
Abstract
Settleable particulate matter (SPM) samples are characterised by having very similar qualitative compositions. Therefore, there is a need for a sensitive analysis method that can provide valid quantitative information when facing new research challenges. Establishing a valid and repeatable quantitative mineralogical analysis method is essential in order to establish limits to the concentration of certain mineral phases to, in turn, protect human health and ecosystems. An analytical and sample preparation protocol that can achieve the homogenisation of the standard and test samples has been designed, as well as the repeatability of the results obtained, with the following achievements: improving the samples’ level of homogeneity and the sensitivity in the detection of minority crystalline phases, and decreasing the likelihood of overlap. This study shows the possibility of applying the matrix-flushing adiabatic method to both oriented and randomly distributed samples, as well as the possibility of applying it interchangeably when preparing samples with finite or infinite thickness.
Similar content being viewed by others
Introduction
Focusing on the study of settleable atmospheric particulate, different authors have conducted work aimed at characterising this type of atmospheric contaminant in the ceramic cluster of Castellón province (Spain) to establish its origin (natural or anthropogenic). This area is located in the Mediterranean coastal basin, which has major problems regarding high concentrations of particulate matter in the ambient air, and is also located next to an industrial centre focused on the treatment of ceramic raw mineral materials such as ceramic red clays and silicates. It is a strategic zone in the framework of EU pollution control together with Modena (Italy). Based on some studies and on the data shown online by the regional authorities derived from the air quality networks in these areas, it can be deduced that metals (and other chemical components) and the particulate matter are the two parameters of most concern regarding EU legal requirements. This can be seen in the literature1,2,3. Other studies, such as those conducted by several authors based on episodes of ‘red rain’4,5, have made it possible to establish, depending on the smectite and kaolinite content, the specific geographical origin of the ‘Saharan dust’. These authors also assign a clear cross-border origin to mineral phases such as palygorskite, specifically in North Africa, whereas chlorite is typical of local sources4. Correlations between mineral species and certain ranges of particle sizes have been established by many authors6,7,8,9. This particulate material includes settleable particles larger than 32 μm that remain airborne for relatively short periods of time and can then settle. Thus, the effects are most pronounced in the vicinity of the emitting sources. Pardo et al.10 indicated that the concentration levels of particulate sediments are within the allowable limits. Only twice were these values exceeded because of high precipitation and particulate intrusion from North Africa. The main mineral phases in the settleable particulate matter (SPM) were quartz, illite and kaolinite, resulting from the milling and grinding of raw materials by the ceramic industry, mining, agriculture and other human activities11,12.
This line of work, as in many fields of research, requires analysing a large number of samples to be able to establish links between their composition and other measurable variables13. SPM samples are characterised by having very similar qualitative compositions. This creates the need for a sensitive analysis (SA) method that, quickly, accurately and with the least amount of sample possible (a limiting factor in the sampling time invested), can provide valid quantitative information when tackling new research challenges14,15. This can include, for example, studying (for a given range of particle sizes) the day-to-day evolution of the concentration levels of a specific mineral phase and its variation depending on the direction and strength of the breeze, among other factors. Establishing a valid and repeatable quantitative method of mineralogical analysis is essential to be able to establish limits to the concentration of specific mineral phases to, in turn, protect human health and ecosystems16,17,18,19. Establishing legal limits to the concentration of particles is insufficient in an advanced and committed society.
