
Abstract
Apical periodontitis (AP) is a prevalent immunoinflammatory disease affecting adults worldwide, this disease is also often co-occurring with high exposure to cigarette smoke. While the harmful effects of secondhand smoke (ShS) are well-documented, its interaction with AP and systemic health implications remain underexplored. This study investigated the combined effects of ShS and AP on disease progression and lung health in a rat model. Twenty-eight female Wistar rats were assigned to four groups: control (no ShS, no AP), control-AP (AP without ShS), ShS (ShS without AP), and ShS-AP (ShS with AP). ShS exposure involved daily inhalation of smoke from up to four cigarettes for 10 weeks, with AP induced via pulp exposure in the lower first molar. Post-euthanasia, jaws and lung tissues were analyzed. Micro-computed tomography confirmed ShS exposure significantly increased the volume and area of apical lesions. Oxidative stress levels in the lung tissue were highest in the ShS-AP group, along with increased total oxidant activity and reduced antioxidant enzyme activity. AP and ShS together were associated with pronounced alveolar destruction and chronic airway remodeling in the lungs. These findings suggest a synergistic interaction between AP and ShS, exacerbating both local and systemic effects. This underscores the critical need to address the interplay between oral and systemic health, particularly in the context of environmental exposures like ShS.
Similar content being viewed by others


Introduction
The harmful effects of smoking on overall health are well-documented, including negative impacts on bone metabolism1,2. These effects can impair the repair capacity of apical periodontitis (AP)3,4,5, a common oral inflammatory condition, by increasing apical bone damage and elongating healing time6,7. Smoking reduces blood supply and limits the distribution of nutrients and oxygen, triggering an inflammatory response that exacerbates tissue damage8. Recent systematic reviews3,9 highlight a higher prevalence of apical lesions in smokers compared to non-smokers, emphasizing the role of smoking as a significant risk factor for AP progression. Despite the well-known effects of smoking, the specific impact of secondhand smoke (ShS) on oral diseases remains unexplored. Research has predominantly focused on periodontitis10,11,12,13,14 and periimplantitis15,16 in animals and humans. Emerging evidence, however, demonstrates that ShS inhalation triggers inflammatory responses in AP, which is characterized by elevated cytokine levels and increased resorptive activity7,8,17,18.
Recently, systemic impacts of AP have also been investigated, in respect to potential repercussions on organs such as the kidneys19, heart20, and liver21. These systems can be worsened by nicotine18, a major component of cigarette smoke, which can trigger pro-inflammatory cytokine release22. Tobacco smoke exposure also leads to an increase in the recruitment of neutrophils and macrophages in lung tissue23 and triggers airway remodeling and emphysema24, the primary target of ShS. Additionally, cigarette smoke alters the redox balance in lung tissue by upregulating oxidative components and suppressing antioxidant defenses, leading to oxidative stress (OS)25. OS is also a hallmark of AP, which contributies to osteoclast activation and cytokine secretion26. Evidence suggests that OS induced by AP may be associated with systemic diseases27,28. This raises the possibility of a cumulative effect when combined with ShS exposure. However, no studies have investigated the relationship between AP and ShS in the context of lung damage, thus creating a critical gap in understanding their combined systemic effects.
Given the well-established association between ShS and lung injury, this study addresses the emerging evidence linking AP to oxidative stress and inflammatory modulation to a possible additive impairing effect on lung tissue. This study aimed to assess the combined effect of ShS and AP on the development of AP in rats exposed to ShS using micro-computed tomography (micro-CT) analysis. Additionally, it aimed to evaluate lung tissue damage and airway remodeling through histopathological and OS analyses. Understanding this association may provide insights into the systemic consequences of AP in the context of environmental exposures like ShS, highlighting the need for integrated approaches to oral and systemic health.
Results
Clinical observations
No animals were lost during the experimental period. Animals in the ShS-AP group weighed significantly less than those in the control group (p < 0.05). The weight gain curve showed a slight reduction after 4 weeks for the ShS and ShS-AP groups, with a significant difference for the ShS-AP group (p < 0.05). Animals with induced AP but without ShS exposure had a higher daily solid intake (p < 0.05). In contrast, those exposed to ShS and ShS-AP had lower solid intake compared to both control groups (p < 0.05). Daily liquid intake decreased in the groups exposed to ShS (p < 0.05). Body weight and liquid/solid consumption data are presented in Fig. 1 and Table 1.

