary Information; Table S2.
Assessment of LSC viability - MTT assay
Cells were cultured in 96-well plates at a density of 104 per well. Then, the cells were added to the culture medium and incubated for the specified time. The negative control was cells treated with 0.1% Triton X-100. In the next step, MTT (3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide, Sigma-Aldrich, USA) solution was added to the wells, and the cells were incubated for 3 h at 37 °C in 5% CO2. Subsequently, the medium was discarded and 100 µl DMSO (Sigma-Aldrich, USA) was added to each well to dissolve the formazan crystals. Absorbance was measured using VICTOR Nivo plate reader (Perkin Elmer, USA) at 570 nm. The absorbance value obtained in the study group was compared with control value, which was taken as 1.0.
Cell proliferation - Colony-Forming assay
Control (CTRL) and BAC-treated LSCs (BAC) were tested with the colony-forming assay (CFA). To perform CFA, cells were seeded at a density of 1,000 cells per well of 6-well plates and incubated in the control medium or 0.0002% BAC for 10 days at 37 °C in 5% CO2 in the incubator. Both groups were analyzed in six replicates. The culture medium was changed every two days. Then, the cells were fixed with 4% formaldehyde for 25 min and stained with 0.5% crystal violet solution for 10 min. The stained colonies were counted and classified into holoclones, meroclones, and paraclones. Holoclones are colonies with high proliferative potential, meroclones are colonies with intermediate growth capabilities, and paraclones are colonies with limited growth potential, reflecting varying abilities to form colonies in culture. The colony-forming efficiency (CFE) was calculated according to the formula below:
Cell proliferation and migration - Scratch assay
A scratch assay was performed to evaluate the effects of BAC on LSC proliferation and migration. For this purpose, the CTRL and BAC groups were seeded into 12-well plates and cultured until reaching 100% confluence. The cells were then scratched with 200 µl micropipette tip, rinsed with PBS, and cultured for 24 h. The wound area was observed and photographed every 6 h until the complete closure of at least one cell culture. Image J program (v. 1.52a, USA) was used to quantify the wound area. Normalization in a scratch assay was performed by fold change. It is a method used to standardize the data and allow for meaningful comparisons across different experimental conditions or time points21. Experiments were done in five biological and two technical replicates (n = 10).
Cell cycle – Flow cytometry
The cells representing individual phases of the cell cycle (G0/G1, S, and G2/M phases) were qualified and quantified by flow cytometry with the use of FxCycle PI/RNase Staining Solution (Thermo Fisher Scientific, USA). This solution allows propidium iodide (PI) to intercalate with DNA, thereby producing a fluorescent signal with excitation/emission peak wavelengths of 535/617 nm. After a 48 h incubation with 0.0002% BAC solution, the cells were fixed in 100% ethanol and then treated with the staining solution for 30 min at room temperature. The samples were then examined at 488 nm and 532 nm excitation wavelengths and compared with untreated cells. All groups were analyzed in triplicate (n = 3).
Statistical analysis
Statistical analysis was performed using Statistica 13.3 software. Study groups were compared using ANOVA with post hoc Tukey or Student’s t-test for normally distributed data and Mann–Whitney U test for non-normally distributed data. The level of significance was set at 5% (α = 0.05) for all statistical tests.
Results
LSC characteristics
The isolated LSC cells were mostly cuboidal in shape, typical of epithelial cells, but this shape changed during the culture and between the third and fifth passage some of the cells took on more branched shapes typical of mesenchymal cells (Fig. 2).

Isolation and culture of LSCs. (A) A portion of the limbal tissue in explant culture of LSCs. Scale bar = 15 μm. (B) LSC culture after first passage. (C) LSC culture from 3 to 5 passage; (B and C) Scale bar = 30 μm.
The cells were characterized by the expression of ABCG2, and TP63 genes and the lack of the expression of KRT3 and KRT12 genes (Fig. 3A). The expression of cytokeratins CK3 and CK12 was not detected either at mRNA (KRT3 and KRT12 genes, respectively) or at protein level (Fig. 3B), which suggested that the isolated cells represent stem population.

(A) Agarose gel electropherogram of amplification products (passage 1). Lane 1: DNA ladder (M), lanes 2–3: KRT3 (101 bp), lanes 4–5: KRT12 (190 bp), lane 6: DNA ladder (M), lanes 7–8: TP63 (96 bp), lanes 9–10: ABCG2 (144 bp), lanes 11–12: ACTβ (131 bp), lane 13: DNA ladder (M). NC, negative control. (B) Visualization of lack of expression of CK3, and CK12 and expression of positive markers p63 and ABCG2, in LSCs cultured in a medium supplemented with H/I/E. FITC-conjugated specific antibodies (green); DAPI-stained cell nuclei (blue). IC, isotype control. Scale bars = 15 μm.
