
Abstract
Apolipoprotein A-IV (apoA-IV), the largest member of the exchangeable apolipoprotein family, is a common constituent of amyloid deposits in renal and cardiac amyloidosis. In this study, we characterized the aggregation propensity of the apoA-IV N-terminal fragment to form amyloid fibrils using a variety of biophysical techniques. Thioflavin T fluorescence assay, circular dichroism measurement, and microscopic observations revealed that the N-terminal 1−70 amino acid fragment of apoA-IV readily forms amyloid fibrils by a transition from a random coil to a β-sheet-rich structure. Sequence-based analysis indicated that residues 7−16 and 38−42 are the major aggregation-prone segments within the N-terminal 1−70 residues of apoA-IV. Consistent with this, deletion of these residues strongly inhibited the β-transition and fibril formation of apoA-IV 1−70. Kinetic and thermodynamic analyses of fibril formation by the apoA-IV 1−70 fragment demonstrated that primary nucleation is the dominant step in fibril formation, for which the activation energy barrier is entirely entropic. In addition, we found that the presence of heparin, a representative glycosaminoglycan, accelerated fibril formation kinetics and enhanced the yield of apoA-IV 1−70 fibrils, and the positively charged residues K58-K59 play a critical role in heparin interaction. Overall, our results suggest that the strong amyloid-forming propensity of the N-terminal fragment of apoA-IV may play a key role in amyloid deposition associated with apoA-IV amyloidosis.
Similar content being viewed by others


Introduction
Apolipoprotein A-IV (apoA-IV) is a 43-kDa lipid-binding protein that shares structural features with other exchangeable apolipoproteins1,2,3. In humans, apoA-IV is synthesized in the small intestine and secreted into the gastric lymphatics, where it is primarily associated with chylomicrons4. Unlike other apolipoproteins such as apolipoprotein A-I (apoA-I), the major protein component of plasma high-density lipoprotein (HDL), apoA-IV binds weakly to lipoprotein particles and can readily dissociate from their surface, thereafter circulating in the plasma both as a lipid-free protein and in association with HDL5. Despite the abundance of apoA-IV in circulation, its exact function remains unclear. It has been shown that apoA-IV has diverse biological functions including regulation of lipoprotein metabolism6,7, promotion of reverse cholesterol transport8, protection against lipid oxidation and atherosclerosis9,10, inhibition of platelet aggregation and thrombosis11, and control of glucose homeostasis12. In addition, it has been reported that apoA-IV is involved in amyloid β metabolism to facilitate its clearance in the brain13.
Previous structural studies have indicated that lipid-free apoA-IV is composed of a large helical bundle domain with the N- and C-termini in close proximity3,14,15. It has been proposed that the interaction between the N- and C-termini in apoA-IV prevents the protein from unfolding in response to lipid3,16,17. Indeed, disruption of this interaction significantly increases the lipid-binding ability of apoA-IV to solubilize lipid vesicles17. X-ray crystallography and small-angle x-ray scattering studies have indicated that the central core domain of apoA-IV adopts an elongated helical structure that dimerizes via domain swapping of a helical segment18,19. The reciprocating helix interaction in the core domain has been proposed to play an important role in the structure and lipid-binding ability of apoA-IV20.
Many exchangeable apolipoproteins, such as apoA-I and apoA-IV, have been increasingly recognized as amyloid precursor proteins in hereditary and acquired systemic amyloidosis21,22. In apoA-IV amyloidosis, amyloid fibril formation by wild-type apoA-IV appears to occur in different organs and tissues, including the heart23,24,25 and kidneys26,27. In cardiac amyloidosis, an N-terminal 70-residue fragment of apoA-IV has been reported to be deposited as a fibril24. In agreement with this, bioinformatics analysis of amyloid propensity prediction for apoA-IV has indicated that major amyloidogenic segments are located in the N-terminal and C-terminal parts of the protein21. Additionally, apoA-IV is known as one of the amyloid signature proteins found in most types of amyloid deposits28,29. However, detailed characterization of the fibril-forming properties of apoA-IV has not been reported to date.
In the present study, we biophysically characterized the fibril-forming properties of the N-terminal 1‒70 fragment of human apoA-IV to elucidate the molecular mechanism underlying apoA-IV amyloid deposition. We also examined the effects of heparin, a representative glycosaminoglycan (GAG), on the fibril-forming behavior of the apoA-IV 1‒70 fragment.
Results
Fibril-forming properties of apoA-IV 1‒70
We first characterized the structural stability and fibril-forming propensity of full-length apoA-IV. Circular dichroism (CD) measurements demonstrated that full-length apoA-IV (376 amino acid residues) is less stable but has a more highly organized helical structure than apoA-I (Supplementary Fig. S1A, B and Table S1)3. In keeping with this, apoA-IV exhibited lower 8-anilino-1-naphthalenesulfonic acid (ANS) fluorescence than apoA-I (Supplementary Fig. S1C), indicating that apoA-IV has fewer exposed hydrophobic surfaces30. Thioflavin T (ThT) fluorescence demonstrated that full-length apoA-IV did not exhibit a fibril-forming propensity under our experimental conditions (Supplementary Fig. S1D).
Because an N-terminal 70-residue fragment of apoA-IV has been reported to be deposited as fibrils24, we performed a sequence-based analysis to predict the local solubility and amyloid aggregation propensity of the N-terminal 1‒70 residues of human apoA-IV using CamSol31 and AmylPred232 (Fig. 1A). The results revealed that there are two major aggregation-prone segments, residues 7−16 (VATVMWDYFS) and 38−42 (LNALF), that are predicted to have low solubility and high amyloid propensity. In keeping with this, a previous bioinformatics analysis using AmylPred2 and PASTA2.033 indicated that there are two amyloidogenic segments within the N-terminal residues 1−96: one major segment consisting of residues 7−16 and the other consisting of residues 40−4121.