Graphic representation of variations of weight per group through weeks. (a) Comparison of animal weight between groups; (b) Curve of weight gain. Asterisks indicate significant differences between intervention groups compared to control (p < 0.05); ns indicate non-significant differences between groups. “○, □, ◇, ▽”: Individual weight of rats across groups. “-” of vertical lines: median. Top of vertical lines: Maximum value. Bottom of vertical lines: minimum value.
Micro-CT assessment
Results related to the volume and area of apical lesions are summarized in Table 2 and Fig. 2. The group exposed to ShS-AP had more extensive lesions in volume and area than the control-AP group (p < 0.05). No differences were found between the control and ShS groups (p > 0.05).

Representation of periodontal ligament or apical lesions, including (a) sagittal, (b) axial, and (c) 3D reconstruction views.
Lung oxidative stress markers
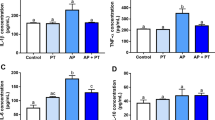
The levels of total oxidant status (TOS) significantly increased in the control-AP, ShS, and ShS-AP groups compared to the control group (p < 0.01). Conversely, total antioxidant status (TAS) levels significantly decreased in the ShS-AP group compared to the other experimental groups (p < 0.01). The oxidative stress index (OSI) levels significantly increased in the ShS-AP group (p < 0.01). The OS results are summarized in (Fig. 3).

The level of OS in the lungs of rats, both exposed and unexposed to ShS, with or without induced AP. (a) TOS levels, (b) TAS levels, and (c) OSI levels.
Lung morphology
In the lung parenchyma, a significant cumulative increase in mean linear intercept (MLI) was observed (p < 0.05). In contrast, an opposite trend was noted for the mean transsectional wall length (LMW) and the volume density of alveolar septa (VVSEP), both of which showed a significant decrease and were inversely proportional to MLI. However, ShS and ShS-AP groups did not differ from each other in these parameters (p > 0.05). Bronchial analysis showed an enlarged bronchial wall thickness (BWT) area in animals exposed to ShS and ShS-AP compared to control groups (p < 0.05), but no difference was observed between the ShS groups (p > 0.05). The histopathological results are summarized in (Fig. 4).


(a) Representative images of significant airspace enlargement and morphological destruction in rat lung tissue after inhalation of cigarette smoke, with or without apical periodontitis (HE staining, magnification ×200). These were assessed using histopathologic assessment, including (b) mean linear intercept values, (c) mean transsectional wall length, (d) volume density of alveolar septa, and (e) bronchial wall thickness. Data are presented as representative images or expressed as mean ± standard deviation. *p ≤ 0.05, **p ≤ 0.005, ***p ≤ 0.0005, ****p ≤ 0.0001.
Discussion
This study reports that AP associated with ShS exposition led to a greater apical lesion and increased pulmonary damage in rats. Therefore, the null hypothesis was rejected. Wistar rat models have been used in the past to explore the link between AP and nicotine7,8,17,18. Previous studies employing a nicotine injection model revealed that animals exposed to nicotine exhibited larger apical lesions compared to control groups, as assessed by micro-CT5,18. However, this injection model does not effectively simulate smoke exposure because it does not encompass all the toxic components of cigarettes8.
It was previously reported that animal models exposed to 4 cigarettes per day have cotinine levels comparable to those of humans exposed to ShS29. Recent studies associating ShS and AP used a regimen of 20 cigarettes daily for 50 days7,8,17, which may overestimate a ShS model. To better simulate a passive smoker model, we reduced the number of cigarettes used per day. Despite the well-known dose-dependent nature of cigarette harm on bone tissue1, our study showed significant apical bone destruction even with lower smoke exposure than the current literature7,8,17. The lack of significant differences in periodontal ligament parameters between the control and ShS groups can be attributed to the absence of localized inflammatory stimulus.
Concerning the pulmonary alterations, the histopathological analysis revealed that the combined effects of AP and ShS exacerbate a condition similar to emphysema. Imaging evaluation showed progressive dilation of airspaces distal to the terminal bronchioles and destruction of alveolar septa. Although emphysema diagnosis involves other clinical characteristics, our observations meet the criteria for emphysema30 and were identified in all of the groups with AP. Additionally, we found an imbalance between oxidative and antioxidative profiles of the ShS-AP group, evidenced by OSI in the lung tissue. Other parameters related to OS analysis have been previously used in AP models31,32,33. However, the OSI from TAS/TOS ratio is a highly reproducible tool associated with inflammation and bone remodeling in endodontics34.