We observed simultaneous stable mRNA expression of LESC markers in LSC cells isolated from whole corneoscleral rims after passages 3 and 5 (Supplementary Information, Figure S1), along with expression of mesenchymal stem cell (MSCs) markers remaining at a fairly high but stable level (Supplementary Information, Figure S2). These results indicate that the cultured limbal cell population was composed to some extent of limbal mesenchymal stem cells (LMSCs), but both the LSC phenotype and the proportion of MSCs in this population did not change until the fifth passage of cell culture.
The effect of culture medium supplements on cells
Hydrocortisone, insulin, and EGF, whether administered individually or in combination, did not affect LSC viability and appeared to be nontoxic to the cells. All observed changes were statistically insignificant (Fig. 4). Each of these factors plays a key but different role in ensuring appropriate conditions for the proliferation, metabolism, growth, and functioning of the cells. We chose the combination of H/I/E as a supplement enriching the standard medium in both the control and BAC groups.

LSC viability in the medium containing: 0.45 µg/ml hydrocortisone (H); 5 µg/ml insulin (I), and 20 ng/ml EGF (E); or combined (H/E; I/E; H/I/E) compared to a standard medium containing DMEM/F12, 10% FBS, and 1% AA (SM), based on MTT assay (passage 2nd ). NC, negative control (cells treated with 0.1% Triton X). The bars represent the mean percentage ± standard deviation (SD) of the control group (100%). ANOVA with the Tukey post hoc test. *p < 0.001 as compared to SM.
Evaluation of the BAC toxic dose
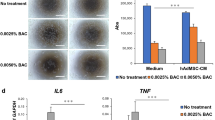
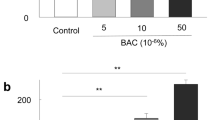
We showed a dose-dependent and statistically significant decrease in LSC viability after exposure to different concentrations of BAC (Fig. 5). Cell viability decreased by 11.1% and 31.9% at the doses of 0.00002% and 0.0002%, respectively compared to CTRL. The higher doses caused almost complete cell death. The difference of 67.8% between the dose ranges of 0.00002–0.0002% vs. 0.002–0.02% was the most noticeable and statistically significant (p < 0.001). Based on these results, the concentration of 0.0002% BAC was selected to further experiments.

LSC viability after 48 h exposure to different doses of BAC, based on MTT assay (passage 3rd ). The bars represent the mean percentage ± standard deviation (SD) of the control group (1.00). CTRL, control; NC, negative control. ANOVA with the Tukey post hoc test. *p < 0.001 as compared to CTRL, **p < 0.001 as compared to the indicated values.
Clonogenicity, proliferation, and migration of control and BAC-treated LSCs
One of the characteristics of LSCs was their ability to form colonies confirmed with CFA assay (Fig. 6). The CFA was analyzed using classifications based on colony morphology and size. Holoclones were identified as large, round colonies with a dense cellular arrangement and smooth perimeters. Meroclones were classified as intermediate-sized colonies with moderate density and irregular perimeters, showing significant cell clustering. Paraclones were recognized as small, irregularly shaped colonies with loosely packed cells and rough perimeters (Fig. 6A and B), and counted in CTRL and BAC-treated cultures (Fig. 6C).

Colony-forming efficiency based on the total number of colonies (total), holoclones, (holo-), meroclones (mero-), and paraclones (para-) (passage 4th ). (A) A representative image of the holoclone (I), meroclone (II) and paraclone (III) in untreated cells (CTRL). (B) A representative image of the colony-forming assay and (C) the percentage of colonies in control (CTRL) and BAC-treated culture (BAC). The bars represent means ± SD. Student’s t-test. *p < 0.001.
The CFE was 5.8% in total, of which 16% were holoclones considered mostly as undifferentiated cells. The BAC-treated group completely lost the colony-forming ability. This suggested that LSCs lose their self-renewal capacity under the BAC influence.
The proliferation and migration of LSCs were analyzed using the wound healing test (scratch assay). Significantly higher proliferation and migration rates were observed for control LSCs as compared to the BAC group at all time points (Fig. 7).

The LSC proliferation and migration test based on scratch assay (passage 4th). (A) A representative image of a scratched LSC monolayer in the control group (CTRL) and cells treated with 0.0002% BAC at 0 h, 6 h, 12 h, 18 h, and 24 h. The scratch area is marked black. (B) The scratch surface area as a multiple of the area measured at time point 0 h. Mann–Whitney U Test. *p < 0.05, **p < 0.01, ***p < 0.001 as compared to CTRL; n = 10.