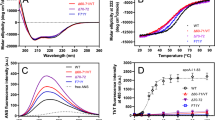
Amyloid-forming property of the N-terminal 1‒70 residues of human apoA-IV. (A) Amyloid propensity and solubility prediction profiles of apoA-IV 1‒70. The solubility score was obtained by the CamSol method31,81. Amyloid propensity prediction was generated using the consensus algorithm AmylPred232. The hydropathy was calculated as described by Kyte and Doolittle82 using a sliding window of nine residues. The predicted aggregation-prone and low-solubility regions 7VATVMWDYFS16 and 38LNALF42) are highlighted in gray. (B) Kinetics of the formation of amyloid fibrils monitored by ThT fluorescence of the apoA-IV 1‒70 fragment with increasing initial monomer concentrations. a. u., arbitrary units. ApoA-IV solution in 10 mM Tris buffer (150 mM NaCl, 0.02% NaN3) was incubated at pH 7.1, 37 ºC in the presence of 10 µM ThT. (C) Effects of the initial monomer concentration of apoA-IV 1‒70 on the lag time (left) and the apparent rate constant for the growth of fibrils (right). (D) Far-UV CD spectra of apoA-IV 1‒70 in the monomer (before incubation, dashed line) and fibrillar (after 24 h incubation, solid line) forms pelleted by centrifugation at 80,000 g for 30 min. The protein concentration was 50 µg/ml. (E) TEM and TIRFM images of apoA-IV 1‒70 after 24 h incubation. The scale bars represent 200 nm (TEM) and 10 μm (TIRFM), respectively. (F) Fibril formation kinetics of apoA-IV 1‒70 (○) and its Δ7‒16 (∆) and Δ38‒42 (∇) deletion variants. ApoA-IV solution in 10 mM Tris buffer (150 mM NaCl, 0.02% NaN3) was incubated at pH 7.1, 37 ºC in the presence of 10 µM ThT. (G) Far-UV CD spectra of apoA-IV 1‒70 Δ7‒16 and Δ38‒42 before (0 h, dashed line) and after incubation (168 h, solid line).
We hence assessed the fibril-forming propensity of the apoA-IV 1‒70 fragment. The ThT fluorescence assay showed that the apoA-IV 1‒70 fragment resulted in a large increase in ThT fluorescence intensity in a time-dependent manner, depending on the initial protein concentration (Fig. 1B). As shown in Fig. 1C, the lag time decreased with increasing protein concentration, whereas the apparent rate constant for fibril growth remained largely unchanged at all concentrations tested. This suggests that saturation of the fibril growth step occurs at high protein concentrations34,35. We note that application of AmyloFit analysis (https://www.amylofit.ch.cam.ac.uk)36 to the kinetic data shown in Fig. 1B indicated that saturating elongation and fragmentation is plausible model for the fibril formation process of apoA-IV 1‒70 (Supplementary Fig. S2).
To determine the secondary structural changes in apoA-IV 1‒70 fragment during incubation, CD spectra were collected (Fig. 1D). While a monomeric apoA-IV 1‒70 fragment predominantly existed as a random coil structure before incubation, it exhibited a single minimum spectrum around 216 nm after incubation, implying conversion to a β-sheet-rich structure. Transmission electron microscopy (TEM) and total internal reflection fluorescence microscopy (TIRFM) demonstrated that the apoA-IV 1‒70 fragment formed ThT reactive, thin and straight fibrils after incubation (Fig. 1E). In addition, deletion of the major aggregation-prone segments of residues 7−16 or 38−42 almost completely inhibited the increase in ThT fluorescence (Fig. 1F) and conversion of the CD spectra (Fig. 1G) after incubation, indicating that both residues are critical for the β-transition and fibril formation of the apoA-IV 1‒70 fragment.
Effects of seed fibrils and temperature on the fibril-forming kinetics of apoA-IV 1‒70
To understand the molecular mechanism underlying fibril formation by apoA-IV 1‒70, we examined the effect of preformed seeds on the fibril formation kinetics. Addition of preformed fibrils greatly accelerated fibril formation by apoA-IV 1‒70 (Fig. 2A), indicating bypassing of the conversion of soluble proteins into amyloid nuclei37,38. As shown in Fig. 2B, an increase in the seed concentration gradually reduced the lag time without significantly affecting the apparent rate constant of fibril growth, suggesting that primary nucleation is the predominant process during the lag phase of fibril formation by apoA-IV 1‒7039.

Effects of the seed and temperature on the aggregation kinetics of the apoA-IV 1‒70 fragment. (A) ThT fluorescence kinetics for fibril formation by apoA-IV 1‒70 in the presence of various concentrations of seed. ApoA-IV monomer solution (100 µg/ml) in 10 mM Tris buffer (150 mM NaCl, 0.02% NaN3) was incubated at pH 7.1, 37 ºC in the presence of 10 µM ThT. a. u., arbitrary units. (B) Effects of seed concentration on the lag time (left) and apparent rate constant for the growth of fibrils (right) of apoA-IV 1‒70. (C) ThT fluorescence kinetics for fibril formation by apoA-IV 1‒70 at different temperatures. The solid lines are the fitted curves by the Finke‒Watzky equation. ApoA-IV solution (100 µg/ml) in 10 mM Tris buffer (150 mM NaCl, 0.02% NaN3, pH 7.1 at 37 ºC) was incubated in the presence of 10 µM ThT. a.u., arbitrary units. (D) Comparison of the rate constants of nucleation (k1) and fibril growth (k2) for the fibril formation by apoA-IV 1‒70. (E) Eyring plots of the rate constants of k1 and k2 for the amyloid fibril formation by apoA-IV 1‒70.
We further conducted a thermodynamic analysis of the fibril formation characteristics of apoA-IV 1‒70 by monitoring ThT fluorescence kinetics at different temperatures38,40,41. Figure 2C shows the time course of the ThT fluorescence change with the fitted curves by the Finke‒Watzky equation for a temperature range of 27‒42 ºC. We note that although the ThT fluorescence intensity and pH of Tris buffer slightly decreased with increasing temperature, such slight changes in the buffer pH did not affect the aggregation kinetics of apoA-IV 1‒70 (Supplementary Fig. S3). Comparison of the obtained rate constants for nucleation (k1) and fibril growth (k2) at each temperature indicated that the rate constant of fibril growth greatly increased with temperature, whereas the rate constant of nucleation remained almost unchanged (Fig. 2D). The linear plots based on the Eyring equation for each rate constants of k1 and k2 (Fig. 2E) provide the activation enthalpy (ΔH*) and entropy (ΔS*) for the nucleation and fibril growth steps in fibril formation by apoA-IV 1–70. Table 1 summarizes the ΔH* and ΔS* values together with the activation Gibbs free energy ΔG*. Interestingly, the activation enthalpy for nucleation was almost negligible, i.e., the free energy barrier for nucleation in fibril formation by apoA-IV 1–70 is entirely entropic. In contrast, the fibril growth step was enthalpically and entropically unfavorable, indicating that the free energy barrier for fibril growth consists of both enthalpic and entropic barriers, as reported for other amyloidogenic proteins38,40,41,42. These results suggest that apoA-IV 1–70 exhibits distinct thermodynamic characteristics in the nucleation step of fibril formation.