The impairment of antioxidant defenses and excessive production of reactive oxygen species (ROS) that drive OS are key contributors to mitochondrial dysfunction and intracellular damage35. Cigarette smoke is well-known for inducing the production of ROS, which contributes to lung remodeling and impaired function36. Elevated ROS production also plays a critical role in inflammatory conditions and is implicated in bone remodeling within apical lesions37. Since both AP and ShS generate ROS by NADPH oxidase26,38, their synergistic effects may share similar pathways. Cytokines released in AP (e.g. tumor necrosis factor [TNF], interleukins [IL6, IL17], and interferon-gamma [IFNG])39 can circulate and sensitize lung tissue, amplifying ROS production. Simultaneously, ShS introduces free radicals and reactive nitrogen species directly into the lungs40, which interact with AP-induced marker alterations and potentially exacerbate oxidative damage and enhanced tissue remodeling.
The effect of AP in increased systemic OS has been described for other targets, such as the submandibular glands of rats41. Additionally, AP is also linked to molecular damage in cardiac tissue, by the impairment of antioxidant parameters such as superoxide anion radical, nitrite, hydrogen peroxide, and lipid peroxidation42. Regarding chronic exposure to cigarette smoke, especially under a chronic inflammation model as AP19,43, hematological parameter alterations in rats have been previously described8. Altered levels of red blood cells, hemoglobin, and neutrophils in the blood of these animals have been linked to a systemic immunomodulatory response8. However, few studies assess the real impact of the association between both AP and ShS on systemic health8,18. To the best of our knowledge, this is the first study to evaluate the synergistic effects of cigarette smoke and AP on lung tissue remodeling and oxidative stress.
The histopathological analysis of the lung in this study used a semi-automated tool to assess pulmonary emphysema, to ensure efficiency and accuracy44. This analysis allows manual selection of non-parenchymal areas and inflammatory exudates, which is commonly neglected in automatic methods44. This reduced bias and lead to more reliable results. Our results showed an increase in MLI, which is related to the average size of airspaces due to alveolar destruction and fusion45. In contrast, the decreased values of LMW and VVSEP indicate changes in the thickness and density of interalveolar septa, respectively. Previously, a similar pattern of MLI and LMW was found in animals exposed to ShS for 7 months30, although the authors did not assess AP. Additionally, a key finding in the current study is that AP alone can cause lung damage without ShS exposure. This was indicated by changes in MLI, LMW, and VVSEP. These results support the idea of a cumulative effect of AP and ShS on morphological changes identified in both the bone and periodontal ligament, and by extension, the lung tissue, which can be directly correlated with our results of OS.
The increased BWT in ShS-exposed groups is supported by other animal studies which also report airway tissue remodeling associated with BWT46,47. Although AP alone did not enhance BWT, this might be because only one tooth was AP-induced, which did not create a significant inflammatory load. It has been reported that a higher number of teeth exposed to AP is associated with greater systemic inflammatory damage48. Further studies should investigate the combined effects of AP and ShS on bronchial remodeling in respect to the number of teeth with AP and prolonged exposure durations. These investigations should also focus on immunoregulatory conditions, and the molecular pathways involved in cytokine signaling to clarify their roles.
The findings highlight the role of ShS in worsening AP progression in rats. Additionally, combined exposure to ShS and AP increased the oxidative stress index in lung tissue and promoted changes in lung tissue similar to emphysema with parenchymal destruction and airway remodeling. These results emphasize the detrimental impact of ShS on lung health, particularly when combined with AP.
Methods
Animals
All experimental procedures in animals were conducted under approval by the Ethics Committee for Animal Use at UNIUBE, Uberaba, Brazil (Protocol No 001/2021). The protocols were strict followed with the regulations of the national council for the control of animal experimentation (CONCEA), formulated by the Ministry of Science, Technology and Innovation of Brazil. The manuscript of this animal study has been written according to Animals in Research: Reporting In Vivo Experiments (ARRIVE) and Preferred Reporting Items for Animal Studies in Endodontology (PRIASE) guidelines49,50.