The impact of BAC on inflammation-related gene expression
Independently of the LSCs cultures in the CTRL and BAC groups, a group containing 2 µg/ml LPS was created, as a positive control for triggering inflammation. The expression of proinflammatory IL1β was upregulated (p < 0.001) and the expression of CASP1 (p < 0.01), and IL18 (p < 0.001) was downregulated in the BAC group compared to the CTRL group. The expression of IL4 and IL10 was not detected. There were not statistically significant differences in the expression of NLRP3 and TIMP1 among the groups. The expression of CASP1 (p < 0.001) and IL18 (p < 0.001) genes significantly increased, but IL1β (p < 0.001) decreased in the LPS group compared to the BAC group. A significantly increased expression of CASP1 (p < 0.01) and IL6 (p < 0.001) was also noticed in the LPS group compared to CTRL (Fig. 8A). The expression of MMP3 was upregulated (p < 0.001) in the LPS group compared to the CTRL and BAC groups. The expression of MMP9 (p < 0.05) was also upregulated in the LPS group compared to the BAC group. We did not detect statistically significant differences in the expression of MMP3 and MMP9 between the CTRL and BAC-treated groups (Fig. 8B).

The fold change of expression of genes related to (A) inflammation and NLRP3 pathway; (B) MMP activation, in LSC cells (passage 5th). CTRL, control; BAC, benzalkonium chloride-treated group; LPS, lipopolysaccharide-treated group for 48 h. The bars represent means ± SD. ANOVA. *p < 0.05, **p < 0.01, ***p < 0.001; n = 6.
The effect of BAC on apoptosis-related gene expression
In the BAC-treated groups the expression of apoptosis-related genes BAX, CASP3, and CASP7 significantly increased, while the expression of BCL2 was downregulated. We did not detect statistically significant differences in the expression of TP53 between the CTRL and BAC-treated groups (Fig. 9A).

The fold change of expression of genes related to (A) apoptosis and (B) the cell cycle in LSCs (passage 5th). CTRL, control; BAC, benzalkonium chloride-treated group for 48 h. The bars represent means ± SD. Student’s t- test. *p < 0.05, **p < 0.01, ***p < 0.001; n = 6.
The effect of BAC on LSC cell cycle
Under the influence of BAC, the expression of RB1 (p < 0.001) was upregulated, and the expression of E2F2 (p < 0.001) was downregulated. Cyclin-related genes, namely CDKN2C (p < 0.001), CCNE1 (p < 0.01), and CCNE2 (p < 0.01) were upregulated, whereas the expression of CCND1 and CCND2 genes remained unchanged, and the expression of CDKN2A was not detected (Fig. 9B). However, cell cycle analysis showed no statistically significant differences between the CTRL and BAC groups (Fig. 10).

LSC cell cycle analysis (passage 5th). (A) Histograms presenting the proportion of the number of LSCs in different phases of the cell cycle. (B) Percentage of the number of cells in cell cycle phases. CTRL, control; BAC, benzalkonium chloride-treated group for 48 h. The bars represent means ± SD. Student’s t-test; n = 3.
Discussion
Eye drops are commonly used in patients suffering from diseases that damage the eye. These pharmaceuticals often contain preservatives and substances that protect against the growth of microorganisms, such as benzalkonium chloride. Unfortunately, prolonged exposure of the ocular surface to BAC is potentially harmful4. The corneal epithelium can be reconstituted thanks to the proliferative and differentiating properties of limbal epithelial stem cells22. However, LESCs are exposed to the toxic effects of BAC simultaneously with the corneal epithelium, and accompanying cells, as LMSCs.
There is no gold standard for collecting LESCs from tissue samples. These cells can be obtained from the corneal limbus by the enzymatic isolation or explant culture methods. Both can be modified at any stage of the procedure, resulting in divergent protocols23,24,25. New studies are constantly emerging on the methods to optimize the process of isolating and culturing LESCs, limbal niche cells (LNCs), and other populations residing in the limbus for research or therapeutic purposes26,27,28,29. The isolation effectiveness depends on the donor’s age and the tissue storage conditions from the moment of tissue collection to the initiation of culture30. In vitro, the cell population of limbal cells isolated from the whole corneoscleral rims consists to some extent of LESC progeny, i.e., transitional-amplifying stem cells and limbal mesenchymal stem cells. Moreover, mesenchymal cells tend to replace epithelial cells during cell culturing after isolation or epithelial cells undergo epithelial-to-mesenchymal transition during the first few passages, especially when a cellular population is obtained via explant culture and high serum concentrations are used17,18. As a result, a more appropriate term to describe the final cultured cell population seems to be limbal stem cells (LSC). This issue should be taken into account in research and we aimed to characterize the phenotype of human LSCs influenced by BAC in vitro.
Our approach to LSC isolation was based on the explant culture, in which the cells migrate from the tissue section within a few days. A major advantage of explant culture is the elimination of additional chemicals in cell culture, facilitating its clinical translation23,24,25. After the first passage, the obtained LSCs showed cuboidal epithelial morphology and expressed ABCG2 and TP63 genes and appropriate proteins, but not CK3 and CK12 encoded by KRT3 and KRT12 genes, respectively. The latter are negative markers for LESCs expressed only in the central and peripheral corneal epithelial cells but absent in the limbal region23,31. The intense expression of molecular markers p63 and ABCG2 proteins is specific for most LSCs, including LMSCs and was observed by us consistently across all cell samples, both after passage 3 and 5, confirmed their identity32,33. It may also be advisable to assess the presence of CK14, CK15, and CK19 molecular markers that are stably expressed in the corneal epithelium or vimentin, which is specific to the basal layer28,34. These observations confirmed that the LSC population is not homogeneous and fully identical to LESC (it consists of at least LESCs and LMSCs).
