
Abstract
THRB encodes thyroid hormone receptor β which produces two human isoforms (TRβ1 and TRβ2) by alternative splicing. The first THRB variant associated with autosomal dominant macular dystrophy (ADMD), NM_001354712.2:c.283 + 1G > A, was recently described. This study aims to refine the ophthalmologic phenotype, report a novel THRB variant, and investigate the impact of these splicing variants at the protein level. THRB variants were identified by re-analysis of next-generation sequencing data from the FJD database. Family segregation was performed using Sanger sequencing. Clinical data were collected from self-reported ophthalmic history questionnaires and ophthalmic exams. Functional splicing test was performed by in vitro minigene approach. We identified 12 patients with ADMD from 3 families carrying variants in THRB. Two families carried the variant NM_001354712.2:c.283 + 1G > A, and one the novel variant NM_001354712.2:c.283G > A. Patients exhibited common ophthalmologic findings with disruption of subfoveal ellipsoid layers, and variable onset of symptoms. Splicing assays showed complete exon 5 skipping or a 6 bp deletion in both variants. Our results support the association of THRB with ADMD. The high intra-familial variability could be influenced by phenotype modifiers. Aberrant TRβ1/TRβ2 proteins could lead to a gain-of-function mechanism. Including THRB in inherited retinal dystrophy genetic panels could enhance diagnoses and clinical patient management.
Similar content being viewed by others

Introduction
Inherited retinal dystrophies (IRD) are a group of rare diseases that significantly impact vision and overall quality of life. These conditions are the consequence of the dysfunction or degeneration of photoreceptors (rods and cones) and/or retinal pigmented epithelium (RPE) cells, leading to visual impairment or blindness1,2. Currently, 328 causative genes of IRD are known (RetNet, last accessed April 2025).
THRB (MIM*190160), located on 3p24.2, encodes the thyroid hormone receptor beta (TRβ). The structure of the protein is composed of the N-terminal A/B domain, the DNA-binding domain (DBD), the hinge domain, and the C-terminal ligand-binding domain (LBD) which include a ligand-dependent activation function (AF-2)3,4. Human THRB is expressed as two isoforms produced by alternative splicing, TRβ1 and TRβ2. TRβ1 (ENST00000646209.2 / NM_001354712.2) consists of 11 exons (461 amino acids). TRβ2 which is expressed in only a few tissues and has limited sequence data in databases, complicating its interpretation, has been described as an isoform consisting of 7 exons (476 amino acids) (ENST00000280696.9 / -) or, more recently, 9 exons (430 amino acids) (- / NM_001354714.2, June 2024). The difference between TRβ1 and TRβ2 lies in the A/B domain, which is shorter in the TRβ2 isoform. Thus, the N-terminal modulates the isoform-specific activity/function by controlling the interaction with other ligands in a cell type-dependent or promoter-specific fashion5. The expression of TRβ2 is limited to the hypothalamus, pituitary, cochlea, and retina6, and it plays an important role in retinal cones development7,8. TRβ1 is widely distributed in other tissues, and it is also expressed in retina and anterior ocular tissues9. Different studies with animal models, such as mice and zebrafish, or human retinal organoids, suggest that THRB mutations may be associated with human cone disorders, as this gene regulates the expression of cone opsins10,11,12. Specifically, these studies show that cone dysfunction occurs when the TRβ2 isoform is affected and not TRβ19,10.
According to the Human Gene Mutation Database (HGMD® 2025.1, last accessed April 2025), 196 pathogenic variants have been reported for THRB. Variants in this gene have mainly been associated with thyroid hormone resistance (RTHβ) phenotypes (99.5% of reported variants), characterized by increased serum thyroid hormone levels (T3 and T4) and normal or slightly increased serum thyrotropin (TSH)13. The vast majority are missense variants (87%)14, and only two variants are located in the A/B domain. Interestingly, a clinical observation study involving 31 patients with RTHβ reported functional defects of their photoreceptors, leading to worse colour sensitivity in RTHβ patients than controls15.
Furthermore, a splicing variant (NM_001354712.2:c.283 + 1G > A) has been recently reported to cause autosomal dominant macular dystrophy without thyroid phenotype in 3 unrelated Spanish families16, being in so far the only reported variant in THRB associated with ophthalmic phenotype. Fernández-Suárez et al.16 suggested that this variant exclusively affected TRβ1 isoform, since it was not present in TRβ2 (ENST00000280696.9). However, using the updated TRβ2 isoform (NM_001354714.2), the variant may affect both TRβ1 and TRβ2. In addition, the variant is located in the first coding exon of TRβ2 (exon 3) (NM_001354714.2). (Fig. 1).

