
Abstract
As a newly discovered histone modification, abnormal lactation has been found to be present in and contribute to the development of various cancers. The aim of this study was to investigate the potential role between lactylation and the prognosis of breast cancer patients. Lactylation-associated subtypes were obtained by unsupervised consensus clustering analysis. Lactylation-related gene signature (LRS) was constructed by 15 machine learning algorithms, and the relationship between LRS and tumor microenvironment (TME) as well as drug sensitivity was analyzed. In addition, the expression of genes in the LRS in different cells was explored by single-cell analysis and spatial transcriptome. The expression levels of genes in LRS in clinical tissues were verified by RT-PCR. Finally, the potential small-molecule compounds were analyzed by CMap, and the molecular docking model of proteins and small-molecule compounds was constructed. LRS was composed of 6 key genes (SHCBP1, SIM2, VGF, GABRQ, SUSD3, and CLIC6). BC patients in the high LRS group had a poorer prognosis and had a TME that promoted tumor progression. Single-cell analysis and spatial transcriptome revealed differential expression of the key genes in different cells. The results of PCR showed that SHCBP1, SIM2, VGF, GABRQ, and SUSD3 were up-regulated in the cancer tissues, whereas CLIC6 was down-regulated in the cancer tissues. Arachidonyltrifluoromethane, AH-6809, W-13, and clofibrate can be used as potential target drugs for SHCBP1, VGF, GABRQ, and SUSD3, respectively. The gene signature we constructed can well predict the prognosis as well as the treatment response of BC patients. In addition, our predicted small-molecule complexes provide an important reference for personalized treatment of breast cancer patients.
Similar content being viewed by others

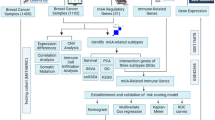

Introduction
Breast cancer remains the leading malignancy affecting women globally, with approximately 2.3 million new cases reported in 2022, underscoring its persistent clinical and societal burden1. Although advances in early detection and therapeutic strategies have reduced mortality rates2,3,4, precise prognostic biomarkers are still urgently needed to optimize personalized treatment and improve survival outcomes. The construction of prognostic signatures can more accurately predict the prognosis of patients and optimize treatment strategies, which in turn improves the quality of patient survival.
Epigenetic modifications (including DNA methylation, histone modification, and non-coding RNA regulation) can drive tumorigenesis by altering gene expression and have therefore become central to cancer research5. In breast cancer, these epigenetic alterations are associated with tumorigenesis, progression, and drug resistance6,7,8. Recently, a new form of histone modification, lactylation, has emerged as an exciting area of research. Lactic acid-induced lactylation modifies histone lysine residues and has been shown to directly affect gene transcription9. Unlike traditional histone modifications, lactylation has a unique role in regulating gene expression in response to metabolic changes, especially under hypoxic or glycolytic conditions10.
Recent studies have shown that lactic acid-induced lactylation modifications play an important role in promoting aspects of tumor development11. It has been found that lactylation levels are elevated in ocular melanoma and that patients with high histone lactylation levels have a poorer prognosis12. Another study found that increased lactylation of H3 histones promotes hepatocellular carcinoma (HCC) progression by promoting liver cancer stem cell (LCSC) proliferation13. The metabolism-dependent and disease-specific modulation pattern of this modification makes it a promising candidate biomarker, but its prognostic significance in breast cancer remains largely unknown.
In this study, we constructed a lactylation-associated gene signature containing six genes in different cohorts by multiple machine learning and analyzed its relationship with immune cell infiltration as well as drug sensitivity. In addition, we validated the expression of key genes by PCR and predicted drugs targeting these genes. These results provide new ideas for prognostic prediction and personalized treatment of breast cancer.
Results
Identification of lactylation-related subtypes
First, we obtained 98 genes that intersect LRGs with DEGs (Fig. 1A). Afterwards, 18 prognostic LRGs were obtained by one-way Cox analysis and Kaplan–Meier analysis (Fig. 1B). Figure 1C demonstrated the expression of the 18 prognostic LRGs in tumor tissues and paracancerous tissues. Based on the expression profiles of the 18 prognostic LRGs, we used an unsupervised consensus clustering algorithm to categorize the BC samples into two subtypes (Fig. 1D,E). The CDF curve showed the smoothest curve at K = 2 (Fig. 1F). Meanwhile, the PCA plot also showed a good differentiation between the two subtypes (Fig. 1G). The results of survival curves showed that BC patients with L1 subtype had a worse prognosis (Fig. 1H).