Furthermore, the studied LSCs showed a tendency to form holoclones containing cells with a high proliferation rate and ability of self-renewal. The CFE value depends on the cell culture conditions and averages 4–12%35,36. The higher percentage of holoclones appearing in the CFA reflects the better quality of the LESC population in the culture dish22. Finally, the presence of cells isolated from the corneal limbal region having epithelial-like morphology, expressing specific markers and showing 5.8% CFE confirmed the successful isolation of LSCs containing LESC cells and characterized by good quality.
The composition of the culture medium for LESCs is not clearly defined in the literature, so it is useful to determine it before the experiment. To identify the optimal cell culture conditions, we investigated the effects of some bioactive compounds typically added to the culture media on LSCs. The basal medium for LESCs/LSCs is DMEM/F12, FBS, and an antibiotic solution with other components, for example hydrocortisone, insulin, EGF, cholera toxin, and dexamethasone37,38. Hydrocortisone plays a role in maintaining epithelial colonies and also supports cell proliferation39. Several factors can potentially modulate cell migration and growth, e.g., insulin-like growth factor (IGF), EGF, and hepatocyte growth factor (HGF)40,41. Insulin and IGF act similarly by having an affinity for both of their receptors42. In our study, hydrocortisone, insulin, and EGF, administered separately and together, did not affect LSC viability. Assuming a more comprehensive effect of the three analyzed supplements on the cultured cells, we chose the combination of H/I/E as a standard supplement to the control medium in the experiments conducted with LSCs and BAC.
Several studies have investigated different ranges of BAC concentration, usually chosen based on the total amount of BAC present in the medicines of certain interest as well as the time- and dose-dependent toxic effects of BAC4,43,44,45. It was shown that BAC causes morphological changes, apoptosis, oxidative stress, and induction of pro-inflammatory genes in corneal epithelial cells44. Here we observed a dose-dependent decrease in LSC viability under BAC exposure. We have shown that there is indeed a very clear impact of all the tested BAC concentrations (ranging from 0.00002 to 0.02%) on LSC viability, but there was also a significant difference in LSC viability between the dose ranges of 0.00002–0.0002% and 0.002–0.02%. The use of lower doses of BAC in studies is intended to simulate more realistic conditions that may occur in the body. Dilution by the tear film can have a significant impact on the toxicity of a BAC dose, especially in the context of using lower doses in in vitro studies. In a study by Datta et al., a concentration of 0.0002% was determined as the IC50 in the context of inhibition of mitochondrial O2 consumption leading to cellular dysfunction and eventual cell death11. Based on the observations in our study presented above, the concentration of 0.0002% BAC was selected to further experiments.
The scratch assay assesses both cell migration and proliferation, which are crucial in evaluating the toxicity of a substance. BAC may impair cell migration, slowing or preventing wound closure, or it might inhibit cell proliferation, reducing the number of cells available to fill the gap. We demonstrated a reduction in the rate of LSC proliferation and migration under the BAC influence at each of the time points studied in scratch assay. Similar findings were made at 24 h on corneal epithelial cells (CECs), which are not stem cells, and with higher BAC concentrations (0.002–0.2%)46. Furthermore, we observed that LSCs lose their ability to form colonies when exposed to BAC. The CFE showed living cells in the BAC group without any type of colony. We confirmed that BAC reduces the proliferative potential of LSCs also through observations of cell cycle dysregulation at the molecular level. This could potentially result in inhibition of corneal epithelial regeneration in vivo. BAC is also known to disrupt the cell cycle and cause its arrest in the G0/G1 phase in somatic cells, i.e., in lung epithelial cells47. At the molecular level, the cell cycle promotion depends on the expression of the E2F2, CCND1, CCND2, CCNE1, and CCNE2 genes. CDKN2C and Rb1 products inhibit the cell cycle progression. Rb1 exerts its inhibitory effect on the cell cycle by binding to and inhibiting the activity of the E2F transcription factor. The E2F2 factor belongs to the E2F family and is essential for G1/S phase transition and apoptosis regulation48. We observed a statistically significant higher level of RB1 and CDKN2C genes expression and a significant decrease in E2F2 gene expression in the LSC group after 48 h incubation with BAC. However, a significant increase in the expression of the CCNE1 and CCNE2 genes in the BAC group may indicate an attempt to suppress the negative impact of BAC by stem cells. Stem cells possess unique properties different from somatic cells, which help them recover from unfavorable environments49. This is especially true since, regardless of the changes in the expression of some genes observed at the mRNA level, the flow cytometric analysis of cell cycle phases showed no statistically significant differences between the control and BAC-treated groups. This result confirms that BAC is not completely neutral to the LSC cell cycle activity.