Schematic representation of the TRβ1 and TRβ2 isoforms of THRB. The red circle with an “M” letter represents the reported variant NM_001354712.2:c.283 + 1G > A. (A) TRβ1 (NM_001354712.2) contains 11 exons and 461 amino acids. (B) TRβ2 (ENST00000280696.9) contains 7 exons and 476 amino acids and (C) TRβ2 (NM_001354714.2) contains 9 exons and 430 amino acids. The variant NM_001354712.2:c.283 + 1G > A is located in the donor splicing site of TRβ1 exon 5, and in the donor splicing site of TRβ2 (NM_001354714.2) exon 3 (NM_001354714.2:c.190G > A). However, it does not affect the isoform TRβ2 (ENST00000280696.9). aa: amino acids; A/B: N-terminal A/B domain; DBD: DNA-binding domain; Hinge: hinge domain; LBD: C-terminal ligand-binding domain; AF2: ligand-dependent activation function.
The aim of this work is to refine the ophthalmic phenotype, describe a novel variant in THRB associated to IRD, and study the consequences of the variants at the protein level, thus confirming the role of this gene in the etiopathogenesis of IRD.
Results
Clinical data
Table 1 summarizes clinical data available from 12 patients of 3 unrelated families carrying pathogenic THRB variants in our IRD-cohort. All patients with available ophthalmologic data presented with ocular symptoms at the moment at their first visit. In addition, 9 patients completed the self-reported questionnaire, of whom 8 reported visual acuity loss and 1 dyschromatopsia as first symptom. The hormone testing was performed in patients III:1 and IV:2 from Family 1, II:4 from Family 2, and IV:1, IV:7, IV:9, IV:12 and V:3 from Family 3 with negative results. In the remaining patients, although no hormone testing was performed, no endocrine disorders were observed.
Additionally, the proband from Family 1 (patient IV:2) reported that his parents were from a village with fewer than 5000 inhabitants located in Badajoz, Extremadura, Spain. The index case from Family 2 (patient II:4) reported that her affected father was also from that village in Badajoz. We have performed an inbreeding analysis for these families carrying the same variant (NM_001354712.2:c.283 + 1G > A), which showed an Identity-By-Descent (IBD) value of 0, indicating no detectable familial relationship up to the first cousin level.
The proband of Family 1 (IV:2) exhibited visual acuity loss followed by dyschromatopsia in early childhood, and night blindness later in life. In his initial examination in clinic in 2018 his visual acuity was 20/30 bilaterally and dropped in his last visit in 2024 to 20/50 (right eye) and 20/60 (left eye). The SD-OCT Spectralis scans on his last visit showed central disruption of outer retinal layers and collapsed overlying inner retinal layers in both eyes, whereas Optos pseudocolour fundus autofluorescence depicted central hypoautofluorescence in both eyes, larger on the left eye, with adjacent hyperautofluorescence temporally to these areas (Fig. 2A). Pattern electroretinogram (P-ERG) responses were consistent with severe, bilateral macular dysfunction, whereas on full field electroretinograms (FF-ERG), dark adapted (DA) 3 and 10 and light adapted (LA) 3 ERG responses were within normal limits; however, the peak time in flicker 30 Hz responses were delayed, suggesting generalized cone dysfunction bilaterally (Fig. 3A), which correlates with his symptoms of dyschromatopsia, and visual acuity drop. His father (III:1-Family 1) had been diagnosed previously in another center with macular dystrophy; he complained of central visual acuity loss as well as central visual field loss, and OCT scans were reported to show disruption of subfoveal ellipsoid layers in both eyes. The proband’s second aunt (III:4-Family 1) and second cousin (IV:4-Family 1) had also been seen previously in another center and diagnosed with a retinal dystrophy; however, no further clinical information is available for III:4. The latest best corrected visual acuity (BCVA) of IV:4 was reported to be was 20/30 (right eye) and 20/40 (left eye), and it was said to present with fundoscopic macular alterations and ellipsoid zone disruption on OCT scans. Patient IV:2-Family 1 also presented undefined behavior disorder and epilepsy secondary to perinatal hypoxia.

Ophthalmologic findings in patients with pathogenic variants in THRB in our IRD-cohort. (A) Patient IV:2 from Family 1: A1, A2, A4 and A5 correspond to Optos pseudocolour fundus autofluorescence images of the right eye (A1, A2) and left eye (A4, A5). A3 and A6 correspond to SD-OCT of the right and left macula, respectively. (B) Patient III:4 from Family 2: B1, B2 (right eye) and B4, B5 (left eye) represent her Optos pseudocolour wide-field images, and B3 (right eye) and B6 (left eye) correspond to her SD-OCT Spectralis scans. (C) Patient IV:1 from Family 3: SD-OCT Spectralis scans images are shown of the right (C1, C2) and left (C4, C5) eye. C3 and C6 represent SD-OCT scans of both maculae. (D) Patient III:7 from Family 3: Optos pseudocolour wide-field images of the right (D1, D2) and left (D4, D5) eye are shown. D3 and D6 correspond to SD-OCT Spectralis scans of the right and left macula, respectively. (E) Patient V:3 from Family 3: E1, E2 and E4, E5 correspond to Optos pseudocolour wide-field images of the right (E1, E2) and left (E4, E5) maculae. E3 and E6 are SD-OCT Spectralis scans of the right and left macula, respectively. (F) Patient IV:12 from Family 3: Optos pseudocolour wide-field imaging of the right (F1, F2) and left (F4, F5); and SD-OCT Spectralis scans of the right (F3) and left (F6) macula, showing granularity of the ellipsoid zone subfoveally in both eyes. (G) Patient III:4 from Family 3: Both images represent SD-OCT scans of the right (G1) and left (G2) maculae. (H) Patient IV:7 from Family 3: Cirrus HD OCT images of the right (H1) and left (H2) eyes.