The sample size calculation was performed using SigmaPlot v. 12 software (Systat Software). One-way ANOVA was used with α error probability = 0.05, and power (1-β error probability) = 0.80. The size of specific effect for each variable was calculated from a previous study5. Seven animals per group were indicated. Twenty-eight 60-day-old Wistar female rats (Rattus Norvegicus), weighing ± 150 g were acquired from ANILAB (Paulínia, SP, Brazil). Animals were housed in polypropylene cages in a temperature-controlled environment (22–25 °C) under 12-h light and dark cycles with air humidity of approximately 55%, and with free access to food and water. Clinical monitoring was conducted daily to detect any signs of animal distress. The rats were weighed once a week throughout the experiment using a semi-analytical balance. To compare the weight gain curve, rats that were not exposed to ShS and without AP (control) were observed. To measure daily solid intake, the weight difference of the feed at the end of the day was compared to its weight after daily replenishment. The same method was used for liquid intake but with considering the volume. The animals were also cared for by facility staff under veterinary supervision.
Smoke inhalation model
The animals were randomly signed to four experimental groups (n = 7): (1) rats not exposed to ShS and without AP (control), (2) rats not exposed to ShS and with AP (control-AP), (3) exposed to ShS without AP (ShS), (4) exposed to ShS with AP (ShS-AP).
Exposure to cigarette smoke, aiming to replicate secondhand smoke (ShS), was done as described previously29. Briefly, a transparent acrylic box with four cylindrical inhalation chambers (7.0 cm in diameter, 23.0 cm in length, total volume of 885 cm³) was utilized. One end of each chamber allowed for animal access, while the opposite end was connected to a peristaltic pump. This pump drew in smoke and distributed it into the chambers. During exposure, animals were secured in place with a restrainer, and positioned with their noses directed towards the smoke. Marlboro cigarettes (Phillips Morris, Richmond, VA, US) were used as the smoke source, containing 0.8 mg nicotine, 10 mg tar, and 10 mg carbon monoxide per unit.
Before smoke exposure, animals acclimated to the chambers for one week without cigarette smoke. In the second week, smoke inhalation began with two cigarettes per day (morning and afternoon, at 6-hour intervals). In the third week, exposure increased to four cigarettes per day (two in the morning, two in the afternoon, at 6-hour intervals) until euthanasia (10 weeks of exposure, 266 cigarettes in total). Control animals were placed in smoke-free chambers to undergo the same handling stress.
Apical periodontitis induction
In the eighth week, AP was induced by exposing the pulps of the lower right first molars. Under anesthesia (intramuscular ketamine/xylazine at doses of 70 and 6 mg/kg, respectively), a round bur with a diameter of 0.1 mm (Dentsply Sirona, Ballaigues, Switzerland) operating at high speed with continuous irrigation was used to expose the pulp. Following pulp exposure, a 10 K-file (Dentsply Sirona) was introduced to verify access51, and the pulp chambers were left open to the oral environment for 4 weeks to allow AP to develop. At the end of the experiment, euthanasia was performed with an excessive dose of sodium thiopental administered intraperitoneally, followed by cervical dislocation for confirmation.
Sample collection and processing
The jaws from animals were collected post-euthanasia and stored in a 4% paraformaldehyde buffered solution at neutral pH for 48 h. After sectioning, they were stored in a phosphate-buffered saline (PBS) solution for micro-CT analysis. The right lungs of the animals were frozen in nitrogen for 24 h and stored in a -80 °C freezer for oxidative parameter analysis. The left lung was fixed in 4% paraformaldehyde for 48 h and embedded in paraffin for histopathological assessment.
Micro-CT assessment
The jaws were scanned using a SkyScan 1272 micro-CT device (Bruker, Kontich, Belgium) with an Al 1 mm filter, operating at 80 kV voltage, uA current, and a resolution of 9 μm. A 360-degree scan was performed. The datasets obtained from the scans were reconstructed using NRecon software (v.1.6.10.4, Bruker) with universal correction for ring artifacts set at 4. DataViewer software (v.1.5.2.4, Bruker) was used to obtain an axial view of datasets for periradicular assessment. Volumetric analysis was conducted in CTAn software (v.1.15.4, Bruker) based on the grey values of structures, as described previously5. The control and ShS groups without AP were also analyzed to confirm the development of AP and the preservation of the periodontal ligament, lamina dura, and alveolar bone. Volume (mm2) and area (mm2) values of apical lesions were obtained. For the three-dimensional representation, the apical lesion, dentin, and enamel structures were segmented in CTAn and then imported into Meshmixer software (v.3.5.464, Autodesk Inc., California, US).