Moreover, an analysis of gene expression related to apoptosis provided unequivocal information that apoptosis begins to progress under the influence of BAC. In our study, the expression of the anti-apoptotic gene BCL2 was significantly reduced, and the expression of the pro-apoptotic BAX, CASP3, and CASP7 genes was significantly higher after BAC treatment. The balance between the expression of the pro-apoptotic BAX gene and the anti-apoptotic BCL2 gene determines whether cells undergo apoptosis. These changes corresponded to observations likely lead to decreased cell proliferation and cell cycle dysregulation reflecting a transition towards a more apoptotic state. This result confirms that BAC is not completely indifferent to the cell cycle activity of LSCs, but the observed differences in gene expression, especially those related to apoptosis, although statistically significant, are relatively small and do not exclude a stronger effect at higher doses.
It should be taken into consideration that DED is accompanied not only by apoptosis and local damage to the corneal epithelium but also by inflammation. These processes interact with each other and create a vicious circle of DED. Recent studies indicate a mechanism of action of BAC in corneal damage through activation of the NLRP3 pathway associated with inflammation and pyroptosis50. Similarly to LPS (a strong pro-inflammatory agent), the activation of the NF-κβ transcription factor by BAC may result in the activation of the NLRP3 inflammasome complex. This leads to the activation of CASP1 and the production of proinflammatory cytokines such as IL1β and IL18. MMP3 and MMP9 are also implicated in the pathogenesis of inflammations. Increased expression of MMP3 and MMP9 in response to NF-κβ activation contributes to tissue damage. MMP3 and MMP9 activity can be inhibited by TIMP151. Also, some other pro-inflammatory cytokines, such as IL6 and TNFα, and anti-inflammatory cytokines, such as IL4 and IL10, are also involved in the development of inflammation, which is a complex process52,53. The induction of IL6 by IL1β is known to highlight its role as one of the first cytokines secreted during early inflammation, suggesting that elevated IL1β levels may precede full activation of the inflammatory process, potentially contributing to the subsequent elevation of IL6 levels in later stages54. The application of BAC at doses in the range of 0.001–0.1% to immortalized human conjunctival and corneal epithelial cells induced significant amounts of IL-1α and TNFα55. In the cultured LSCs we observed an increase in IL1β gene expression, but not IL4 and IL10 genes after BAC treatment. The expression of CASP1 and IL18 genes was decreased. Moreover, BAC did not activate MMP3 or MMP9. Taken together, the observed changes could potentially lead to reduced inflammasome activity. These results correspond with some in vivo studies that have found no inflammatory changes under the influence of a higher dose of BAC56, longer exposure time, and development of large epithelial defects.
Conclusions
Benzalkonium chloride at a concentration of 0.0002% has a toxic effect against LSCs and reduces their metabolism, migration, and proliferation as well as dysregulates the cell cycle. LSCs under the influence of BAC lose their clonogenic potential. BAC induces apoptosis but does not trigger an inflammatory response. It cannot be ruled out that this latter effect of BAC, not observed in this study, will become apparent in vitro after using higher doses of BAC contained in eye drops.
Data availability
Data is provided within the manuscript.
References
Li, W., Hayashida, Y., Chen, Y. T. & Tseng, S. C. Niche regulation of corneal epithelial stem cells at the limbus. Cell. Res. 17, 26–36. https://doi.org/10.1038/sj.cr.7310137 (2007).
Lin, Z. et al. A mouse model of limbal stem cell deficiency induced by topical medication with the preservative benzalkonium chloride. Invest. Ophthalmol. Vis. Sci. 54, 6314–6325. https://doi.org/10.1167/iovs.12-10725 (2013).
Schermer, A., Galvin, S. & Sun, T. T. Differentiation-related expression of a major 64K corneal keratin in vivo and in culture suggests limbal location of corneal epithelial stem cells. J. Cell Biol. 103, 49–62. https://doi.org/10.1083/jcb.103.1.49 (1986).
Hedengran, A. et al. Benzalkonium chloride-preserved anti-glaucomatous eye drops and their effect on human conjunctival goblet cells in vitro. Biomed. Hub. 6, 69–75. https://doi.org/10.1159/000517845 (2021).
Coursey, T. G. & de Paiva, C. S. Managing Sjögren’s syndrome and non-Sjögren syndrome dry eye with anti-inflammatory therapy. Clin Ophthalmol. 8, 1447–1458. https://doi.org/10.2147/OPTH.S35685 (2014).
Ahn, J. & Choi, M. Binding affinity of benzalkonium chloride on contact lens surfaces and the effects on their physical properties. Colloids Surf. B Biointerfaces. 203, 111744. https://doi.org/10.1016/j.colsurfb.2021.111744 (2021).