Electrophysiologic testing performed at patients carrying THRB variants in our IRD-cohort. (A) Electrophysiology testing of patient IV:2-Family 1, compatible with symmetric, bilateral, severe maculopathy. (B) Electrophysiology testing of patient II:4-Family 2, suggestive of macular dysfunction. (C) Electrophysiology testing of patient IV:1-Family 3, compatible with bilateral maculopathy. (D) Electrophysiology testing of patient III:7-Family 3, compatible with bilateral maculopathy, in line with the patient´s symptoms of dyschromatopsia and visual acuity loss. (E) FF-ERG of patient V:3-Family showing responses well within normal limits. However, P-ERG is lacking to correctly assess macular dysfunction.
The index case of Family 2 reported visual acuity and central visual field loss from her first decade of life, followed by dyschromatopsia and photophobia later in life. Her visual acuity on her latest clinical visit was 20/200 bilaterally, and her SD-OCT Spectralis scans depicted granular appearance and mild disruption of the central ellipsoid subfoveally in both eyes. Optos pseudocolour wide-field images showed unremarkable findings and autofluorescence showed central hyperautofluorescence (Fig. 2B). P-ERGs and FF-ERGs were available for this patient, which showed delayed peak time of the a-wave in light adapted (LA) 3 ERG and delayed peak time in light adapted (LA) flicker 30 Hz bilaterally, suggesting generalized cone dysfunction in both eyes, along with delayed a-wave peak time in dark adapted (DA) 0.01 and 10 ERGs, and increased b/a ratio in dark adapted (DA) 3 ERGs bilaterally, which could be in line with generalized rod dysfunction in both eyes. P-ERGs responses were within normal limits in the left eye, but in the right eye were suggestive of macular dysfunction (Fig. 3B). Both her sister and father were said to present with similar symptoms as hers (her sister having been diagnosed with macular dystrophy in another center); however, neither of them was available for clinical examination in our clinic or segregation analysis.
Family 3, with a total of 7 affected relatives with molecular study, showed a high variability with a wide range of disease onset from the first to the seventh decade. The proband (IV:1) reported visual acuity loss at the age of 35, followed by dyschromatopsia and photophobia. His latest BCVA was 20/100 bilaterally. SD-OCT Spectralis scans depicted mild granular appearance of the ellipsoid zone with subfoveal disruption bilaterally. Optos pseudocolour wide-field images showed unremarkable findings and autofluorescence showed central hyperautofluorescence in both eyes (Fig. 2C). FF-ERGs responses did not show evidence of generalized photoreceptor dysfunction, whereas P-ERGs were in line with macular dysfunction (Fig. 3C). Patient III:7 from Family 3 reports to wear glasses for myopia from the age of 7; at the age of 50 he started noticing altered colour vision, and recently, visual acuity loss likely secondary to cataract formation; his latest visual acuity was 20/25 in both eyes. SD-OCT Spectralis scans showed mild disruption of the ellipsoid zone subfoveally in both eyes. Optos pseudocolour wide-field images showed unremarkable findings and autofluorescence showed central hyperautofluorescence in both eyes (Fig. 2D). FF-ERGs were well within normal limits whereas P-ERGs were compatible with severe macular dysfunction bilaterally (Fig. 3D). The patient V:3 was 9 at the time of her latest examination; she reports stationary low visual acuity (latest BCVA 20/50), photophobia and photopsia. Her SD-OCT Spectralis findings showed granular appearance of the ellipsoid zone with central disruption bilaterally. Optos pseudocolour wide-field images showed unremarkable findings and autofluorescence showed central hyperautofluorescence in both eyes (Fig. 2E). No P-ERGs were performed, but her FF-ERGs did not show any evidence of generalized photoreceptor dysfunction bilaterally (Fig. 3E). The patient IV:12 complained of visual acuity loss from the age of 26, and later developed photophobia, dyschromatopsia, central visual field loss and mild nyctalopia. Her latest BCVA was 20/30 in both eyes. Her SD-OCT Spectralis depicted granular appearance of the ellipsoid zone with central disruption bilaterally. Optos pseudocolour wide-field images showed unremarkable findings and autofluorescence showed mild loss of the normal central macular hypoautofluorescence (Fig. 2F). The patient III:4 started noticing visual acuity loss at age of 65. His latest BCVA was reported to be 20/40 (right eye) and 20/80 (left eye). SD-OCT showed moderate disruption of the subfoveal outer retinal layers, especially the ellipsoid layer, with a granular aspect (Fig. 2G). No electrophysiologic data is available for patients IV:12 and III:4 from Family 3. The patient IV:7 reported visual acuity loss, night blindness, dyschromatopsia and photophobia at the age of 28; her last visual acuity was 20/50 in both eyes. Her Cirrus HD OCT scans depicted granular appearance of the ellipsoid zone subfoveally accompanied by mild disruption (Fig. 2H). No Optos pseudocolour wide-field images or electrophysiologic testing were performed for this patient. The patient IV:9 had been seen in another hospital and was reported to show ellipsoid zone disruption subfoveally with granular appearance in both eyes; however, further clinical data is lacking for this patient.