Effect of heparin on the fibril-forming properties of apoA-IV 1‒70
GAGs are involved in the formation of amyloid deposits in a variety of human diseases43,44. In particular, heparan sulfate and its highly sulfated derivative heparin accelerate the formation of amyloid fibrils in various amyloidogenic proteins45,46. Therefore, we investigated the effect of heparin on the fibril-forming behavior of the apoA-IV 1‒70 fragment.
The ThT fluorescence kinetic assay demonstrated that heparin significantly accelerated the rate of apoA-IV 1‒70 fibril formation with marked increases in the ThT fluorescence signal (Fig. 3A), whereby the lag time tended to decrease and the apparent rate constant for fibril growth increased with increasing heparin concentration (Fig. 3B). Quantification of the fibril fraction of apoA-IV 1‒70 at the end of incubation revealed that the presence of heparin significantly increased the fibril yields after incubation (Fig. 3C). As shown in Fig. 3D, a transition in the CD spectrum was observed for apoA-IV 1‒70 after incubation in the presence of heparin, similar to fibrils formed without heparin (Fig. 1D). Indeed, attenuated total reflection-Fourier transform infrared (ATR-FTIR) spectra of apoA-IV 1‒70 fibrils formed in the absence and presence of heparin exhibited similar spectral conversion from the band at approximately 1660 cm[–1 to that at approximately 1625 cm[–1, indicating the transition of random coil to β-sheet-rich structure (Fig. 3E). In addition, the apoA-IV 1‒70 fibrils formed in the absence and presence of heparin exhibited similar stabilities against urea-induced disaggregation monitored by ThT dissociation from fibrils (Fig. 3F). However, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay demonstrated that the presence of heparin increased cell toxicity induced by apoA-IV 1‒70 fibrils in HEK293 cells (Fig. 3G), despite the similar secondary structures and stabilities of the fibrils examined.

Effect of heparin on the fibril-forming property of the apoA-IV 1‒70 fragment and the structure, stability, and cytotoxicity of the formed fibrils. (A) ThT fluorescence kinetics for fibril formation by apoA-IV 1‒70 in the presence of various concentrations of heparin. ApoA-IV solution (100 µg/ml) in 10 mM Tris buffer (150 mM NaCl, 0.02% NaN3) was incubated at pH 7.1, 37 ºC in the presence of 10 µM ThT. (B) Effects of heparin concentration on the lag time (left) and apparent rate constant for the growth of fibrils (right) of apoA-IV 1‒70. (C) Quantification of the fibril fraction of apoA-IV 1‒70 after incubation in the absence or presence of heparin. (D) Far-UV CD spectra of apoA-IV 1‒70 in the monomer (before incubation, dotted line) and fibrillar (after 24 h incubation, solid line) forms in the presence of heparin. Fibrillar forms were pelleted by centrifugation at 80,000 g for 30 min. The protein concentration was 50 µg/ml. (E) ATR-FTIR spectra of apoA-IV 1‒70 before (dotted line) and after (solid lines) incubation in the absence or presence of heparin. a. u., arbitrary units. (F) Urea-induced disaggregation of apoA-IV 1‒70 fibrils. Changes in the ThT fluorescence spectra of the fibrils formed in the presence of heparin at a weight ratio of 1/10 (heparin/protein) with increasing concentrations of urea (left). The protein concentration was 100 µg/ml. a.u., arbitrary units. Comparison of the urea-induced disaggregation curves for apoA-IV 1‒70 fibrils formed in the absence and presence of heparin (right). (G) Effect of heparin on the cytotoxicity of apoA-IV 1‒70 fibrils. ApoA-IV 1‒70 monomer (before incubation) or fibrils (after 24 h incubation) formed in the absence or presence of heparin were added to the medium of HEK293 cells at a final concentration of 1 µM of protein. Heparin was added to apoA-IV 1‒70 at a weight ratio of 1/10 (heparin/protein) before [“(1‒70 + heparin) fibril”] or after (“1‒70 fibril + heparin”) incubation. *, p < 0.05; ***, p < 0.001 versus “1‒70 fibril”.
To identify possible heparin-binding site(s) within the N-terminal 1‒70 residues of apoA-IV, we used the ClusPro server (https://cluspro.org)47,48 to construct a heparin docking model of the apoA-IV 1‒70 fragment. The resultant docking model demonstrated that heparin can bind to residues K58–K59 in apoA-IV 1–70, with the largest cluster size of 885 and the lowest score of − 430.7 (Fig. 4A), suggesting that the positively charged residues K58 and K59 are most likely involved in the heparin binding of apoA-IV 1‒70. This is supported by the finding that the K58E substitution altered the predicted heparin-binding site from E58–K59 to residues N22–K24, with a lower cluster size of 425 and an increased score of − 112.8 (Fig. 4B).

Interaction of the apoA-IV 1‒70 fragment with heparin. (A and B) Docking model simulations of binding of apoA-IV 1‒70 (A) and the apoA-IV 1‒70/K58E variant (B) to a heparin tetrasaccharide structure generated by the ClusPro web server47. (C and D) BLI sensorgrams for the binding of apoA-IV 1‒70 (C) and the 1‒70/K58E variant (D) to immobilized heparin on a streptavidin sensor chip. The dotted lines represent globally fitted curves using a 1:1 Langmuir binding model. (E) Quantification of the fibril fraction of apoA-IV 1‒70/K58E variant after 120 h incubation in the absence or presence of heparin.