The prior development of a methodology to extract several groups of particles of different sizes from each sample and the selection of a diffraction surface consisting of a round, flat glass with 47 mm of diameter and 5 mm of thickness9,15,16 as an alternative to the permeable membrane (filters) used in previous studies1,2,17, have made it possible to perform XRD analyses capable of revealing a mineralogical composition in which the following crystalline phases can be clearly distinguished: for the part rich in large-sized particles, low amounts of illite/muscovite (K(Al, Mg)3SiAl10(OH)), kaolinite (Al2Si2O5(OH)4), plagioclase ((NaCa)(AlSi)4O8), potassium feldspar (KAlO3SiO2) and hematite, and higher proportions of quartz (SiO2), calcite (CaCO3) and dolomite (CaMg(CO3)2). For the part rich in smaller-sized particles, clayey minerals such as illite/muscovite, kaolinite and, in rare cases, chlorite ((Si2-xAlx)O5(Mg3-xAlx)(OH)4), as well as quartz, hematite and, to a much lesser extent, calcite and dolomite. It is worth noting that not detecting phases such as halite (NaCl), gypsum (CaSO4·2H2O) or the ammonium sulphates mohrite and mascagnite, previously identified by Boix1 by using high- and medium-volume samplers or cascade impactors, reveals the inability of the settleable material sampling equipment and the sample preparation technique to measure the soluble crystalline compounds.
Factors such as the use of constants listed in the literature9,20,21,22,23, which are only suitable for formulations of polycrystalline samples with no preferential orientation, and/or the poor resolution of diffraction diagrams, add a high level of uncertainty when calculating the percentages of mineral phases, the variability of chemical and mineralogical profiles24,25 or high levels of heterogeneity26,27,28. Calculated using the adiabatic approach of the matrix-flushing method29,30, the percentages considered must not be interpreted as absolute values, but as mere indicators of the existence of significant differences between locations.
Without an advanced methodology validated by the scientific community, it is not feasible to carry out serious studies on air quality in terms of the characterisation of atmospheric mineral particulate matter and its quantification29,30,31. This adds to the uncertainty produced by the quantification of particles of natural and anthropogenic origin, the influence of the composition of bulk precipitation32 or the contribution of Saharan dust deposition33.
This and many other reasons (particle size distribution, compositional homogeneity, etc.) make it necessary to conduct a new study capable of optimising as much as possible, and in the least amount of time, the resolution of the diffraction diagrams of SPM15. In a parallel and complementary way to achieving this objective, it is also necessary to develop a quantification technique that makes it possible to obtain much more realistic results that are consistent with the percentages of mineral phases present in the material analysed24,25.
Materials and methods
Sampling
In order to obtain a group of representative samples, it has been necessary to establish a small network formed by seven British standard type captors. The strategic placement of these devices in towns such as Castellon (an important urban nucleus), Grao of Castellon (an industrial and harbour area), l’Alcora and Onda (two important industrial areas) has enabled the collection of the SPM after a simultaneous exposure for approximately 30 days15,16. This collection process is carried out by washing the particles deposited on the surface of the collecting funnel, using distilled water.
Particle size analysis using laser diffraction
Laser diffraction measures particle behaviour by how they scatter light. This phenomenon, which plays out in all spatial directions, has an intensity pattern that depends on the particles’ size (smaller ones scatter light at large angles, whereas larger ones scatter light at much smaller angles). The size distribution (Vi) is linked to the data obtained (Dj) using the following Eq. (1):
where ‘i’ are the band size ratios, ‘j’ expresses the element detector and ‘Uij’ describes how the particles in the ‘i’ band sizes scatter light towards the detector, ‘j’. Thus, ‘Vi’ is the relative volume of the material within band size ‘i’. It is worth noting that the new ISO 13320 standard establishes the distribution of Mie-theory, and not Fraunhofer’s approach, as advisable to measure particles smaller than 50 µm with laser diffraction equipment. The size distribution determinations were performed by laser dispersion using a Malvern Mastersizer 200E (100 nm to 1000 µm range). This analytical equipment makes it possible to calculate the average particle size, among other calculations of interest15.