Genetic analysis and variants interpretation
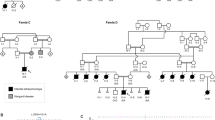
We identified two variants in THRB gene in these 3 families from our IRD cohort, the variant NM_001354712.2:c.283 + 1G > A, previously reported as pathogenic15, and the novel variant NM_001354712.2:c.283G > A. Both were predicted as pathogenic and were not found in 4675 pseudocontrols from the Fundación Jiménez Díaz (FJD) database.
Variant c.283 + 1G > A was carried by 4 affected relatives from Family 1 (Fig. 4A and D), and the index case of Family 2 (Fig. 4B and D). The novel variant c.283G > A is located in exon 5 (TRβ1), 1 bp away from the splice site, and was found in 7 family members of Family 3 (Fig. 4C and D).

Pedigrees and segregation of families carrying disease-causing variants in THRB. (A) Family 1 carrying the variant c.283 + 1G > A, (B) Family 2 carrying the variant c.283 + 1G > A, (C) and Family 3 carrying the variant c.283G > A. (D) Sanger chromatogram. Arrows indicate the proband in each family; m: mutant alleles; wt: wild-type alleles.
These variants were not identified in the gnomAD population databases. Splicing predictions support a disruption of the same canonical donor splicing site in both variants. Variant c.283 + 1G > A has been reported in ClinVar as pathogenic16, and in silico predictors for missense variants classify the variant c.283G > A as damaging (Table 2).
Minigene splice assay results
SpliceAI17 predicted a 0.98 and a 0.73 decrease in the probability of activation of canonical donor splice site, and a 0.24 and a 0.08 increase in the probability of activation of a cryptic donor 6 bp upstream, in the variants c.283 + 1G > A and c.283G > A, respectively (Table 2). Minigene splice assay was performed using DNA from a control individual (wt), patient IV:2-Family 1, carrying the variant c.283 + 1G > A (m1), and patient IV:1-Family 3, harboring the novel missense variant c.283G > A (m2). The minigene results showed the expected 368 bp fragment in the wt minigene, whereas the m1 and m2 minigenes revealed two bands; a large-size fragment and an additional small-sized fragment (105 bp). The small bands corresponded to the full skipping of exon 5 (Fig. 5A). Sanger sequencing revealed that the large-size fragment of the m1 and m2 minigenes corresponded to a 362 bp fragment, due to a deletion of 6 bp in the last part of exon 5, producing the protein change p.Cys93_Gly95delinsTrp. While the m1 minigene only showed to give rise to the protein change p.Cys93_Gly95delinsTrp and the skipping of exon 5. The m2 minigene showed both a deletion of 6 bp, leading to, p.Cys93_Gly95delinsTrp, correctly spliced transcripts including a missense variant, p.Gly95Arg, as well as the skipping of exon 5 (Fig. 5B).

Minigene splice assay results. (A) Gel image of PCR product. (B) Sanger chromatogram and schematic representation of PCR products. NTC: no-template control; wt: wild-type; m: mutant. The original gel image can be found in Supplementary Fig. 1.
Discussion
In this study, we describe two variants in THRB, one of them novel, in three Spanish families with autosomal dominant macular dystrophy.
This work supports the pathogenicity associated with the variant c.283 + 1G > A, so far identified only in three previously reported families16. Additionally, we describe a novel variant, c.283G > A, that affects a neighboring nucleotide to the one affected by c.283 + 1G > A. Both variants are predicted to have a strong disrupting effect on the canonical donor splicing site.
These variants were found in a cohort of 5222 families with IRD recruited by a single center (FJD, Madrid, Spain). The variant c.283 + 1G > A, which has only been described to date in other Spanish families15, whose precise origin was not reported. The presence of c.283 + 1G > A only in the Spanish population, along with its identification in only two families from the same confirmed geographical origin within such a sizable cohort, suggests a likely founder effect.