Baudouin, C., Labbé, A., Liang, H., Pauly, A. & Brignole-Baudouin, F. Preservatives in eyedrops: the good, the bad and the ugly. Prog Retin Eye Res. 29, 312–334. https://doi.org/10.1016/j.preteyeres.2010.03.001 (2010).
Rouen, P. A. & White, M. L. Dry eye disease: prevalence, assessment, and management. Home Healthc. Now. 36, 74–83. https://doi.org/10.1097/NHH.0000000000000652 (2018).
Stapleton, F. et al. TFOS DEWS II epidemiology report. Ocul Surf. 15, 334–365 https://doi.org/10.1016/j.Jtos.2017.05.003 (2017).
Han, Y. et al. Berberine ameliorate inflammation and apoptosis via modulating PI3K/AKT/NFκB and MAPK pathway on dry eye. Phytomedicine 121, 155081. https://doi.org/10.1016/j.Phymed.2023.155081 (2023).
Datta, S., Baudouin, C., Brignole-Baudouin, F., Denoyer, A. & Cortopassi, G. A. The eye drop preservative venzalkonium chloride potently induces mitochondrial dysfunction and preferentially affects LHON mutant cells. Invest. Ophthalmol. Vis. Sci. 58, 2406–2412. https://doi.org/10.1167/iovs.16-20903 (2017).
Secker, G. A. & Daniels, J. T. Limbal epithelial stem cells of the cornea. StemBook (Harvard Stem Cell Institute, 2009).
Byrd, L. B. & Martin, N. Corneal ulcer. StatPearls. (StatPearls Publishing, 2023).
Pauly, A. et al. New tools for the evaluation of toxic ocular surface changes in the rat. Invest. Ophthalmol. Vis. Sci. 48, 5473–5483. https://doi.org/10.1167/iovs.06-0728 (2007).
Yim, N. H., Park, E., Cho, W. K., Kim, Y. H. & Ma, J. Y. Therapeutic effects of Acer palmatum thumb. Leaf extract (KIOM-2015E) on Benzalkonium chloride-induced dry eye in a mouse model. Int. J. Mol. Sci. 23, 14964. https://doi.org/10.3390/Ijms232314964 (2022).
Wang, G. et al. Exosomes derived from mouse adipose-derived mesenchymal stem cells alleviate benzalkonium chloride-induced mouse dry eye model via inhibiting NLRP3 inflammasome. Ophthalmic Res. 65, 40–51. https://doi.org/10.1159/000519458 (2022).
Zhang, X. et al. Comparison of cell-suspension and explant culture of rabbit limbal epithelial cells. Exp. Eye Res. 80, 227–233. https://doi.org/10.1016/j.exer.2004.09.005 (2005).
Li, W., Hayashida, Y., He, H., Kuo, C. L. & Tseng, S. C. The fate of limbal epithelial progenitor cells during explant culture on intact amniotic membrane. Invest. Ophthalmol. Vis. Sci. 48, 605–613. https://doi.org/10.1167/iovs.06-0514 (2007).
Ayaki, M., Iwasawa, A. & Inoue, Y. Toxicity of antiglaucoma drugs with and without benzalkonium chloride to cultured human corneal endothelial cells. Clin. Ophthalmol. 4, 1217–1222. https://doi.org/10.2147/OPTH.S13708 (2010).
Sikora, B., Skubis-Sikora, A., Prusek, A. & Gola, J. Paracrine activity of adipose derived stem cells on limbal epithelial stem cells. Sci. Rep. 11, 19956. https://doi.org/10.1038/s41598-021-99435-1 (2021).
Bahar, E. & Yoon, H. Modeling and predicting the cell migration properties from scratch wound healing assay on cisplatin-resistant ovarian cancer cell lines using artificial neural network. Healthcare 9, 911. https://doi.org/10.3390/healthcare9070911 (2021).
Pellegrini, G. et al. From discovery to approval of an advanced therapy medicinal product-containing stem cells, in the EU. Regen Med. 11, 407–420. https://doi.org/10.2217/rme-2015-0051 (2016).
Zhang, Z. H., Liu, H. Y., Liu, K. & Xu, X. Comparison of explant and enzyme digestion methods for ex vivo isolation of limbal epithelial progenitor cells. Curr. Eye Res. 41, 318–325. https://doi.org/10.3109/02713683.2015.1014566 (2016).
Stasi, K. et al. Optimal isolation and xeno-free culture conditions for limbal stem cell function. Invest. Ophthalmol. Vis. Sci. 55, 375–386. https://doi.org/10.1167/iovs.13-12517 (2014). Massaro-Giordano, G.; Gearhart, J.D.
Li, D. Q. et al. Single-Cell transcriptomics identifies limbal stem cell population and cell types mapping its differentiation trajectory in limbal basal epithelium of human cornea. Ocul Surf. 20, 20–32. https://doi.org/10.1016/j.jtos.2020.12.004 (2021).