Identification of lactylation-related subtypes in BC. (A) Venn diagram showing intersecting genes of LRGs, TCGA-DEGs, GSE86166, METABRIC, and GSE96058. (B) Univariate Cox analysis of LRGs. (C) Prognostic LRGs expression in normal and cancer tissues. (D) The consensus clustering matrix to classify BC patients into two categories. (E, F) CDF distribution plots and Delta area plots of the consensus clustering analysis (K = 2–9). (G) Principal component analysis revealed a clear distinction between emulsification-related subtypes. Blue points represent L1 subtypes and red points represent L2 subtypes. (H) Prognostic analysis of lactylation-related subtypes. **** P < 0.0001.
Functional enrichment analysis of lactylation-related subtypes
Supplementary Fig. 1A demonstrates the differential expression of 18 prognostic LRGs between subtypes. By using the limma R package, we obtained the differentially expressed genes between the different subtypes (Supplementary Fig. 1B). The results of KEGG showed that the differentially expressed genes were mainly enriched in cell cycle, PPAR signaling pathway, and regulation of lipolysis in adipocytes (Supplementary Fig. 1C). The results of GSVA-KEGG showed that the L1 and L2 subtypes were mainly enriched in ubiquitin-mediated proteolysis, cell cycle, DNA replication, ERBB signaling pathway, and fatty acid pathway, DNA replication, ERBB signaling pathway, fatty acid metabolism, and VEGF signaling pathway (Supplementary Fig. 1D). The results of GO showed that the differentially expressed genes were mainly enriched in the biological processes related to cell cycle and DNA replication (Supplementary Fig. 1E). The results of GSVA-GO showed that the L1 and L2 subtypes differed in the regulation of ubiquitin-dependent protein catabolic process, immune response, histone phosphorylation, and cell cycle-related biological processes (Supplementary Fig. 1F).
Screening of core genes based on machine learning
Through one-way Cox analysis of differentially expressed genes among different subtypes, we obtained 54 genes that were closely associated with prognosis (Supplementary Table S1). After that, the core genes were further screened by three machine learning algorithms. 12 genes were filtered by the Lasso algorithm (Fig. 2A, B). 6 genes were screened by Coxboost (Fig. 2C). 22 genes were selected by RSF (Fig. 2D). Finally, we extracted the intersection genes of the three algorithms for constructing the LRS (Fig. 2E).

Screening core genes by machine learning. (A) Screening key genes by the Coxboost algorithm. (B) Screening of key genes by RSF algorithm. (C) Plot of ten-fold cross-validations. (D) Plot of LASSO coefficient. (E) Venn diagram showing the intersection genes of the three algorithms.
Construction of a lactation-related gene signature
Figure 3A shows the C-index of gene signatures constructed by 15 machine learning algorithms in different datasets, and we chose the GBM algorithm with the highest C-index to construct the LRS. BC patients were categorized into high-LRS and low-LRS groups based on the median of LRS. Compared with the low-LRS group, the mortality rate was higher in the high-LRS group (Supplementary Fig. 2A-D). In the TCGA-BRCA, METABRIC, GSE96058, and GSE86166 cohorts, BC patients in the high-LRS group had a poorer prognosis (Fig. 3B, D, F, H). In the TCGA-BRCA cohort, the AUC values of LRS to predict 1-, 3-, and 5-year overall survival (OS) in BC patients were 0.837, 0.790, and 0.799 (Fig. 3C). In the METABRIC cohort, the AUC values of LRS for predicting 1-, 3-, and 5-year OS in BC patients were 0.653, 0.606, and 0.587 (Fig. 3E). In the GSE96058 cohort, the AUC values of LRS predicting 1-, 3-, and 5-year OS in BC patients were 0.799, 0.697, and 0.641 (Fig. 3G). In the GSE86166 cohort, the AUC values of LRS predicting 1-, 3-, and 5-year OS in BC patients were 0.596, 0.685, and 0.664 (Fig. 3I).