Oxidative stress parameters
Lung tissue samples weighing 80 mg each were homogenized in a 1:9 ratio following the manufacturer’s instructions for TAS and TOS kits (Elabscience, Houston, Texas, US). The homogenate was centrifuged at 10.000 rpm for 10 min at 4 ºC. The supernatant was collected, and the total protein concentration was determined using the Pierce™ BCA Protein Assay Kit (Thermo Scientific, Waltham, Massachusetts, US) for TOS calculation. Optical density readings for TAS and TOS were taken using a microplate reader (Biochrom, Cambridge, UK). TAS results were reported in mmol Trolox Equiv./L, while TOS results were reported in µmol H2O2 Equiv./L. Additionally, the OSI was calculated as the ratio between TOS and TAS to provide a more precise indicator of OS through the following formula: OSI (arbitrary units) = [(TOS, µmolH2O2equiv./L)/(TAS, µmol Trolox equiv./L)×100]52.
Histomorphometric analyses
The fixed samples were embedded in paraffin. For each animal, at least three semi-serial sections were obtained (5 μm interval). Three different parameters were evaluated. For alveolar morphometry, four slides per lung were stained with hematoxylin and eosin (H&E). Stained sections were scanned with a slide scanner (Aperio ScanScope AT-Turbo, Leica Biosystems, Wetzlar, Germany). Quantitative alveolar morphometry was assessed with a self-designed semi-automated Fiji-plugin (ImageJ; http://fiji.sc/Fiji) on 10 randomly selected fields per lung44. Calculations were made through counting of MLI, LMW, and VVSEP45. BWT was also measured using ImageJ software and expressed as a ratio of the total airway area to lumen area, in mm²/µm. A standardization of 100–300 μm mean diameter was established for the selection of bronchial fields. Data for all histopathological evaluations were presented as the mean of all measured fields.
Statistical analysis
Data were collected and analyzed using GraphPad Prism 9 software (GraphPad Software, Massachusetts, US) after Shapiro-Wilk normality test. For parametric data, a one-way analysis of variance (ANOVA) was conducted with a significant level set at α = 0.05. Homogeneity of variances was verified using the Brown-Forsythe test, and corrections were applied using Bartlett’s test where necessary. Nonparametric data were analyzed using the Kruskal-Wallis test. Tukey post-hoc tests were employed for multiple comparisons. The null hypothesis was that chronic exposure to ShS neither affects the volume of the apical lesion nor the OS parameters or morphology in lung tissue.
Data availability
The data that support the findings of this study are available from the corresponding author upon request.
References
Gruber, M. D. et al. The effects of nicotine- and cigarette-related products on osteogenesis, bone formation, and bone mineralization: A systematic review. Neurosurgery 93, 247–256. https://doi.org/10.1227/neu.0000000000002412 (2023).
Xie, G. et al. Smoking and osteoimmunology: Understanding the interplay between bone metabolism and immune homeostasis. J. Orthop. Transl. 46, 33–45. https://doi.org/10.1016/j.jot.2024.04.003 (2024).
Aminoshariae, A., Kulild, J. & Gutmann, J. The association between smoking and periapical periodontitis: a systematic review. Clin. Oral Investig. 24, 533–545. https://doi.org/10.1007/s00784-019-03094-6 (2020).
Paljevic, E., Brekalo Prso, I., Hrstic, J. V. & Pezelj-Ribaric, S. Persic Bukmir, R. Impact of smoking on the healing of apical periodontitis after nonsurgical endodontic treatment. Eur. J. Dent. 18, 124–130. https://doi.org/10.1055/s-0043-1761451 (2024).
Pinto, K. P. et al. Effects of alcohol and nicotine consumption on the development of apical periodontitis in rats: a correlative micro-computed tomographic, histological and immunohistochemical study. Int. Endod J. 53, 1238–1252. https://doi.org/10.1111/iej.13344 (2020).
Lopez-Lopez, J. et al. Tobacco smoking and radiographic periapical status: a retrospective case-control study. J. Endod 38, 584–588. https://doi.org/10.1016/j.joen.2012.02.011 (2012).
Vasques, A. M. V. et al. Bone resorption in apical periodontitis enhanced by cigarette smoke inhalation: histometric, immunohistochemical, and microtomographic analysis in rats. J. Endod 50, 493–498. https://doi.org/10.1016/j.joen.2024.01.005 (2024).