Xiao, Y. T., Qu, J. Y., Xie, H. T., Zhang, M. C. & Zhao, X. Y. A comparison of methods for isolation of limbal niche cells: maintenance of limbal epithelial stem/progenitor cells. Invest. Ophthalmol. Vis. Sci. 61, 16. https://doi.org/10.1167/Iovs.61.14.16 (2020).
Polisetti, N., Sharaf, L., Schlötzer-Schrehardt, U., Schlunck, G. & Reinhard, T. Efficient isolation and functional characterization of niche cells from human corneal limbus. Int. J. Mol. Sci. 23, 2750. https://doi.org/10.3390/ijms23052750 (2022).
Netto, A. R. T., Hrusa, M. D., Bartz-Schmidt, K. U., Schnichels, S. & Hurst, J. Two methods for the isolation and cultivation of porcine primary corneal cells. Methods Protoc. 6, 50. https://doi.org/10.3390/mps6030050 (2023).
Santra, M. et al. Good manufacturing practice production of human corneal limbus-derived stromal stem cells and in vitro quality screening for therapeutic Inhibition of corneal scarring. Stem Cell. Res. Ther. 15, 11. https://doi.org/10.1186/s13287-023-03626-8 (2024).
Romo-Valera, C. et al. Characterisation of corneas following different time and storage methods for their use as a source of stem-like limbal epithelial cells. Exp. Eye Res. 211, 108720. https://doi.org/10.1016/j.Exer.2021.108720 (2021).
Zhu, L. et al. Cotransplantation of limbal epithelial and stromal cells for ocular surface reconstruction. Ophthalmol. Sci. 2, 100148. https://doi.org/10.1016/j.Xops.2022.100148 (2022).
Shukla, S. et al. Limbal epithelial and mesenchymal stem cell therapy for corneal regeneration. Curr. Eye Res. 45, 265–277. https://doi.org/10.1080/02713683.2019.1639765 (2020).
Shaharuddin, B. et al. Human limbal mesenchymal stem cells express ABCB5 and can grow on amniotic membrane. Regen Med. 11, 273–286. https://doi.org/10.2217/rme-2016-0009 (2016).
Guo, Z. H., Zeng, Y. M. & Lin, J. S. Dynamic spatiotemporal expression pattern of limbal stem cell putative biomarkers during mouse development. Exp. Eye Res. 192, 107915. https://doi.org/10.1016/j.exer.2020.107915 (2020).
Echevarria, T. J., Chow, S., Watson, S., Wakefield, D. & Di Girolamo, N. Vitronectin: a matrix support factor for human limbal epithelial progenitor cells. Invest. Ophthalmol. Vis. Sci. 52, 8138–8147. https://doi.org/10.1167/iovs.11-8140 (2011).
Park, K. S. et al. The side population cells in the rabbit limbus sensitively increased in response to the central cornea wounding. Invest Ophthalmol Vis Sci. 47, 892–900. https://doi.org/10.1167/iovs.05-1006 (2006).
Can, G. D., Akdere, Ö. E., Can, M. E. & Gümüşderelioğlu, M. A simple and efficient method for cultivation of limbal explant stem cells with clinically safe potential. Cytotherapy 21, 83–95. https://doi.org/10.1016/j.jcyt.2018.11.005 (2019).
Rheinwald, J. G. & Green, H. Serial cultivation of strains of human epidermal keratinocytes: the formation of keratinizing colonies from single cells. Cell 6, 331–343. https://doi.org/10.1016/s0092-8674(75)80001-8 (1975).
Kim, M. S. et al. Development of a chemically defined in vitro culture system to effectively stimulate the proliferation of adult human dermal fibroblasts. Exp. Dermatol. 24, 543–545. https://doi.org/10.1111/Exd.12695 (2015).
Lee, J. H. et al. Induced expression of insulin-like growth factor-1 by amniotic membrane-conditioned medium in cultured human corneal epithelial cells. Invest. Ophthalmol. Vis. Sci. 47, 864–872. https://doi.org/10.1167/Iovs.05-0596 (2006).
Loo, S., Kam, A. & Tam, J. P. Hyperstable EGF-like bleogen derived from cactus accelerates corneal healing in rats. Front. Pharmacol. 13, 942168. https://doi.org/10.3389/fphar.2022.942168 (2022).
Cai, W. et al. Domain-dependent effects of insulin and IGF-1 receptors on signalling and gene expression. Nat. Commun. 8, 148892. https://doi.org/10.1038/Ncomms14892 (2017).
Xu, M., Sivak, J. G. & McCanna, D. J. Ocular toxicology: synergism of UV radiation and benzalkonium chloride. Cutan. Ocul. Toxicol. 39, 370–379. https://doi.org/10.1080/15569527.2020.1833027 (2020).
Vitoux, M. A. et al. Benzalkonium chloride-induced direct and indirect toxicity on corneal epithelial and trigeminal neuronal cells: proinflammatory and apoptotic responses in vitro. Toxicol. Lett. 319, 74–84. https://doi.org/10.1016/j.toxlet.2019.10.014 (2020).