The standard materials used in this study were the following: for SiO2, a fine, washed and calcined quartz of manufacturer Merck. For Al2O3, a standard corundum (X-Ray Diffraction Standard 676) from the National Institute of Standards and Technology (USA). We also used an γ-alumina from Merck, which was then calcined in a laboratory. For CaCO3, a precipitated calcium carbonate of manufacturer Normapur. For CaMg(CO3)2, a commercial dolomite supplied by manufacturer Lladró. For Al2Si2O5(OH)4, a commercial kaolinite supplied by kaolin manufacturer CAOBAR. For K(Al, Mg)3SiAl10(OH), a muscovite supplied by GEONATURA.
Managing the compositional homogeneity of the calibration mixtures
A homogenisation method with which to ensure the greatest reliability and repeatability of the experimental results is needed. Each of the pure phases that composed the calibration mixtures were weighed with the highest accuracy (up to the fourth decimal number) on the same watch glass (48 mmφ), using a Sartorius electronic scale. This scale-desiccant cabinet configuration allows us, to the extent possible, to avoid weight variations due to air currents (this structure provides optimal sealing for the measuring equipment) and/or changes to the relative humidity because, inside the cabinet, we set a (more or less constant) percentage of around to 35–40%. After the previous operation, and always on the same watch glass, we added a very small amount of hematite to the end material that comprised the calibrating sample. This gave an initially white powder a light pinkish colour, which made it possible to (visually) establish the level of homogeneity achieved by the mixture. After completing the first dry homogenisation, adding a minimal amount of distilled water (approximately three drops) allowed us to, with the help of a fine glass rod, scatter the particles, thus favouring the thorough mixing of the components. Once the suspension was dry, the colour of the solid material adhered to the watch glass was more or less uniformly pink, showing some anomalously clear (white) areas that were indicative of heterogeneity. This process had to be repeated. To do so, following a second dry homogenisation, we again added a small amount of distilled water and then scattered the particles with the same rod as before. Once the suspension was dry, the pink colour of the solid adhered was evenly distributed, therefore completing the mixture of the different phases composing the calibration sample. As a visual examination may not be fully convincing, we had to apply an analytical technique in order to confirm the material’s homogeneity.
Analysis of the level of homogeneity with scanning electron microscopy (SEM). Mapping the elemental distribution
Electron microscopy is one of the most decisive physical-chemical techniques in the study of materials’ microstructure. Scanning electron microscopy (SEM) is based on reconstructing images based on the sequential detection of electrons resulting from the interaction between an electron beam and the matter (the sample). The scanning electron microscopy coupled with energy dispersive X-ray (SEM-EDX) analyses were performed with a Zeiss Schottky SEM. The accelerating voltage was 20 kV. The beam current was adjusted to 2 nA to obtain the maximum signal without saturating the detector. The choice of parameters and the possibility of using lower-efficiency detectors was explored with a Quanta 200 tungsten filament SEM equipped with a 30 mm2 EDS detector (Brucker XFlash 4030). The acquisition parameters used were: a resolution of 1024 × 768, a beam current of 2 nA and an acquisition time of ~ 1 h. Certified standards were used for all elements of interest: calcium silicate (CaSiO3) for silicon (Si) and calcium (Ca), magnesia (Al2O3) for aluminium (Al), silica (SiO2) for silicon (Si) and magnesium oxide (MgO) for magnesium (Mg) (standards from Micro-Analysis Consultants Ltd).
Electron backscatter diffraction (EBSD) is a relevant tool, because it shows material patterns of a significant size due to either poorly milled particles and/or lumps composed of groups of small particles. The use of X-ray microanalysis enables the creation of elemental distribution maps (mapping) corresponding to specific areas of the sample’s surface, which makes it possible to observe the distribution of certain elements on it. Therefore, from a sample composed of a mixture of phyllosilicates (illite/muscovite and kaolinite) and quartz, carbonates (calcite and dolomite) and corundum, the following chemical elements were selected as representative of each of these phases: Ca as characteristic of carbonates, Mg for dolomite (although it can also be present in illite-muscovite), Al for corundum (also present in the structure of phyllosilicates) and Si for quartz (also present in phyllosilicates). At this point, it became necessary to check for complete spatial randomness (CSR) in the mappings to verify (or discard) the interpretations27. Due to the significant amount of computational time required for calculating the K (r) function (L(r)-r), only a detailed study of Si and Ca was conducted.