We complement the description of the THRB pathogenic genomic variability with the phenotypes of the 12 affected patients from three families. NGS was performed in five of the studied patients (IV:2-Family 1; II:4-Family 2; IV:1-Family 3; IV:7-Family 3; and IV:9-Family 3), with no other possible causal genes for their IRD identified using NGS. Despite the similarity among the reported symptoms in our patients (visual acuity loss, dyschromatopsia, photophobia), the age of onset could vary widely (early childhood to seventh decade of life). Also, the severity of symptoms may also differ among patients, and this may not always be age-dependent in our cohort. However, most patients showed similar pseudocolour fundus imaging, OCT and autofluorescence findings, seemingly unrelated to age of severity of symptoms. These findings are in line with those of Fernández-Suárez et al.16, supporting the hypothesis that phenotype modifiers could be influencing the clinical manifestation of THRB-associated conditions or contributing to the observed phenotypic variability. In addition, the relative amount of mutant and wild-type protein produced may vary between individuals, influencing the severity of symptoms and the age of disease onset. Consequently, individuals with higher levels of aberrant protein could present with more severe phenotypes, while those with lower levels might exhibit milder manifestations. However, directly quantifying mutant and wild-type protein levels in the ocular tissue in patients presents a significant challenge. Additional studies are required to validate this hypothesis.
Most variants associated with endocrine phenotype in THRB are located at C-terminal/LBD region, as reviewed by Fernández-Suárez et al.16, using HGMD-pro data18. But the variants c.283 + 1G > A and c.283G > A are in the N-terminal A/B domain of the TRβ1 and TRβ2 isoforms, where they are predicted to have a deleterious effect. Furthermore, no endocrine disorders were observed in patients with THRB variants from our IRD-cohort. Therefore, the location of the IRD-causing variants at N-terminal A/B domain highlights its importance in the visual function. Recent studies showed that light/dark adaptation is coordinated by Müller glial cells and photoreceptors via thyroid hormone signaling pathway in mouse models19. Interestingly, the THRB gene regulates the expression of CYP27C1 (cytochrome P450 enzyme)20, necessary for the conversion of vitamin A1 into A2 in zebrafish21,22. However, further studies are still needed to elucidate the role of THRB in the human retina. Understanding the molecular mechanisms in which this gene is involved could have therapeutic implications for patients with THRB-associated macular dystrophy.
To investigate how many variants in THRB lead to an endocrine phenotype without retinal problems while the two variants we described are associated with retinal phenotype without endocrine issues, we can look more in details about the consequences of the variants at the protein level. The variants found in IRD patients showed both a complete exon 5 skipping or a 6 bp deletion while a missense variant was only found in the second variant (c.283G > A). The exon 5 skipping implies the deletion of multiple amino acids in TRβ1 (p.Glu8_Lys94del) and leads to the removal of the start codon of TRβ2 (NM_001354714.2), since TRβ1 exon 5 corresponds to the TRβ2 (NM_001354714.2) first coding exon (Fig. 1). The 6 bp deletion results in a small indel, p.Cys93_Gly95delinsTrp in TRβ1 and p.Cys62_Gly64delinsTrp in TRβ2 (NM_001354714.2). However, if TRβ2 corresponds to ENST00000280696.9, it would remain intact. Since multiple loss-of-function variants affecting both TRβ1 and TRβ2 are known to be pathogenic for autosomal dominant thyroid phenotype, it is unlikely that the cause of the IRD is the decreased expression of TRβ2 (NM_001354714.2) due to the exon 5 skipping. Therefore, we hypothesize that either p.Glu8_Lys94del or p.Cys93_Gly95delinsTrp in TRβ1 or p.Cys62_Gly64delinsTrp in TRβ2 create a gain-of-function for which the pathogenesis mechanism is unknown at the moment.
In conclusion, THRB should be included in the targeted panel sequencing of IRD patients, given its association with autosomal dominant macular dystrophy. Moreover, this study highlights the importance of NGS data reanalysis from uncharacterized patients.
Materials and methods
Subjects and clinical data collection
Twelve affected patients from 3 unrelated families were included in this study. These families were identified from the Genetics Department of the FJD University Hospital, Spain, which have collected and maintains a cohort of 5222 unrelated IRD families23,24, 2838 of them with next-generation sequencing (NGS) available, and the rest with other molecular approaches.
All patients completed and signed an informed consent. The FJD Research Ethics Committee approved the study project (Approval No.: PIC172-20_FJD), that fulfills the tenets of the Declaration of Helsinki and its further reviews.
Eight of the 12 patients included in the study had been previously seen in the Ophthalmology Department of the FJD (IV:2 from Family 1, II:4 from Family 2, III:4, III:7; IV:1, IV:7, IV:12 and V:3 from Family 3), and detailed clinical examination, including best-corrected visual acuity, fundus examination, and multimodal imaging, were performed when possible. Electrodiagnostic testing was performed when possible, following the ISCEV (International Society for Clinical Electrophysiology of Vision) standard protocol. Clinical data from 3 of the 12 patients was obtained from clinical reports from other centers (III:1 and IV:4 from Family 1, and IV:9 from Family 3). The patient III:4 from Family 1 was reported to have IRD in her daughter’s report (patient IV:4), but no detailed examination is available.