Vasques, A. M. V. et al. Inflammatory profile of apical periodontitis exacerbated by cigarette smoke inhalation: histological and immunohistochemical analysis in rats. Int. Endod J. 56, 465–474. https://doi.org/10.1111/iej.13883 (2023).
Pinto, K. P. et al. Does tobacco smoking predispose to apical periodontitis and endodontic treatment need? A systematic review and meta-analysis. Int. Endod J. 53, 1068–1083. https://doi.org/10.1111/iej.13316 (2020).
Akinkugbe, A. A., Slade, G. D., Divaris, K. & Poole, C. Systematic review and meta-analysis of the association between exposure to environmental tobacco smoke and periodontitis endpoints among nonsmokers. Nicotine Tob. Res. 18, 2047–2056. https://doi.org/10.1093/ntr/ntw105 (2016).
Oliveira, L. M., Oliveira, M. D. M., Ardenghi, T. M. & Zanatta, F. B. Is secondhand smoke exposure associated with poor periodontal status in children and adolescents? A systematic review and meta-analysis. Eur. Arch. Paediatr. Dent. 23, 513–525. https://doi.org/10.1007/s40368-022-00709-7 (2022).
Pesce, P. et al. Evaluation of periodontal indices among non-smokers, tobacco, and e-cigarette smokers: a systematic review and network meta-analysis. Clin. Oral Investig. 26, 4701–4714. https://doi.org/10.1007/s00784-022-04531-9 (2022).
Sutton, J. D., Ranney, L. M., Wilder, R. S. & Sanders, A. E. Environmental tobacco smoke and periodontitis in U.S. non-smokers. J. Dent. Hyg. 86, 185–194 (2012).
Sutton, J. D., Martinez, S., & Gerkovich, M. M. Environmental tobacco smoke and periodontitis in United States non-smokers, 2009 to 2012. J. Periodontol. 88, 565–574. https://doi.org/10.1902/jop.2017.160725 (2017).
Correa, M. G. et al. Histometric evaluation of bone around titanium implants with different surface treatments in rats exposed to cigarette smoke inhalation. Clin. Oral Implants Res. 20, 588–593. https://doi.org/10.1111/j.1600-0501.2008.01695.x (2009).
Nociti, F. H. Jr., Cesar, N. J., Carvalho, M. D. & Sallum, E. A. Bone density around titanium implants May be influenced by intermittent cigarette smoke inhalation: a histometric study in rats. Int. J. Oral Maxillofac. Implants 17, 347–352 (2002).
da Silva, A. C. R. et al. Effects of cigarette smoke inhalation on the immune-inflammatory profile of experimental apical periodontitis in rats. Int. Endod J. 56, 1559–1570. https://doi.org/10.1111/iej.13981 (2023).
Pinto, K. P. et al. Chronic alcohol and nicotine consumption as catalyst for systemic inflammatory storm and bone destruction in apical periodontitis. Int. Endod J. 57, 178–194. https://doi.org/10.1111/iej.13994 (2024).
Lamba, J. et al. Association of apical periodontitis with different stages of chronic kidney disease measured by glomerular filtration rate and systemic markers: an observational study. J. Endod 49, 1472–1479. https://doi.org/10.1016/j.joen.2023.08.012 (2023).
Barcelos, R. C. S. et al. Apical periodontitis induces changes on oxidative stress parameters and increases Na(+)/K(+)-ATPase activity in adult rats. Arch. Oral Biol. 118, 104849. https://doi.org/10.1016/j.archoralbio.2020.104849 (2020).
Xiao, S. et al. Is oxidative stress involved in the hepatic inflammatory response to apical periodontitis? A comparative study in normal and hyperlipidaemic rat. Int. Endod J. 56, 722–733. https://doi.org/10.1111/iej.13907 (2023).
Brembach, T. C., Sabat, R., Witte, K., Schwerdtle, T. & Wolk, K. Molecular and functional changes in neutrophilic granulocytes induced by nicotine: a systematic review and critical evaluation. Front. Immunol. 14, 1281685. https://doi.org/10.3389/fimmu.2023.1281685 (2023).
Pham, A. K. et al. Differential lung inflammation and injury with tobacco smoke exposure in Wistar Kyoto and spontaneously hypertensive rats. Inhal. Toxicol. 32, 328–341. https://doi.org/10.1080/08958378.2020.1805052 (2020).
Liang, G. B. & He, Z. H. Animal models of emphysema. Chin. Med. J. (Engl.) 132, 2465–2475. https://doi.org/10.1097/CM9.0000000000000469 (2019).