Calibration sample Preparation and application of the matrix-flushing method
Considering that samples that foster the formation of oriented aggregates are the most suitable for applying a semi-quantitative analysis technique, the goal was to describe a methodology aimed at determining their most appropriate preparation (calibration and test samples). To do so, we tested two types of preparations:
Experiment 1: A 48 mm round, flat glass sample holder was obtained and a small depression was made in its centre. The material was placed on said depression and, with the help of a small spatula, uniformly distributed on it. The part of the sample that overflowed when exerting pressure with a sheet of glass was forced to become a flat surface on the same level as the rest of the sample carrier. In this specific case, we can consider that the sample had, for the X-rays, infinite thickness.
Experiment 2: A 48 mm round, flat glass sample holder was used. The material was placed directly on the smooth surface of the sample carrier. Adding a minimal amount of distilled water (around three drops), and with the help of a fine glass rod, we then carefully spread the sample until it was evenly distributed around the area of influence of the X-rays. It was then dried and ready for its analysis using X-ray diffraction. In this specific case, we can consider that the sample had, for the X-rays, finite thickness.
To verify the validity of this method on oriented samples and ascertain the most appropriate method of preparation, with and without finite thickness, we prepared 16 calibration samples (eight for each experiment). The working conditions were: incident radiation—Cu, Kα (1.54052 Å); operation voltage and amperage—40 kV and 30 mA; step size—0.05°; time per step—2.5 s; Mode—continuous recording. We obtained a normal diagram from 4° to 65° of 2θ, using the following combination of apertures: incidence (2 mm); divergence (2 mm); anti-scattering (0.2 mm) and for the detector (0.6 mm). The diffractometer was handled at all times through a Pentium 100 PC, using the SIEMENS DIFFRAC plus v software 2.0. After obtaining the diffractograms for each sample, we removed the background and, using the EVA assessment program of the company Socabim and the JCPDS files, identified the mineral phases present. We also determined the RA, average intensity values measured in cps·dg, for each of the previously selected diffraction peaks. The instrumental operation conditions were the same in both experiments. However, the method of preparation and the type of corundum used were different (calcined Merck corundum as the standard phase in Experiment 1 and standard corundum as the standard phase in Experiment 2).
The ambiguity and misleading concepts on the X-ray detection limit in the current literature raise the urgent need to establish a method that is accurate and easy to use. Detection limit is defined as the concentration equivalent to three times the statistical fluctuation of the background intensity. The background intensity is calculated from the standard samples.
Results and discussion
The average particle diameter values (Table 1) make it possible to deduce that the pure quartz, calcite, dolomite and kaolinite phases have optimal attributes, with a D[4,3] of around 10 μm. Regarding the corundum, it showed a normal particle size distribution with an average particle diameter of around 3 μm to 4 μm. As it has already been milled when supplied, its normal distribution curve cannot be rectified. Regarding the illite-muscovite phase, it was impossible to improve the milling conditions to decrease its size given the layered structure of the standard material used, which prevents the effective milling of this phase. Therefore, we achieved sufficiently homogeneous particle sizes to perform calibration mixtures despite having a D[4,3] greater than the range proposed in the bibliography (1 μm to 5 μm).
Figure 1 shows the X-ray diffraction patterns, for different particle sizes, of two samples collected in Onda and l’Alcora (Spanish ceramic cluster, NE Spain). The patterns of significant size were formed either by poorly milled particles (it is impossible to ensure that the material is fully milled) and/or lumps composed of groups of small particles. The homogenisation technique developed can favour the formation of these groupings (Fig. 2).