In addition, a self-reported ophthalmic history recorded from questionnaires was obtained from 9 of these patients, including the age of onset of subjective symptoms.
Hormone testing, including the measurement of thyroid-stimulating hormone (TSH), T3 and T4 levels, was performed when possible (III:1 and IV:2 from Family 1; II:4 from Family 2; and IV:1, IV:7, IV:9, IV:12 and V:3 from Family 3).
Inbreeding analysis
The inbreeding analysis was performed using PLINK whole-genome association analysis toolkit as described in Iancu IF et al., 25.
Molecular diagnosis
THRB variants were identified by means of a re-analysis of NGS data, including clinical exome sequencing (CES) and whole exome sequencing (WES), from the FJD cohort25. NGS re-analysis was performed using a DNASeq bioinformatics pipeline available at https://github.com/TBLabFJD/VariantCallingFJD based on bwa, Genome Analysis Toolkit (GATK) best practices and Ensembl Variant Effect Predictor (VEP) annotation. The FJD genomic database included variant frequencies of 10 172 index rare diseases patients, including 2838 with IRD and 7334 with non-ophthalmogenetic diseases (considered here as “pseudocontrols”). Furthermore, segregation and confirmation studies in affected families were performed by Sanger sequencing.
Analysis of variants
The pathogenicity of the variants was established according to the American College of Medical Genetics and Genomics (ACMG) guidelines26, in silico pathogenicity predictions and family segregation. Additionally, the variants were explored in three databases: (i) Human Gene Mutation Database, HGMD® 2025.1, (https://digitalinsights.qiagen.com/products-overview/clinical-insights-portfolio/human-gene-mutation-database/), (ii) ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/), and (iii) Leiden Open Variation Database, LOVD v.3.0, (https://www.lovd.nl/).
The potential effect of the variants was assessed in silico using functional pathogenicity, conservation, and splicing predictors including in MobiDetails27, such as spliceAI17, MaxEntScan28, SPiP29, dbscSNV30 and HexoSplice31.
Minigene assay
The DNA from a healthy individual (Control), patient IV:2-Family 1 and patient IV:1-Family 3 were used to assess the pathogenic impact on splicing of the THRB variants (c.283 + 1G > A and c.283G > A). For PCR amplification, primers were designed with attB-1 and attB-2 sites at their 5’ ends (forward primer: 5’-ggggacaagtttgtacaaaaaagcaggctaagtgggactggcgaaagta-3’; reverse primer: 5’-ggggaccactttgtacaagaaagctgggtgccaaaacttacaacccctct-3’). Both forward and reverse primers anneal ~ 280 bp away from the canonical splice sites of exon 5. The amplified PCR product was purified and cloned first into the pGEM®-T Vector (Promega AG), then transferred into the pDONR 201 vector using the Gateway® BP Clonase™ II Enzyme Mix (Invitrogen), and finally shuttled into the pCI-neo-RHO exon3,5/DEST destination vector (a kind gift by Dr. Frans P. M. Cremers, Radboud University Medical Center, Nijmegen, The Netherlands) using Gateway® LR Clonase™ II Enzyme Mix (Invitrogen).
HEK-293 cells were grown in 6-well plates with 2 ml of Dulbecco’s Modified Eagle’s Medium (DMEM) supplemented with 10% fetal bovine serum (FBS) at 37°C in a humidified atmosphere of 5% CO2. The cells (confluency of ∼70%) were transfected with 500 ng of each plasmid in the presence of Lipofectamine 2000 (Thermo Fisher Scientific) prepared in Opti-MEM® (Thermo Fisher Scientific). After 15 hours, transfection medium was replaced with fresh DMEM with 10% FBS. After 48 hours post-transfection, total RNA was extracted using the Illustra RNAspin Mini Kit (GE Healthcare, Opfikon, Switzerland). Then, 1 µg of total RNA was reverse transcribed into cDNA using the High-Capacity cDNA Reverse Transcription Kit (Thermo Fisher Scientific). Finally, PCR amplification was performed using 10 ng of cDNA as template, and primers binding to the exon 3 (forward primer: 5’-tacatgttcgtggtccacttc-3’) and exon 5 (reverse primer: 5’-atggtggtgagcatgcagt-3’) of RHO gene. PCR product was analyzed by electrophoresis on a 2% agarose gel.
Plasmids and PCR products nucleotide sequence were confirmed by Sanger sequencing (Microsynth AG, Balgach, Switzerland).
Data availability
Genomic data are available on the European Genome-phenome Archive (EGA, https://ega-archive.org/), ID: EGAD50000001255.