To further evaluate the association and dissociation kinetics of the interactions of apoA-IV 1‒70 variants with heparin, we performed bio-layer interferometry (BLI) analyses using biotinylated heparin immobilized on streptavidin sensor chips. As shown in Fig. 4C, D; Table 2, global fitting to the sensorgram curves with the Langmuir 1:1 binding model demonstrated that apoA-IV 1‒70 binds to heparin with a submicromolar KD value. Moreover, the K58E mutation significantly decreased the heparin binding of apoA-IV 1‒70, with drastically reduced association and dissociation rate constants. In addition, the yield of fibrils formed by the K58E variant was similar, irrespective of the presence of heparin (Fig. 4E), in contrast to apoA-IV 1‒70 (Fig. 3C). These results suggest that the positively charged region around residues K58 and K59 is involved in interaction with heparin, thereby promoting fibril formation by the apoA-IV 1‒70 fragment.
Discussion
Exchangeable apolipoproteins are highly susceptible to misfolding and aggregation into amyloid fibrils, which may be due to their partially folded, labile conformation in the lipid-free form21,49. Amyloidosis associated with apoA-I is a well-recognized phenomenon and involves amyloid fibril deposition of the N-terminal 80−100 amino acid-residue fragments of mutated apoA-I50,51 or oxidized forms of wild-type apoA-I52,53. Indeed, we and others have demonstrated that the N-terminal fragment of apoA-I has a strong propensity to form amyloid fibrils in vitro54,55,56. Similarly, the N-terminal 70-residue fragment of apoA-IV has been reported to be deposited as an amyloid fibril in apoA-IV-associated amyloidosis24. However, the amyloid-forming propensity of the apoA-IV N-terminal fragment has not been characterized to date.
The present study demonstrates that the apoA-IV 1‒70 fragment indeed has a strong ability to form amyloid fibrils concomitant with transition to β-structure in vitro (Fig. 1). Sequence-based predictions of local solubility and amyloid propensity indicate that there are two major aggregation-prone segments comprising residues 7‒16 (VATVMWDYFS) and 38−42 (LNALF) in the N-terminal 1‒70 amino acid residues of apoA-IV (Fig. 1A). Consistent with this, deletion of these residues greatly inhibited the β-transition and fibril formation of apoA-IV 1‒70 (Fig. 1F, G). Seeded aggregation kinetics experiments (Fig. 2A, B) suggest that primary nucleation is the dominant process during the lag phase of fibril formation by apoA-IV 1‒70. The relatively hydrophilic nature of the N-terminal 1‒70 residues of apoA-IV57 may favor primary nucleation over surface-catalyzed secondary nucleation58,59.
Thermodynamic analysis of the fibril formation kinetics provided further molecular insights into the fibril formation process of apoA-IV 1‒70. Despite the similar values of activation Gibbs free energy barriers, the contributions of activation enthalpy and entropy to the free energy barrier are quite different for the nucleation and fibril growth processes: the free energy barrier for nucleation is entirely entropic, whereas that for fibril growth consists of both enthalpic and entropic barriers (Table 1). Because the unfavorable enthalpy barrier for nucleation is thought to stem from breakage of molecular interactions and desolvation of hydrophobic regions of the protein molecule in the transition state41,60, the negligible activation enthalpy of nucleation may indicate weak molecular interactions and less hydrophobic exposure in the monomeric state of apoA-IV 1‒70. In this regard, the large unfavorable activation entropy for nucleation likely originates from the small contribution of desolvation to the activation entropy in the nucleation process of apoA-IV 1‒70. In the fibril growth process, unfavorable activation enthalpy and entropy were observed, whereby the activation entropy largely contributed to the free energy barrier. This may be explained by the possibility that resolvation of the fibril surface dominates desolvation of the existing nucleus during fibril growth40,41.
It is well known that heparan sulfate and its highly sulfated derivative heparin are involved in the formation of amyloid fibrils by many amyloidogenic proteins including amyloid β61, serum amyloid A62, transthyretin63, α-synuclein64,65, and apoA-I66,67. The present results demonstrate that heparin significantly accelerated the rate of fibril formation by the apoA-IV 1‒70 fragment with increasing fibril yield (Fig. 3A‒C). In contrast, the presence of heparin did not affect the secondary structure or stability of the apoA-IV 1‒70 fibrils (Fig. 3D‒F). It is plausible that heparin provides templates for nuclear formation and fibril growth46,65,68, resulting in faster aggregation and fibril formation by apoA-IV 1‒70. However, it is unlikely that incorporation of heparin into the apoA-IV 1‒70 fibrils occurs, in contrast to the case of α-synuclein, for which heparin causes the structural rearrangement of α-synuclein fibril assembly64,69. In this regard, the increased cytotoxicity of fibrils in the presence of heparin (Fig. 3G) may be due to the increased amount of apoA-IV 1‒70 fibrils induced by heparin. We note that the prefibrillar state of apoA-IV 1‒70 after incubation for 6 h had no effect on the cell growth, in contrast to the strong toxicity of fibrillar state after 24 h incubation (Supplementary Fig. S4). These results suggest that the formation of fibril structure is essential for the cytotoxicity of apoA-IV 1‒70 as observed for the N-terminal 1‒83 fragment of apoA-I35,40.
Heparin-binding sites on various proteins contain clusters of basic amino acid residues capable of binding to negatively charged heparin70. Molecular docking simulations revealed that heparin predominantly bound to the positively charged K58 and K59 residues of the apoA-IV 1‒70 fragment (Fig. 4A, B). In support of this, the K58E mutation in apoA-IV 1‒70 greatly reduced the association and dissociation rate constants for binding to heparin (Fig. 4C, D) and diminished the enhancing effect of heparin on the yield of formed fibrils (Fig. 4E). Further studies are needed for more extensive characterization of the interaction between apoA-IV and heparin. Regarding lipid interactions, we note that α-helix formation upon lipid binding completely inhibited fibril formation by the apoA-IV 1−70 fragment (Supplementary Fig. S5) as previously reported for the N-terminal fragment of apoA-I71. This suggests that biological membranes may not be involved in amyloid fibril formation induced by apoA-IV.
In conclusion, we demonstrated for the first time that the N-terminal 1‒70 amino acid residue fragment of apoA-IV has a strong propensity to form amyloid fibrils and that two major aggregation-prone residues 7−16 and 38−42 play critical roles in the β-transition and fibril formation. In addition, the presence of heparin significantly accelerated the rate of apoA-IV 1‒70 fibril formation with increasing the yield of fibrils, whereby the positively charged residues K58 and K59 are the predominant site for heparin interaction. These results explain why cardiac apoA-IV amyloid fibrils consist of an N-terminal 70-residue fragment and may support the hypothesis that the proteolytic cleavage of full-length apoA-IV occurs before aggregation, leading to the release of the N-terminal fragment to form amyloid deposits. Thus, our findings provide novel insights into the molecular mechanisms underlying amyloid deposition in apoA-IV-associated amyloidosis.