Diffractograms made on a flat glass support: (a) particulate matter collected at the Mestre Caballero school (Onda, NE Spain); (b) particulate matter collected at town hall of L’Alcora (NE Spain). FG or G—Enriched fraction in large-size particles, FM or M—Enriched fraction in intermediate-sized particle and FF or F—Enriched fraction in small-size particles.

Micrograph of one of the mixtures (M-3). Image obtained via EBSD.
Figure 3 shows the mapping of the elemental distribution of Al, Ca, Mg and Si, respectively. This form of mapping represents an alternative way of verifying the level of homogeneity achieved. Statistical techniques can be applied to this isolated process to detect randomness (homogeneity) and to find, where appropriate, lumps or local concentrations (heterogeneity) of the element that is to be analysed. In this sense, considering that a univariate spatial process (mapping a certain element) has a homogeneous nature when the function (L(r)-r), represented by the continuous line, is within the upper and lower confidence limits (discontinuous lines), and heterogeneous when outside the referred limits, in light of the previous graphical representations, it is possible to see (except for the image of the total Si) how the K (r) function reveals a pattern that is indicative of a fairly homogeneous distribution for both elements (Fig. 4).

Mapping of the elemental distribution of Al, Ca, Mg and Si. Image obtained from the EDX analysis of sample M-3.

Graphic results of applying the CSR test to the mappings of the elemental distribution. (a) Ca mapping and (b) Si mapping.
The results largely depend on the graph analysed. Furthermore, analysing the graph of the total L(r)-r is not the same as analysing just a part of this function (1st to 4th quadrants) (Fig. 4). This is where there could be a debate on what part of the image (or its entirety) should be considered representative of the homogeneity of the elements. This type of spatial analysis is a complete and sophisticated (on a theoretical level) analysis and, although the technique was developed several years ago, the use described of it is novel. This proposal can be considered a valid alternative for assessing the compositional homogeneity of a polycrystalline sample. The information provided by the elemental distribution maps together with powerful mathematical analysis tools (spatial statistics in specific processes) undoubtedly require thorough processing with further and more detailed analyses.
Tables 2 and 3 show the most relevant analytical data regarding the X-ray diffraction analysis of Experiment 1 and Experiment 2.
The least-squared linear adjustment applied to the experimental samples was not exactly a straight line that crossed the coordinate origin. However, the experimental validity of the adiabatic principle that can be established between pairs of components28,29 has been proven, as the slope does not change noticeably when the percentage and/or number of pure phases that compose the samples vary (Eq. 2 and Eq. 3):
where ‘Ic’ is corundum intensity, ‘Ii’ is the mineral phase mass intensity, ‘Xc’ is corundum mass fraction and ‘Xi’ represents the phase mass fraction parameters.
The matrix-flushing theory offers a simple intensity-concentration equation without a matrix. The adiabatic principle establishes that the intensity–concentration relationship between each and every pair of components in a multicomponent system is not disrupted by the presence or absence of other components. A key equation is derived, which conducts the decoding process. Both the matrix-flushing theory and the adiabatic principle are experimentally verified.
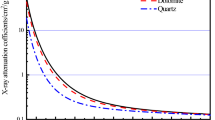
The Rius method34 or the Rietveld method35 require the knowledge of a series of starting parameters, apart from specific computer programs (Menge for the former and DBW for the latter). One of these parameters is the mass absorption coefficients of certain phases, from the mass absorption coefficients of their constitutive element (a precise knowledge of the compound’s formula is required). In the Rietveld case, complicated strategies for refining all of the diffraction spectrum intensity data are necessary34,36. The application of the ALJOR method1 is proposed, which is based on the Matrix–Flushing method, proposed by Chung28,29, but includes important modifications. The advantages of this method are that it can be applied to samples that have non-identified compounds, used to analyse just the phases that share a common interest, and help with the determination of amorphous materials. There are a series of problems when comparing the intensities of a peak of the phase with a peak of a standard added to the sample in a known proportion, such as finding patterns whose reflections do not overlap other phases present in the sample and, once the pattern is added, the possible disablement of the starting sample. Corundum (α-Al2O3) has been selected when performing this research as an ideal standard because of its purity, stability and easy availability, as shown by the Powder Diffraction File. One of the main aspects to deal with (as stated above) before starting the construction of calibration curves, is the selection of reflection planes that are free (or practically free) of overlaps because, if not, the results of the experiment would not be valid.