References
Ayuso, C. et al. Retinitis pigmentosa in Spain. The Spanish multicentric and multidisciplinary group for research into retinitis pigmentosa. Clin. Genet. 48(3), 120–122. https://doi.org/10.1111/j.1399-0004.1995.tb04069.x (1995).
Hartong, D. T., Berson, E. L. & Dryja, T. P. Retinitis pigmentosa. Lancet Lond. Engl. 368(9549), 1795–1809. https://doi.org/10.1016/S0140-6736(06)69740-7 (2006).
Lazar, M. A. Thyroid hormone receptors: Multiple forms, multiple possibilities. Endocr. Rev. 14(2), 184–193. https://doi.org/10.1210/edrv-14-2-184 (1993).
Yen, P. M. Physiological and molecular basis of thyroid hormone action. Physiol. Rev. 81(3), 1097–1142. https://doi.org/10.1152/physrev.2001.81.3.1097 (2001).
Tian, H., Mahajan, M. A., Wong, C. T., Habeos, I. & Samuels, H. H. The N-Terminal A/B domain of the thyroid hormone receptor-beta2 isoform influences ligand-dependent recruitment of coactivators to the ligand-binding domain. Mol. Endocrinol. Baltim. Md. 20(9), 2036–2051. https://doi.org/10.1210/me.2005-0437 (2006).
Jones, I., Ng, L., Liu, H. & Forrest, D. An intron control region differentially regulates expression of thyroid hormone receptor beta2 in the cochlea, pituitary, and cone photoreceptors. Mol. Endocrinol. Baltim. Md. 21(5), 1108–1119. https://doi.org/10.1210/me.2007-0037 (2007).
Suzuki, S. C. et al. Cone photoreceptor types in zebrafish are generated by symmetric terminal divisions of dedicated precursors. Proc. Natl. Acad. Sci. USA 110(37), 15109–15114. https://doi.org/10.1073/pnas.1303551110 (2013).
Aramaki, M. et al. Transcriptional control of cone photoreceptor diversity by a thyroid hormone receptor. Proc. Natl. Acad. Sci. USA. 119(49), e2209884119. https://doi.org/10.1073/pnas.2209884119 (2022).
Ng, L., Liu, H., Liu, Y. & Forrest, D. Biphasic expression of thyroid hormone receptor TRβ1 in mammalian retina and anterior ocular tissues. Front. Endocrinol. 14, 1174600. https://doi.org/10.3389/fendo.2023.1174600 (2023).
Ng, L. et al. A thyroid hormone receptor that is required for the development of green cone photoreceptors. Nat. Genet. 27(1), 94–98. https://doi.org/10.1038/83829 (2001).
Deveau, C. et al. Thyroid hormone receptor beta mutations alter photoreceptor development and function in Danio rerio (zebrafish). PLoS Genet. 16(6), e1008869. https://doi.org/10.1371/journal.pgen.1008869 (2020).
Eldred, K. C. et al. Thyroid hormone signaling specifies cone subtypes in human retinal organoids. Science 362(6411), eaau6348. https://doi.org/10.1126/science.aau6348 (2018).
Pappa, T. & Refetoff, S. Resistance to thyroid hormone beta: A focused review. Front. Endocrinol. 12, 656551. https://doi.org/10.3389/fendo.2021.656551 (2021).
Concolino, P., Costella, A. & Paragliola, R. M. Mutational landscape of resistance to thyroid hormone beta (RTHβ). Mol. Diagn. Ther. 23(3), 353–368. https://doi.org/10.1007/s40291-019-00399-w (2019).
Campi, I. et al. Retinal photoreceptor functions are compromised in patients with resistance to thyroid hormone syndrome (RTHβ). J. Clin. Endocrinol. Metab. 102(7), 2620–2627. https://doi.org/10.1210/jc.2016-3671 (2017).
Fernández-Suárez, E. et al. Expanding the phenotype of THRB: A range of macular dystrophies as the major clinical manifestations in patients with a dominant splicing variant. Front. Cell. Dev. Biol. 11, 1197744. https://doi.org/10.3389/fcell.2023.1197744 (2023).
de Sainte Agathe, J. M. et al. SpliceAI-visual: A free online tool to improve spliceai splicing variant interpretation. Hum. Genomics. 17(1), 7. https://doi.org/10.1186/s40246-023-00451-1 (2023).
Stenson, P. D. et al. The human gene mutation database (HGMD®): Optimizing its use in a clinical diagnostic or research setting. Hum. Genet. 139(10), 1197–1207. https://doi.org/10.1007/s00439-020-02199-3 (2020).
Wei, M. et al. Single-cell profiling reveals Müller glia coordinate retinal intercellular communication during light/dark adaptation via thyroid hormone signaling. Protein Cell. 14(8), 603–617. https://doi.org/10.1093/procel/pwad007 (2023).
Volkov, L. I. et al. Thyroid hormone receptors mediate two distinct mechanisms of long-wavelength vision. Proc. Natl. Acad. Sci. USA 117(26), 15262–15269. https://doi.org/10.1073/pnas.1920086117 (2020).