Materials and methods
Preparation of recombinant apoA-IV proteins
Full-length and the N-terminal 1−70 fragments of apoA-IV variants were expressed in E. coli as fusion proteins with a thioredoxin and His-tag at the N-terminus and were then isolated by Ni-affinity chromatography as previously described72,73. The fused tags were cleaved using HRV 3 C protease (Japan Lamb, Hiroshima, Japan), which produced apoA-IV proteins with two extra N-terminal amino acids, Gly-Pro (Supplementary Fig. S6A). The apoA-IV preparations were at least 95% pure, as assessed by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (Supplementary Fig. S6B). All apoA-IV proteins were solubilized in a 6 M guanidine hydrochloride solution, which was dialyzed into the appropriate buffer before use.
CD spectroscopy
Far-UV CD spectra were recorded at wavelengths of 190–260 nm at 25 °C using a Jasco J-1500 spectropolarimeter (JASCO, Tokyo, Japan), as described40. The α-helix content was derived from the molar ellipticity at 222 nm ([θ]222) using the equation: % α-helix = [(–[θ]222 + 3000)/(36000 + 3000)] × 10074. The thermal denaturation was monitored from the change in molar ellipticity at 222 nm over the temperature range 20–90 °C. The cooperativity index, n, describing the sigmoidicity of the thermal denaturation curve, and van’t Hoff enthalpy of denaturation, ΔHv, were calculated as previously described75.
ATR-FTIR spectrometry
ATR-FTIR spectra were recorded on a Jasco FTIR spectrometer FT/IR-4700 equipped with an ATR PKM-Ge-L reflectance accessory, as described56. An aliquot of the apoA-IV samples (100 µg) in Tris buffer (pH 7.4) was spread on a germanium waveguide and dried under flowing nitrogen gas. The ATR-FTIR spectra were obtained in the wavenumber range of 1,000–3,500 cm‒1 at a resolution of 4 cm‒1 with 256 accumulations under continuous nitrogen purging.
Fluorescence measurements
Fluorescence measurements were carried out using an F-7000 fluorescence spectrophotometer (Hitachi High-Technologies, Tokyo, Japan) and an infinite 200 PRO plate reader (TECAN) at 25 °C. To assess the exposure of hydrophobic sites on the protein, ANS fluorescence spectra were measured from 400 to 600 nm at an excitation wavelength of 395 nm in the presence of 50 µg/ml protein and an excess of ANS (250 µM), as described75. To monitor the kinetics of amyloid fibril formation, apoA-IV solution in 10 mM Tris buffer (150 mM NaCl, 0.02% NaN3, pH 7.1 at 37 °C) were placed into multiple wells of a 96-well black polystyrene plate (250 µl/well) with Teflon beads and incubated at 37 ºC on a microplate shaker with shaking at 150 rpm in the presence of 10 µM ThT. To measure the fluorescence intensity of ThT, the aggregation reaction plate was manually transferred to the reader from the incubator. Each experiment was repeated at least twice with 5 replicates per plate. Time-dependent ThT fluorescence increases were fitted to the sigmoidal equation as follows40,76:
where F is the fluorescence intensity and F0 and Fmax are the initial and maximum baselines at the lag and plateau phases, respectively. kapp is the apparent rate constant for fibril growth and tm is the time at 50% of the maximal fluorescence. The lag time is provided as tm − 2/k.
ThT fluorescence data were analyzed using the Finke‒Watzky two-step model of nucleation followed by autocatalytic growth77,78:
where [A]0 is the initial monomer protein concentration, and k1 and k2 are the rate constants for nucleation and fibril growth, respectively. The thermodynamic parameters for nucleation and fibril growth were given by the Eyring equation:
where kB and h are the Boltzmann and the Planck constants, respectively. The slope and y-intercept of the linear plot according to Eq. 3 provide the activation enthalpy (ΔH*) and entropy (ΔS*), respectively. The activation Gibbs free energy (ΔG*) was calculated from ΔH* and ΔS* according to ∆G* = ∆H* – T∆S*.
For chemical denaturation experiments, apoA-IV 1‒70 fibrils (100 µg/ml) in 10 mM Tris buffer (150 mM NaCl, 0.02% NaN3, pH 7.1 at 37 °C) were incubated overnight at 4 °C with urea at various concentrations in the presence of 10 µM ThT. Disaggregation of fibrils was monitored from the change in ThT fluorescence intensity caused by dissociation of ThT from fibrils.
TEM and TIRFM
TEM and TIRFM were carried out as previously described38,79. Briefly, TEM measurements were performed for the samples negatively stained with a phosphomolybdic acid solution, using a JEOL JEM-1200EX transmission microscope (JEOL, Tokyo, Japan) at an acceleration voltage of 80 kV. For TIRFM, ThT fluorescence images were obtained using an inverted microscope (IX70; Olympus) with exciting the ThT by an argon laser. After cleaning the signals using a band-pass filter, the TIRFM images were visualized using an SIT camera equipped with an image intensifier.
BLI measurements
The kinetics of the binding of apoA-IV 1‒70 variants to heparin were measured using a BLItz system (Sartorius AG, Germany) as described previously80. Porcine intestinal heparin (average molecular weight: 13,000 Da, Celsus Laboratories) was biotinylated and immobilized on a streptavidin biosensor chip. The sensor was dipped into an apoA-IV solution to monitor the binding of apoA-IV to the sensor, and the sensor was then dipped into a buffer to monitor its dissociation. The binding and dissociation rate constants, ka and kd, were determined by global fitting of the obtained kinetic curves to the 1:1 Langmuir binding model using BIAevaluation software (Cytiva, Marlborough USA). The dissociation constant for heparin binding, Kd, was calculated as Kd = kd/ka.
Fibril pelleting assay
To quantify the amount of fibril in the apoA-IV 1‒70 preparations after incubation, the samples were centrifuged at 80,000 g for 30 min to separate the soluble (non-fibril) and fibril fractions. The supernatant (non-fibril fraction) and pellet (fibril fraction) were dissolved in 4 M urea, and the protein content of each fraction was determined by the Lowry method using apoA-IV solutions in 4 M urea as the standard.