Regardless of the type of preparation, the experimental points show an acceptable level of linearity (Table 4), as in all cases the regression coefficient (b) values are much lower than 1, almost close to 0. Furthermore, the addition or non-addition of point (0,0) in the adjustment leaves the slope (m) value calculated for each phase practically unchanged. Relative to the determination coefficient (r2), the results are very good and in line with expectations.
The main difference between both experiments (1 and 2) is the noticeable discrepancy between ‘m’ values. Although it is not especially significant in the specific case of quartz, it is abundantly clear for phyllosilicates and especially for calcite and dolomite. The increased crystallinity of standard corundum compared to the calcined Merck corundum entails that, for identical pure phases and regardless of the type of preparation, the Icor/Iphase ratio must be higher in Experiment 2 than in Experiment 1. Therefore, this phenomenon is not due to the preparation conditions, but simply to the mineralogical properties of the standard used in each case.
After proving the impact of the corundum’s nature on the variation of the slope value, we considered it appropriate to disregard the linear adjustments made after using the Merck corundum (Experiment 1). As it is crystallised in a laboratory following a process of calcination, once the standard sample is used up, it is impossible to ensure the production of a new one with identical properties. This makes it necessary to create other calibration curves. Regular corundum maintains its properties unaltered, thus removing this major drawback. Another benefit of the preparation used in Experiment 2 compared to the one in Experiment 1 is connected to the favourable nature of the intensity values obtained from the calibration mixtures. These values, determined based on the relation of the components’ mass, make it possible to generate, with a minimum number of ‘attempts’ (especially for carbonates), a series of valid experimental points to calculate the adjustment.
Lastly, the discrepancies observed between the slope values for the same phase, with and without the presence of phyllosilicates, can be due to a microstructural issue with the material, whose interpretation requires a much more in-depth analysis.
Conclusions
The methodology proposed has made it possible to learn which mineral phases are present in each group of particle sizes, optimise the techniques for preparing samples for XRD, increase the number of phases detected and consider SPM as a contaminant with a particle size much lower than, in some cases, 100 μm in diameter. The XRD analysis has revealed an increase of certain mineral phases depending on the heightened presence of a certain particle size and the tendency of the amorphous material to gather in the part with a high number of fine particles.
An analytical and sample-preparing protocol capable of achieving homogeneity in the standard test samples and the repeatability of the results obtained has been designed. The level of homogeneity of the samples is guaranteed after mapping the distribution of their elements (Si, Al, Ca and Mg) and is corroborated by applying image analysis combined with techniques of spatial statistics. The level of crystallinity of the standard phase (corundum) markedly affects the slope of the calibration lines. Preparing the samples with a preferential orientation of their particles has enabled a series of benefits when successfully tackling the quantitative analysis: greater capability of increasing sensitivity in the detection of minority crystalline phases and a decrease in the probability of overlap with values that correspond to other standard crystalline phases, including corundum as the internal standard.
The possibility of applying the adiabatic method to both oriented and randomly distributed samples, as well as the possibility of applying it interchangeably when preparing samples with finite or infinite thickness, has been confirmed. It is worth noting that not detecting phases such as the halite, gypsum or the ammonium sulphates mohrite and mascagnite, reveals the inability of the settleable material sampling equipment and the sample preparation technique to measure the soluble crystalline compounds. New analytical protocols are currently being worked on to solve this limitation.
Data availability
Data is provided within the manuscript.