Enright, J. M. et al. Cyp27c1 Red-Shifts the spectral sensitivity of photoreceptors by converting vitamin A1 into A2. Curr. Biol. CB. 25(23), 3048–3057. https://doi.org/10.1016/j.cub.2015.10.018 (2015).
Corbo, J. C. Vitamin A1/A2 chromophore exchange: Its role in spectral tuning and visual plasticity. Dev. Biol. 475, 145–155. https://doi.org/10.1016/j.ydbio.2021.03.002 (2021).
Perea-Romero, I. et al. Genetic landscape of 6089 inherited retinal dystrophies affected cases in Spain and their therapeutic and extended epidemiological implications. Sci. Rep. 11(1), 1526. https://doi.org/10.1038/s41598-021-81093-y (2021).
Perea-Romero, I. et al. Inherited retinal dystrophies in Spain: Three decades of epidemiological, clinical, and genetic study. RANM 139(13903), 274–284. https://doi.org/10.32440/ar.2022.139.03.rev08 (2023).
Iancu, I. F. et al. Aggregated genomic data as cohort-specific allelic frequencies can boost variants and genes prioritization in Non-Solved cases of inherited retinal dystrophies. Int. J. Mol. Sci. 23(15), 8431. https://doi.org/10.3390/ijms23158431 (2022).
Richards, S. et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American college of medical genetics and genomics and the association for molecular pathology. Genet. Med. 17(5), 405–424. https://doi.org/10.1038/gim.2015.30 (2015).
Baux, D. et al. MobiDetails: Online DNA variants interpretation. Eur. J. Hum. Genet. EJHG 29(2), 356–360. https://doi.org/10.1038/s41431-020-00755-z (2021).
Yeo, G. & Burge, C. B. Maximum entropy modeling of short sequence motifs with applications to RNA splicing signals. J. Comput. Biol. J. Comput. Mol. Cell. Biol. 11(2–3), 377–394. https://doi.org/10.1089/1066527041410418 (2004).
Leman, R. et al. SPiP: Splicing prediction pipeline, a machine learning tool for massive detection of exonic and intronic variant effects on mRNA splicing. Hum. Mutat. 43(12), 2308–2323. https://doi.org/10.1002/humu.24491 (2022).
Jian, X., Boerwinkle, E. & Liu, X. In Silico prediction of splice-altering single nucleotide variants in the human genome. Nucleic Acids Res. 42(22), 13534–13544. https://doi.org/10.1093/nar/gku1206 (2014).
Tubeuf, H. et al. Large-scale comparative evaluation of user-friendly tools for predicting variant-induced alterations of splicing regulatory elements. Hum. Mutat. 41(10), 1811–1829. https://doi.org/10.1002/humu.24091 (2020).
Acknowledgements
The authors acknowledge all clinicians from different Spanish hospitals for the recruitment of clinical data. The authors thank the generous support from all patients and their families for consenting to participate in the study.This work was supported by the Instituto de Salud Carlos III (ISCIII) of the Spanish Ministry of Health (PI22/00321), Centro de Investigación Biomédica en Red Enfermedades Raras (CIBERER, 06/07/0036), IIS-FJD BioBank (PT13/0010/0012), the Organización Nacional de Ciegos Españoles (ONCE), European Regional Development Fund (FEDER), and the University Chair UAM-IIS-FJD of Genomic Medicine. L.F.C. is supported by Centro de Investigación Biomédica en Red (CIBER), and P.M. is supported by a Miguel Servet program contract from ISCIII (CPII21/00015). The funding organizations had no role in the design or conduct of this research.
Author information
Authors and Affiliations
Contributions
L.F.C.: Data curation, Investigation, Methodology, Visualization, Writing—original draft; F.B.K: Methodology, Data curation, Writing—review and editing; S.T.S.: Methodology, Data curation, Writing—review and editing; I.M.M.: Methodology, Writing—review and editing; M.Q.: Investigation, Methodology, Visualization, Writing—review and editing; M.U.: Investigation, Methodology, Visualization, Writing—review and editing; E.C.: Methodology, Visualization, Writing—review and editing; M.P.M.G.: Methodology, Visualization, Writing—review and editing; B.G.S.: Methodology, Visualization, Writing—review and editing; P.M.: Data curation, Methodology, Software, Resources, Funding acquisition, Writing—review and editing; C.R.: Resources, Supervision, Writing—review and editing; M.C.: Data curation, Conceptualization, Funding acquisition, Supervision, Writing—review and editing; C.A.: Conceptualization, Funding acquisition, Resources, Supervision, Writing—review and editing.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Fernández-Caballero, L., Blanco-Kelly, F., Swafiri, S.T. et al. Identification of new families and variants in autosomal dominant macular dystrophy associated with THRB. Sci Rep 15, 14904 (2025). https://doi.org/10.1038/s41598-025-97768-9
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-97768-9