Cytotoxicity assay
The cytotoxicity of apoA-IV 1‒70 aggregates against HEK293 cells was measured by a MTT assay, as previously described56. Briefly, HEK293 cells were plated and grown in poly-L-lysine-coated 24-well plates in DMEM containing 2% Fetal Bovine Serum for 24 h, after which they were cultured in the presence of apoA-IV 1‒70 aggregates in 10 mM phosphate-buffered saline (pH 7.4) for 24 h. Determination of cell viability was performed by optical measurements of the reduction of MTT to formazan by living cells.
Statistical analysis
The data were analyzed by one-way ANOVA with Dunnett’s test or Tukey’s multiple comparison test. Results were considered significant at p < 0.05.
Data availability
All the data analyzed in this study are included in the article and the supplementary information.
Abbreviations
- apoA-I:
-
apolipoprotein A-I
- apoA-IV:
-
apolipoprotein A-IV
- ATR-FTIR:
-
attenuated total reflection-Fourier transform infrared spectroscopy
- BLI:
-
bio-layer interferometry
- CD:
-
circular dichroism
- GAG:
-
glycosaminoglycan
- HDL:
-
high-density lipoprotein
- MTT:
-
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
- TEM:
-
transmission electron microscopy
- ThT:
-
thioflavin T
- TIRFM:
-
total internal reflection fluorescence microscopy
References
Segrest, J. P. et al. The amphipathic helix in the exchangeable apolipoproteins: A review of secondary structure and function. J. Lipid Res. 33, 141–166 (1992).
Saito, H., Lund-Katz, S. & Phillips, M. C. Contributions of domain structure and lipid interaction to the functionality of exchangeable human apolipoproteins. Prog Lipid Res. 43, 350–380 (2004).
Pearson, K. et al. Structure of human Apolipoprotein A-IV: A distinct domain architecture among exchangeable apolipoproteins with potential functional implications. Biochemistry 43, 10719–10729 (2004).
Wang, F. et al. Apolipoprotein A-IV: A protein intimately involved in metabolism. J. Lipid Res. 56, 1403–1418 (2015).
Weinberg, R. B. & Spector, M. S. Human Apolipoprotein A-IV: Displacement from the surface of triglyceride-rich particles by HDL2-associated C-apoproteins. J. Lipid Res. 26, 26–37 (1985).
Goldberg, I. J., Scheraldi, C. A., Yacoub, L. K., Saxena, U. & Bisgaier, C. L. Lipoprotein ApoC-II activation of lipoprotein lipase. Modulation by Apolipoprotein A-IV. J. Biol. Chem. 265, 4266–4272 (1990).
Kohan, A. B. et al. Apolipoprotein A-IV regulates chylomicron metabolism-mechanism and function. Am. J. Physiol. Gastrointest. Liver Physiol. 302, G628–636 (2012).
Duka, A. et al. ApoA-IV promotes the biogenesis of apoA-IV-containing HDL particles with the participation of ABCA1 and LCAT. J. Lipid Res. 54, 107–115 (2013).
Qin, X., Swertfeger, D. K., Zheng, S., Hui, D. Y. & Tso, P. Apolipoprotein AIV: A potent endogenous inhibitor of lipid oxidation. Am. J. Physiol. 274, H1836–1840 (1998).
Ostos, M. A. et al. Antioxidative and antiatherosclerotic effects of human Apolipoprotein A-IV in Apolipoprotein E-deficient mice. Arterioscler. Thromb. Vasc Biol. 21, 1023–1028 (2001).
Xu, X. R. et al. Apolipoprotein A-IV binds alphaIIbbeta3 integrin and inhibits thrombosis. Nat. Commun. 9, 3608 (2018).
Wang, F. et al. Apolipoprotein A-IV improves glucose homeostasis by enhancing insulin secretion. Proc. Natl. Acad. Sci. U S A. 109, 9641–9646 (2012).
Cui, Y., Huang, M., He, Y., Zhang, S. & Luo, Y. Genetic ablation of Apolipoprotein A-IV accelerates Alzheimer’s disease pathogenesis in a mouse model. Am. J. Pathol. 178, 1298–1308 (2011).
Tubb, M. R., Silva, R. A. G. D., Fang, J., Tso, P. & Davidson, W. S. A Three-dimensional homology model of Lipid-free Apolipoprotein A-IV using Cross-linking and mass spectrometry. J. Biol. Chem. 283, 17314–17323 (2008).
Walker, R. G. et al. The structure of human Apolipoprotein A-IV as revealed by stable isotope-assisted cross-linking, molecular dynamics, and small angle x-ray scattering. J. Biol. Chem. 289, 5596–5608 (2014).
Pearson, K. et al. Specific sequences in the N and C termini of Apolipoprotein A-IV modulate its conformation and lipid association. J. Biol. Chem. 280, 38576–38582 (2005).
Tubb, M. R. et al. Modulation of Apolipoprotein A-IV lipid binding by an interaction between the N and C termini. J. Biol. Chem. 282, 28385–28394 (2007).
Deng, X. et al. Small-angle X-ray scattering of Apolipoprotein A-IV reveals the importance of its termini for structural stability. J. Biol. Chem. 288, 4854–4866 (2013).
Deng, X. et al. The structure of dimeric Apolipoprotein A-IV and its mechanism of self-association. Structure 20, 767–779 (2012).
Deng, X., Walker, R. G., Morris, J., Davidson, W. S. & Thompson, T. B. Role of conserved proline residues in human Apolipoprotein A-IV structure and function. J. Biol. Chem. 290, 10689–10702 (2015).
Das, M. & Gursky, O. Amyloid-Forming properties of human apolipoproteins: Sequence analyses and structural insights. Adv. Exp. Med. Biol. 855, 175–211 (2015).
Jeraj, N., Hegele, R. A. & Berberich, A. J. Apolipoprotein genetic variants and hereditary amyloidosis. Curr. Opin. Lipidol. 32, 132–140 (2021).
Bergstrom, J. et al. Codeposition of Apolipoprotein A-IV and transthyretin in senile systemic (ATTR) amyloidosis. Biochem. Biophys. Res. Commun. 285, 903–908 (2001).