References
Boix, A., Jordán, M. M., Sanfeliu, T. & Rincón, J. M. Analysis by SEM/EDX of air particles. In Proceedings of ICEM 13, Paris (1994).
Boix, A., Jordan, M. M., Querol, X. & Sanfeliu, T. Characterization of total suspended particles around a power station in an urban coastal area in Eastern Spain. Environ. Geol. 40, 891–896 (2001).
Sanfeliu, T., Jordán, M. M., Gómez, E. T. & Álvarez, C. Caracterización mineralógica Del particulado Atmosférico sedimentable de La provincia de Castellón. Boletín De La. Sociedad Española De Mineralogía. 22-A, 105–106 (1999).
Queralt, I., Domingo, F. & Sole-Benet, A. The influence of local sources on the mineral content of Buld deposition over an altitudinal gradient in the filabres range (SE Spain). J. Geophys. Res. 98, 16761–16768 (1993).
Ávila, A., Queralt, I. & Alarcón, M. Mineralogical composition of African dust delivered by red rains over Northeasters Spain. J. Geophys. Res. 102, 21997–21996 (1997).
Barrie, L. A. Aspects of atmospheric pollutant origin and deposition revealed by multielemental observations at a rural location in Eastern Canada. J. Geophys. Res. 93, 3773–3788 (1998).
Boix, A., Jordán, M. M., Sanfeliu, T. & Justo, A. Dust air pollution in a Mediterranean industrial area. In Proceedings of Air Pollution 94, Barcelona (1994).
-Álvarez, A., Mateu, J., Sanfeliu, T. & Jordán, M. M. Environmental pollution management. In 9th Euroanalysis XI European Conference on Analytical Chemistry (2000).
-Gómez, E. T., Sanfeliu, T., Jordan, M. M., Rius, J. & De La Fuente, C. Geochemical characteristics of particulate matter in the atmosphere surrounding a ceramic industrialized area. Environ. Geol. 45, 536–543 (2004).
Pardo, F. et al. Settlement of particulate matter in a ceramic cluster (NE, Spain) during the years 2000–2005. Appl. Clay Sci. 70, 45–50 (2012).
Sanfelix, V. et al. Geochemical characteristics of mineral air particles emitted inside a clay atomisation plan with introduce waste recycling processes. Environ. Geol. 47, 811–819 (2005).
Sánchez Bisquert, J. M., Peñas Castejón, J. M. & García Fernández, G. The impact of atmospheric dust deposition and trace elements levels on the villages surrounding the former mining areas in a semi-arid environment (SE Spain). Atmos. Environ. 152, 256–269 (2016).
Vicente, A. B., Jordán, M. M., Sanfeliu, T., Sánchez, A. & Esteban, M. D. Air pollution prediction models of particles, as, cd, Ni and Pb in a highly industrialized area in Castellon (NE, Spain). Environ. Earth Sci. 66, 879–888 (2012).
Battaglia, S. Quantitative X-ray diffraction analysis on thin samples deposited on cellulose membrane filters. X-Ray Spectrom. 14, 16–19 (1985).
Jordán, M. M., Álvarez, C. & Sanfeliu, T. A powder diffraction quantification method (ALJOR) for atmospheric particulate matter. Environ. Geol. 42, 810–816 (2002).
Jordán, M. M., Álvarez, C. & Sanfeliu, T. Spherical particles as tracers of atmospheric ceramic industry. Environ. Geol. 51, 447–453 (2012).
Mohapatra, K. & Biswal, S. K. Effect of particulate matter (PM) on plants, climate, ecosystem and human health. Int. J. Adv. Technol. Eng. Sci. 4, 118–12902 (2014).
Machado, M. et al. A new methodology to derive settleable particulate matter guidelines to assist policy-markers on reducing public nuisance. Atmos. Environ. 182, 242–251 (2018).
Negral, L. et al. Settleable matter in a highly industrialized area: Chemistry and health risk assessment. Chemosphere 274, 129751 (2021).