Anwar, H. et al. Analyzing cross-talk of EPO and EGF genes along with evaluating therapeutic potential of cinnamomum verum in cigarette-smoke-induced lung pathophysiology in rat model. Food Sci. Nutr. 11, 1486–1498. https://doi.org/10.1002/fsn3.3188 (2023).
Macedo Signorelli, N. S. et al. Identification of oxidative stress biomarkers in apical periodontitis: A scoping review with bibliometric analysis. Aust Endod J. 50, 742–760. https://doi.org/10.1111/aej.12888 (2024).
Cintra, L. T. et al. Multiple apical periodontitis influences serum levels of cytokines and nitric oxide. J. Endod 42, 747–751. https://doi.org/10.1016/j.joen.2016.01.022 (2016).
Hernandez-Rios, P., Pussinen, P. J., Vernal, R. & Hernandez, M. Oxidative stress in the local and systemic events of apical periodontitis. Front. Physiol. 8, 869. https://doi.org/10.3389/fphys.2017.00869 (2017).
Santiago, H. A., Zamarioli, A., Sousa Neto, M. D. & Volpon, J. B. Exposure to secondhand smoke impairs fracture healing in rats. Clin. Orthop. Relat. Res. 475, 894–902. https://doi.org/10.1007/s11999-016-5184-6 (2017).
Nikula, K. J. et al. A mouse model of cigarette smoke-induced emphysema. Chest 117, 246S–247S (2000).
Hama, S., Takeichi, O., Saito, I. & Ito, K. Involvement of inducible nitric oxide synthase and receptor for advanced glycation end products in periapical granulomas. J. Endod 33, 137–141. https://doi.org/10.1016/j.joen.2006.11.011 (2007).
Jakovljevic, A. et al. Levels of oxidative stress biomarkers and bone resorption regulators in apical periodontitis lesions infected by Epstein-Barr virus. Int. Endod J. 51, 593–604. https://doi.org/10.1111/iej.12886 (2018).
Loureiro, C. et al. Teeth with acute apical abscess vs. teeth with chronic apical periodontitis: a quantitative and qualitative proteomic analysis. Clin. Oral Investig. 27, 591–601. https://doi.org/10.1007/s00784-022-04754-w (2023).
Vengerfeldt, V., Mandar, R., Saag, M., Piir, A. & Kullisaar, T. Oxidative stress in patients with endodontic pathologies. J. Pain Res. 10, 2031–2040. https://doi.org/10.2147/JPR.S141366 (2017).
Zhao, X. et al. Quercetin protects ethanol-induced hepatocyte pyroptosis via scavenging mitochondrial ROS and promoting PGC-1alpha-regulated mitochondrial homeostasis in L02 cells. Oxid. Med. Cell. Longev. 2022 (4591134). https://doi.org/10.1155/2022/4591134 (2022).
Saraiva-Romanholo, B. M. et al. Exposure to sodium hypochlorite or cigarette smoke induces lung injury and mechanical impairment in wistar rats. Inflammation 45, 1464–1483. https://doi.org/10.1007/s10753-022-01625-0 (2022).
Braz-Silva, P. H. et al. Inflammatory profile of chronic apical periodontitis: a literature review. Acta Odontol. Scand. 77, 173–180. https://doi.org/10.1080/00016357.2018.1521005 (2019).
El-Mahdy, M. A. et al. Chronic cigarette smoke exposure triggers a vicious cycle of leukocyte and endothelial-mediated oxidant stress that results in vascular dysfunction. Am. J. Physiol. Heart Circ. Physiol. 319, H51–H65. https://doi.org/10.1152/ajpheart.00657.2019 (2020).
Cavalla, F., Letra, A., Silva, R. M. & Garlet, G. P. Determinants of periodontal/periapical lesion stability and progression. J. Dent. Res. 100, 29–36. https://doi.org/10.1177/0022034520952341 (2021).
Xiong, J. et al. Therapeutic effects of melatonin on the lungs of rats exposed to passive smoking. Respir. Res. 25, 411. https://doi.org/10.1186/s12931-024-03042-3 (2024).
Vazão, A. R. et al. Experimental apical periodontitis alters salivary biochemical composition and induces local redox state disturbances in the salivary glands of male rats. Clin. Oral Investig. 28, 154. https://doi.org/10.1007/s00784-024-05540-6 (2024).