Bergstrom, J. et al. Two different types of amyloid deposits–apolipoprotein A-IV and transthyretin–in a patient with systemic amyloidosis. Lab. Invest. 84, 981–988 (2004).
Martins, E. et al. Cardiac amyloidosis associated with Apolipoprotein A-IV deposition diagnosed by mass Spectrometry-Based proteomic analysis. Eur. J. Case Rep. Intern. Med. 6, 001237 (2019).
Dasari, S. et al. Clinical, biopsy, and mass spectrometry characteristics of renal Apolipoprotein A-IV amyloidosis. Kidney Int. 90, 658–664 (2016).
Sethi, S. et al. Medullary amyloidosis associated with Apolipoprotein A-IV deposition. Kidney Int. 81, 201–206 (2012).
Gottwald, J. & Rocken, C. The amyloid proteome: A systematic review and proposal of a protein classification system. Crit. Rev. Biochem. Mol. Biol. 56, 526–542 (2021).
Misumi, Y. et al. Binding of serum-derived amyloid-associated proteins to amyloid fibrils. Amyloid 30, 67–73 (2023).
Stryer, L. The interaction of a naphthalene dye with apomyoglobin and apohemoglobin. A fluorescent probe of non-polar binding sites. J. Mol. Biol. 13, 482–495 (1965).
Sormanni, P., Aprile, F. A. & Vendruscolo, M. The camsol method of rational design of protein mutants with enhanced solubility. J. Mol. Biol. 427, 478–490 (2015).
Tsolis, A. C., Papandreou, N. C., Iconomidou, V. A. & Hamodrakas, S. J. A consensus method for the prediction of ‘aggregation-prone’ peptides in globular proteins. PLoS One. 8, e54175 (2013).
Walsh, I., Seno, F., Tosatto, S. C. & Trovato, A. PASTA 2.0: an improved server for protein aggregation prediction. Nucleic Acids Res. 42, W301–307 (2014).
Meisl, G. et al. Scaling behaviour and rate-determining steps in filamentous self-assembly. Chem. Sci. 8, 7087–7097 (2017).
Namba, N. et al. Amyloidogenic 60–71 deletion/valthr insertion mutation of Apolipoprotein A-I generates a new aggregation-prone segment that promotes nucleation through entropic effects. Sci. Rep. 13, 18514 (2023).
Meisl, G. et al. Molecular mechanisms of protein aggregation from global fitting of kinetic models. Nat. Protoc. 11, 252–272 (2016).
Cohen, S. I. et al. Proliferation of amyloid-beta42 aggregates occurs through a secondary nucleation mechanism. Proc. Natl. Acad. Sci. U. S. A. 110, 9758–9763 (2013).
Ohgita, T., Namba, N., Kono, H., Shimanouchi, T. & Saito, H. Mechanisms of enhanced aggregation and fibril formation of Parkinson’s disease-related variants of α-synuclein. Sci. Rep. 12 (2022).
Meisl, G. et al. Uncovering the universality of self-replication in protein aggregation and its link to disease. Sci. Adv. 8, eabn6831 (2022).
Mizuguchi, C. et al. Mechanisms of aggregation and fibril formation of the amyloidogenic N-terminal fragment of Apolipoprotein A-I. J. Biol. Chem. 294, 13515–13524 (2019).
Cohen, S. I. A. et al. Distinct thermodynamic signatures of oligomer generation in the aggregation of the amyloid-beta peptide. Nat. Chem. 10, 523–531 (2018).
Morris, A. M. & Finke, R. G. Alpha-synuclein aggregation variable temperature and variable pH kinetic data: a re-analysis using the Finke-Watzky 2-step model of nucleation and autocatalytic growth. Biophys. Chem. 140, 9–15 (2009).
Bellotti, V. & Chiti, F. Amyloidogenesis in its biological environment: challenging a fundamental issue in protein misfolding diseases. Curr. Opin. Struct. Biol. 18, 771–779 (2008).
Iannuzzi, C., Irace, G. & Sirangelo, I. The effect of glycosaminoglycans (GAGs) on amyloid aggregation and toxicity. Molecules 20, 2510–2528 (2015).
Nishitsuji, K. & Uchimura, K. Sulfated glycosaminoglycans in protein aggregation diseases. Glycoconj. J. 34, 453–466 (2017).
Maiza, A. et al. The role of Heparan sulfates in protein aggregation and their potential impact on neurodegeneration. FEBS Lett. 592, 3806–3818 (2018).
Mottarella, S. E. et al. Docking server for the identification of heparin binding sites on proteins. J. Chem. Inf. Model. 54, 2068–2078 (2014).
Kozakov, D. et al. The cluspro web server for protein-protein Docking. Nat. Protoc. 12, 255–278 (2017).
Hatters, D. M. & Howlett, G. J. The structural basis for amyloid formation by plasma apolipoproteins: a review. Eur. Biophys. J. 31, 2–8 (2002).
Rowczenio, D. et al. Amyloidogenicity and clinical phenotype associated with five novel mutations in Apolipoprotein A-I. Am. J. Pathol. 179, 1978–1987 (2011).
Obici, L. et al. Structure, function and amyloidogenic propensity of Apolipoprotein A-I. Amyloid 13, 191–205 (2006).
Wong, Y. Q., Binger, K. J., Howlett, G. J. & Griffin, M. D. Methionine oxidation induces amyloid fibril formation by full-length Apolipoprotein A-I. Proc. Natl. Acad. Sci. U S A. 107, 1977–1982 (2010).
Chan, G. K. et al. Myeloperoxidase-mediated methionine oxidation promotes an amyloidogenic outcome for Apolipoprotein A-I. J. Biol. Chem. 290, 10958–10971 (2015).
Andreola, A. et al. Conformational switching and fibrillogenesis in the amyloidogenic fragment of Apolipoprotein A-I. J. Biol. Chem. 278, 2444–2451 (2003).
Raimondi, S. et al. Effects of the known pathogenic mutations on the aggregation pathway of the amyloidogenic peptide of Apolipoprotein A-I. J. Mol. Biol. 407, 465–476 (2011).
Adachi, E. et al. Dual role of an N-terminal amyloidogenic mutation in Apolipoprotein A-I: destabilization of helix bundle and enhancement of fibril formation. J. Biol. Chem. 288, 2848–2856 (2013).