Davis, B. L. Standarless X-ray diffraction quantitative analysis. Atmos. Environ. 14, 217–220 (1980).
Davis, B. L. A study of the errors in the X-ray analysis procedures for aerosols collected on filter media. Atmos. Environ. 15, 291–296 (1981).
Davis, B. L. Reference intensity quantitative analysis using thin-layer aerosol samples. Adv. X-ray Anal. 27, 339–348 (1984).
Davis, B. L. & Spilde, M. N. Quantitative X-ray powder diffraction analysis applied to transmission diffraction. J. Appl. Cryst. 23, 315–320 (1990).
Davis, B. L., Smith, D. K. & Holomany, M. A. Tables of experimental reference intensity ratios in X-ray quantitative analysis. Powder Diffract. 7, 186–193 (1989).
Manousakas, E. et al. Quantitative assessment of the variability in chemical profiles from source apportionment analysis of PM10 and PM2.5 at different sites within a large metropolitan area. Environ. Res. 192, 110257 (2021).
Martins, E. H., Nogarotto, D. C., Mortatti, J. & Pozza, S. A. Chemical composition of rain water in an urban area of the Southeast of Brazil. Atmos. Pollut Res. 10 (2), 520–530 (2019).
Mateu, J. Second-order characteristics of Spatial market processes with applications. J. Nonlinear Anal. 1, 145–162 (2000).
-Mateu, J., Usó, J. L. & Montes, F. The Spatial pattern of a forest ecosystem. Ecol. Model. 108, 163–174 (1998).
Chung, F. H. Quantitative interpretation of X-ray diffraction patterns of mixtures I. Matrix-flushing method for quantitative multicomponent analysis. J. Appl. Cryst. 7, 519–525 (1974). (1974).
Chung, F. H. Quantitative interpretation of X-ray diffraction patterns of mixtures II. Adiabatic principle of X-ray diffraction analysis of mixtures. J. Appl. Cryst. 7, 526–531 (1974). (1994).
Kleanthous, S., Bari, M. A., Baumbach, G. & Sarachage-Ruiz, L. Influence of particulate matter on the air quality situation in a mediterranean Island. Atmos. Environ. 43, 4745–4753 (2009).
Nickovic, S., Vukovic, A., Vujadinovic, M., Djurdjevic, V. & Pejanovic, G. High-resolution mineralogical database of dust-productive soils for atmospheric dust modeling. Atmos. Chem. Phys. 12, 845–855 (2012).
Klein, H. et al. Saharan dust and ice nuclei over central Europe. Atmos. Chem. Phys. 10, 10211–10221 (2010).
- Paternoster, M. et al. Natural versus anthropogenic influences on the chemical composition of bulk precipitation in the Southern apennines, Italy: A case study of the town of Potenza. J. Geochem. Explor. 145, 242–249 (2014).
Akinbodunse, S. J., Ufer, K., Dohrmann, R. & Mikutta, C. Evaluation of the Rietveld method for determining content and chemical composition of inorganic X-ray amorphous materials in soils. Am. Mineral. 109 (12), 2037–2051 (2024).
Rius, J. A standardless X-ray diffraction method for the quantitative analysis of multiphase mixtures. J. Appl. Cryst., 20, 457–460 (1987). (1987).
Author information
Authors and Affiliations
Contributions
C. A. and M.M.J. Design and Conceptualization. C. A. and F.P. Investigation. M.M.J. Writing—original draft preparation. M.M.J. and F.P. Writing—review and editing the manuscript. M.M.J. supervision. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Consent to participate
All the authors participated in the preparation of the manuscript.
Consent to publish
All authors have read and approved the final manuscript to be published in the Journal.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Jordán, M.M., Pardo, F. & Álvarez, C. Quantitative determination of the mineral content of settleable particulate matter samples. Sci Rep 15, 14369 (2025). https://doi.org/10.1038/s41598-025-97135-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-025-97135-8