Chang, C., Zhang, A. Q., Kagan, D. B., Liu, H. & Hutnik, C. M. Mechanisms of benzalkonium chloride toxicity in a human trabecular meshwork cell line and the protective role of preservative-free Tafluprost. Clin. Exp. Ophthalmol. 43, 164–172. https://doi.org/10.1111/Ceo.12390 (2015).
Moon, J., Ko, J. H., Yoon, C. H., Kim, M. K. & Oh, J. Y. Effects of 20% human serum on corneal epithelial toxicity induced by benzalkonium chloride: in vitro and clinical studies. Cornea. 37, 617–623. https://doi.org/10.1097/ICO.0000000000001475 (2018).
Kanno, S. et al. Plasma membrane damage triggered by benzalkonium chloride and cetylpyridinium chloride induces G0/G1 cell cycle arrest via Cdc6 reduction in human lung epithelial cells. J. Toxicol. Sci. 48, 75–86. https://doi.org/10.2131/jts.48.75 (2023).
Hu, Q. & Huang, T. Regulation of the cell cycle by NcRNAs affects the efficiency of CDK4/6 inhibition. Int. J. Mol. Sci. 24, 8939. https://doi.org/10.3390/ijms24108939 (2023).
He, S., Nakada, D. & Morrison, S. J. Mechanisms of stem cell self-renewal. Annu. Rev. Cell. Dev. Biol. 25, 377–406. https://doi.org/10.1146/annurev.cellbio.042308.113248 (2009).
Lou, Q. et al. Suppression of NLRP3/Caspase-1/GSDMD mediated corneal epithelium pyroptosis using melatonin-loaded liposomes to inhibit benzalkonium chloride-induced dry eye disease. Int. J. Nanomed. 18, 2447–2463. https://doi.org/10.2147/IJN.S403337 (2023).
Zhuang, D., Misra, S. L., Mugisho, O. O., Rupenthal, I. D. & Craig, J. P. NLRP3 inflammasome as a potential therapeutic target in dry eye disease. Int. J. Mol. Sci. 24, 10866. https://doi.org/10.3390/ijms241310866 (2023).
Schultheiß, C. et al. The IL-1β, IL-6, and TNF cytokine triad is associated with post-acute sequelae of COVID-19. Cell. Rep. Med. 3, 100663. https://doi.org/10.1016/j.xcrm.2022.100663 (2022).
Yu, Y. et al. Cytokines interleukin 4 (IL-4) and Interleukin 10 (IL-10) gene polymorphisms as potential host susceptibility factors in virus-induced encephalitis. Med. Sci. Monit. 23, 4541–4548. https://doi.org/10.12659/MSM.904364 (2017).
Oh, K. et al. IL-1β induces IL-6 production and increases invasiveness and estrogen-independent growth in a TG2-dependent manner in human breast cancer cells. BMC Cancer. 16, 724. https://doi.org/10.1186/s12885-016-2746-7 (2016).
Epstein, S. P., Chen, D. & Asbell, P. A. Evaluation of biomarkers of inflammation in response to benzalkonium chloride on corneal and conjunctival epithelial cells. J. Ocul Pharmacol. Ther. 25, 415–424. https://doi.org/10.1089/jop.2008.0140 (2009).
Zhang, R. et al. Dose-dependent benzalkonium chloride toxicity imparts ocular surface epithelial changes with features of dry eye disease. Ocul Surf. 18, 158–169. https://doi.org/10.1016/j.jtos.2019.11.006 (2020).
Acknowledgements
We would like to thank dr hab. Dariusz Dobrowolski from the Clinical Department of Ophthalmology of the District Railway Hospital in Katowice for collecting tissue samples and general cooperation. This work was supported by institutional grands (SUM Katowice) no.: BNW-2-035/N/3/0, and in part by National Science Center grant no.: 2022/06/X/NZ3/00715.
Author information
Authors and Affiliations
Contributions
A.P., B.S., A.S-S., P.C.: conceptualization, methodology; A.P., B.S., A.S-S.: investigation; A.P., B.S.: data curation, writing – original draft; A.P., B.S. and P.C.: resources, funding acquisition; A.P., B.S., A.S-S., P.C.: writing – review & editing; P.C.: supervision. All authors have read and agreed to the published version of the manuscript.
Corresponding author
Ethics declarations
Ethics approval
The study involving human cells was approved by the Bioethics Committee of the Medical University of Silesia (SUM) in Katowice (decisions no. PCN/CBN/0052/KB/26a/22 and PCN/CBN/0052/KB/26b/22).
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Prusek, A., Sikora, B., Skubis-Sikora, A. et al. Assessment of the toxic effect of benzalkonium chloride on human limbal stem cells. Sci Rep 15, 12295 (2025). https://doi.org/10.1038/s41598-025-96919-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-025-96919-2