Construction of the LRS. (A) GBM was selected as the modeling method among14 machine learning methods for scoring. Survival curves of high and low LRS groups in TCGA-BRCA (B), METABRIC (D), GSE96058 (F), and GSE86166 (H) datasets. ROC curves of the LRS predicting 1-, 3-, and 5-year survival in the TCGA-BRCA (C), METABRIC (E), GSE96058 (G), and GSE86166 (I) datasets. **** P < 0.0001.
Relationship between the LRS and clinical characteristics
Compared with the L2 subtype, the LRS was higher in the L1 subtype (Fig. 4A). The LRS had higher AUC values and C-index compared to other clinical characteristics in TCGA-BRCA (Fig. 4B,C). The results of box plots showed that the LRS differed significantly in status, age, stage, and T stage (Fig. 4D-G). In addition, the LRS was also significantly different in different clinical features of METABRIC (Supplementary Fig. 3A-C). The relationship between LRS and clinical characteristics was further assessed by dividing the TCGA-BRCA cohort patients into subgroups based on clinical characteristics. Among all the different subgroups in status, age, stage, and T stage, the high-LRS group had a poorer prognosis (Fig. 4H-M).

Relationship between LRS and clinical features. (A) Differences in LRS between subtypes. (B,C) AUC values and C-index comparison of the LRS and clinical features in the TCGA-BRCA cohort. (D-G) Differences in LRS across status, age, stage, and T stage in the TCGA-BRCA cohort. (L-M) Survival curves of the LRS in different subgroups. * p < 0.05; ** p < 0.01; *** p < 0.001; **** p < 0.0001.
Construction of the nomogram
We assessed the independence of LR socre in the TCGA-BRCA, METABRIC, GSE96058, and GSE86166 cohorts by univariate Cox and multivariate Cox regression. Results showed that the LRS was a predictor independent of other clinical features (Fig. 5A,B; Supplementary Fig. 3D-I). In the TCGA-BRCA cohort, a nomogram that could predict the prognosis of BC patients was constructed by combining age and stage (Fig. 5C). The results of the ROC curve showed that the 1-, 3-, and 5-year AUC values of the nomogram were 0.916, 0.856, and 0.862 (Fig. 5D). In addition, the calibration curve showed that the survival probability predicted by the nomogram was essentially the same as the actual survival probability (Fig. 5E). The results of the DCA decision curve showed that the nomogram was the most effective in predicting the 5-year survival of BC patients (Fig. 5F-H).

Construction of the lactation-associated nomogram. (A,B) Univariate Cox and multivariate Cox regression analyses of LRS and clinical characteristics in the TCGA-BRCA cohort. (C) The lactation-associated nomogram. (D) ROC curves of nomogram predicting 1-, 3-, and 5-year survival. (E) Calibration curves at 1, 3, and 5 years (F-H) DCA decision curves assessing the effectiveness of nomogram in predicting survival at 1, 3, and 5 years.
Analysis of immune cell infiltration
The low-LRS group had a higher ESTIMATE and stromal score compared to the high-LRS group (Fig. 6A). The low-LRS group had a higher abundance of NK cells, B cell naive, and CD56bright natural killer cells, whereas the high-LRS group had higher levels of gamma delta T cells, type II T helper cells, and regulatory T cells (Fig. 6B,C). In addition, we found that the expression levels of immunoinhibitor, immunostimulator, major histocompatibility complex (MHC) molecule, chemokine, and chemokine receptor differed significantly between different LRS groups (Fig. 6D).