Samanovic, A. M. et al. Cardiac, biochemical and histopathological analysis reveals impaired heart function in hypertensive rats with apical periodontitis. Int. Endod J. 54, 1581–1596. https://doi.org/10.1111/iej.13562 (2021).
Khalighinejad, N. et al. Association between systemic diseases and apical periodontitis. J. Endod 42, 1427–1434. https://doi.org/10.1016/j.joen.2016.07.007 (2016).
Salaets, T. et al. A semi-automated method for unbiased alveolar morphometry: validation in a bronchopulmonary dysplasia model. PLoS One 15, e0239562. https://doi.org/10.1371/journal.pone.0239562 (2020).
Hsia, C. C., Hyde, D. M., Ochs, M., Weibel, E.R., ATS/ERS Joint Task Force on Quantitative Assessment of Lung Structure. An official research policy statement of the American Thoracic Society/European Respiratory Society: standards for quantitative assessment of lung structure. Am. J. Respir. Crit. Care Med. 181, 394–418. https://doi.org/10.1164/rccm.200809-1522ST (2010).
Sun, J. et al. Effect of Simvastatin on MMPs and timps in cigarette smoke-induced rat COPD model. Int. J. Chron. Obstruct Pulmon. Dis. 12, 717–724. https://doi.org/10.2147/COPD.S110520 (2017).
Zheng, H. et al. Development and characterization of a rat model of chronic obstructive pulmonary disease (COPD) induced by sidestream cigarette smoke. Toxicol. Lett. 189, 225–234. https://doi.org/10.1016/j.toxlet.2009.06.850 (2009).
Tsosura, T. V. S. et al. Maternal apical periodontitis is associated with insulin resistance in adult offspring. Int. Endod J. 52, 1040–1050. https://doi.org/10.1111/iej.13096 (2019).
Nagendrababu, V. et al. PRIASE 2021 guidelines for reporting animal studies in endodontology: a consensus-based development. Int. Endod J. 54, 848–857. https://doi.org/10.1111/iej.13477 (2021).
Kilkenny, C., Browne, W. J., Cuthill, I. C., Emerson, M. & Altman, D. G. Improving bioscience research reporting: the ARRIVE guidelines for reporting animal research. PLoS Biol. 8, e1000412. https://doi.org/10.1371/journal.pbio.1000412 (2010).
Cintra, L. T. et al. Pulpal and periodontal diseases increase triglyceride levels in diabetic rats. Clin. Oral Investig. 17, 1595–1599. https://doi.org/10.1007/s00784-012-0853-7 (2013).
Aycicek, A. & Erel, O. Total oxidant/antioxidant status in jaundiced newborns before and after phototherapy. J. Pediatr. (Rio J) 83, 319–322. https://doi.org/10.2223/JPED.1645 (2007).
Acknowledgements
This study was funded by the following Brazilian agencies: Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES, finance code 001); National Council for Scientific and Technological Development (CNPq/INCT Saúde Oral e Odontologia, Grant Number 406840/2022-9); Research Support Foundation of the State of Minas Gerais (FAPEMIG, Grant Numbers APQ-02105-18 and APQ-00927-23); and Rede Mineira de Odontologia (FAPEMIG RED00204-23). The authors also thank Maria Eduarda Paz Dotto for technical support.
Author information
Authors and Affiliations
Contributions
Conceptualization, D.C.F., C.C.G.M.; methodology, D.C.F., S.G.S.R., A.P.M., R.C.R.; software, R.C.R., S.A.L.P., P.B.F.S.; validation, S.G.S.R., R.C.R., S.A.L.P., P.B.F.S.; formal analysis, D.C.F.; investigation, D.C.F., S.G.S.R., A.P.M., R.C.R.; resources, S.A.L.P., P.B.F.S., C.C.G.M.; data gathering, D.C.F.; writing—original draft preparation, D.C.F., C.C.G.M.; writing—review and editing, C.C.G.M., A.P.M.; visualization, D.C.F.; supervision, C.C.G.M.; project administration, C.C.G.M., R.C.R., S.A.L.P.; funding acquisition, C.C.G.M., R.C.R., S.A.L.P. All authors have read and agreed to the published version of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Ferraz, D.C., Rende, S.G.S., Melo, A.d.P. et al. Synergistic effect of secondhand smoke and apical periodontitis on lung tissue damage in rats. Sci Rep 15, 13088 (2025). https://doi.org/10.1038/s41598-025-97601-3
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-97601-3