Weinberg, R. B. Differences in the hydrophobic properties of discrete alpha-helical domains of rat and human Apolipoprotein A-IV. Biochim. Biophys. Acta. 918, 299–303 (1987).
Meisl, G. et al. Differences in nucleation behavior underlie the contrasting aggregation kinetics of the Abeta40 and Abeta42 peptides. Proc. Natl. Acad. Sci. U S A. 111, 9384–9389 (2014).
Thacker, D. et al. The role of fibril structure and surface hydrophobicity in secondary nucleation of amyloid fibrils. Proc. Natl. Acad. Sci. U S A. 117, 25272–25283 (2020).
Buell, A. K. et al. Detailed analysis of the energy barriers for amyloid fibril growth. Angew Chem. Int. Ed. Engl. 51, 5247–5251 (2012).
Valle-Delgado, J. J. et al. Modulation of Abeta42 fibrillogenesis by glycosaminoglycan structure. FASEB J. 24, 4250–4261 (2010).
Aguilera, J. J., Zhang, F., Beaudet, J. M., Linhardt, R. J. & Colon, W. Divergent effect of glycosaminoglycans on the in vitro aggregation of serum amyloid A. Biochimie 104, 70–80 (2014).
Noborn, F. et al. Heparan sulfate/heparin promotes transthyretin fibrillization through selective binding to a basic motif in the protein. Proc. Natl. Acad. Sci. U S A. 108, 5584–5589 (2011).
Tao, Y. et al. Heparin induces alpha-synuclein to form new fibril polymorphs with attenuated neuropathology. Nat. Commun. 13, 4226 (2022).
Mehra, S. et al. Glycosaminoglycans have variable effects on alpha-synuclein aggregation and differentially affect the activities of the resulting amyloid fibrils. J. Biol. Chem. 293, 12975–12991 (2018).
Mikawa, S. et al. Heparin promotes fibril formation by the N-terminal fragment of amyloidogenic Apolipoprotein A-I. FEBS Lett. 590, 3492–3500 (2016).
Townsend, D. et al. Heparin and methionine oxidation promote the formation of Apolipoprotein A-I amyloid comprising a-helical and b-sheet structures. Biochemistry 56, 1632–1644 (2017).
Solomon, J. P., Bourgault, S., Powers, E. T. & Kelly, J. W. Heparin binds 8 kda Gelsolin cross-beta-sheet oligomers and accelerates amyloidogenesis by hastening fibril extension. Biochemistry 50, 2486–2498 (2011).
Tao, Y. et al. Time-course remodeling and pathology intervention of alpha-synuclein amyloid fibril by heparin and heparin-like oligosaccharides. Nat. Struct. Mol. Biol. (2024).
Hileman, R. E., Fromm, J. R., Weiler, J. M. & Linhardt, R. J. Glycosaminoglycan-protein interactions: definition of consensus sites in glycosaminoglycan binding proteins. Bioessays 20, 156–167 (1998).
Mizuguchi, C. et al. Amyloidogenic mutation promotes fibril formation of the N-terminal Apolipoprotein A-I on lipid membranes. J. Biol. Chem. 290, 20947–20959 (2015).
Ohgita, T. et al. Novel conformation-selective monoclonal antibodies against apoA‐I amyloid fibrils. FEBS J. 288, 1496–1513 (2020).
Ohgita, T. et al. Intramolecular interaction kinetically regulates fibril formation by human and mouse α-synuclein. Sci. Rep. 13, 10885 (2023).
Tanaka, M. et al. Contributions of the N- and C-terminal helical segments to the lipid-free structure and lipid interaction of Apolipoprotein A-I. Biochemistry 45, 10351–10358 (2006).
Saito, H. et al. Domain structure and lipid interaction in human apolipoproteins A-I and E, a general model. J. Biol. Chem. 278, 23227–23232 (2003).
Nielsen, L. et al. Effect of environmental factors on the kinetics of insulin fibril formation: Elucidation of the molecular mechanism. Biochemistry 40, 6036–6046 (2001).
Morris, A. M., Watzky, M. A., Agar, J. N. & Finke, R. G. Fitting neurological protein aggregation kinetic data via a 2-step, Minimal/Ockham’s Razor model: the Finke-Watzky mechanism of nucleation followed by autocatalytic surface growth. Biochemistry 47, 2413–2427 (2008).
Bentea, L., Watzky, M. A. & Finke, R. G. Sigmoidal nucleation and growth curves across nature fit by the Finke–Watzky model of slow continuous nucleation and autocatalytic growth: explicit formulas for the lag and growth times plus other key insights. J. Phys. Chem. C. 121, 5302–5312 (2017).
Iwahashi, N. et al. Sulfated glycosaminoglycans mediate prion-like behavior of p53 aggregates. Proc. Natl. Acad. Sci. U S A. 117, 33225–33234 (2020).
Ohgita, T. et al. Generation of novel anti-apoE monoclonal antibodies that selectively recognize ApoE isoforms. FEBS Lett. 598, 902–914 (2024).
Sormanni, P. & Vendruscolo, M. Protein solubility predictions using the camsol method in the study of protein homeostasis. Cold Spring Harb Perspect. Biol. 11, a033845 (2019).
Kyte, J. & Doolittle, R. F. A simple method for displaying the hydropathic character of a protein. J. Mol. Biol. 157, 105–132 (1982).
Acknowledgements
This work was partially supported by JSPS KAKENHI (Grant Number, JP22H06556 (H.S.)) and a Nagai Memorial Research Scholarship from the Pharmaceutical Society of Japan (N.N.). The authors thank Dr. Kazuchika Nishitsuji (Wakayama Medical University) for valuable advice.
Author information
Authors and Affiliations
Contributions
N.N. and H.S. designed the study. T.S. recorded the TEM and TIRFM images. T.O. performed the BLI measurements. N.N., T.D., Y.K., and Y.N. performed all of the other experiments. N.N., T.O., and H.S. prepared the manuscript. All of the authors reviewed the results and approved the final version of the manuscript.
Corresponding author
Ethics declarations
Competing interest
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Namba, N., Danjo, T., Kitagawa, Y. et al. Amyloid-forming property of the N-terminal 1−70 residues of human apolipoprotein A-IV. Sci Rep 15, 13203 (2025). https://doi.org/10.1038/s41598-025-97992-3
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-97992-3