Immune cell infiltration analysis. (A) Differences in tumor microenvironment scores (immune, stromal, and estimate score) between different LRS groups. (B,C) Cibersort and ssGSEA algorithms to assess differences in immune infiltration levels between different LRS groups. (D) Differences in expression levels of immunoinhibitor, immunostimulator, major histocompatibility complex (MHC) molecule, chemokine, and chemokine receptor between different LRS groups.
Potential mechanisms between different LRS groups
The results of GSVA-GO showed that the high-LRS group was mainly enriched in biological processes such as cell cycle, histone phosphorylation, and histone methyltransferase activity. While the low-LRS group was mainly enriched in response to iron ions, positive regulation of protein autophosphorylation and negative regulation of epithelial cell migration (Fig. 7A). The results of GSVA-KEGG showed that the high-LRS group was mainly enriched in cell cycle, DNA replication, and galactose metabolism. The low-LRS group was mainly enriched in beta-alanine metabolism, fatty acid metabolism, and TGF beta signaling pathway (Fig. 7B). The results of GSEA-GO analyses showed that the biological processes related to cell cycle were significantly activated, while the biological processes related to immune response were suppressed (Fig. 7C). The results of GSEA-KEGG showed that the cell cycle pathway was significantly activated (Fig. 7D). In addition, LRS was significantly positively correlated with DNA damage, DNA repair, cell cycle, glycolysis, proliferation, and ubiquitination modifications (Supplementary Fig. S4A-H).

Functional enrichment analysis. (A) GSVA analyzes biological processes that differ between LRS groups. (B) GSVA analysis of pathways that differ between LRS groups. (C) GSEA enrichment analysis of biological processes that differ between LRS groups. (D) GSEA enrichment analysis of pathways for differences between different LRS groups.
Drug sensitivity
We analysed differences in sensitivity to chemotherapeutic drugs as well as cell cycle inhibitors between different LRS groups. The results showed that the high-LRS group had better sensitivity to docetaxel, cisplatin, cyclophosphamide, lapatinib, and dinaciclib (Fig. 8A-F). Sensitivity to cell cycle inhibitors (ribociclib, palbociclib, and mitoxantrone) was better in the low-LRS group (Fig. 8G-I).

Sensitivity analysis (IC50) of drugs between high- and low-LRS groups. (A-I) Docetaxel, Cisplatin, Cyclophosphamide, Lapatinib, Dinaciclib, Ribociclib, Palbociclib, Mitoxantrone. * P < 0.05; ** P < 0.01; *** P < 0.001; **** P < 0.0001.
Single-cell and spatial transcriptome analysis of genes in LRS
Figure 9A shows the different cell types in the GSE70658 dataset. The results of single-cell analysis showed that CLIC6, GABRQ, VGF, and SIM2 were predominantly enriched in malignant cells (Fig. 9B-E). SHCBP1 was abundantly expressed in malignant and Mono/Macro cells (Fig. 9F). SUSD3 was abundantly expressed in malignant, Mono/Macro, and CD8Tex cells (Fig. 9G). Figure 10A, B show the distribution of different cells in GSE203612-GSM6177603-NYU-BRCA2. Figure 10C-H shows the distribution of CLIC6, GABRQ, VGF, SUSD3, SHCBP1, and SIM2 in different cells, respectively. Meanwhile, we found that the expression of SUSD3, SHCBP1, and SIM2 was higher in the malignant region than in the normal region (Fig. 10I-K). In addition, we found that SUSD3 did not have a similar distribution to CD8 T cells (Supplementary Fig. S5A), but shares a similar spatial distribution with macrophages and its marker CD68 (Supplementary Fig. S5B).

Single-cell analysis of genes in LRS. (A) Different cell types in the GSE70658 dataset. (B-G) Expression levels of CLIC6, GABRQ, SHCBP1, SIM2, SUSD3, and VGF in different cells.

Spatial transcriptome analysis of genes in LRS. (A-B) Distribution of different cells in GSE203612-GSM6177603-NYU-BRCA2. (C-H) Distribution of CLIC6, GABRQ, VGF, SUSD3, SHCBP1, and SIM2 in different cells. (I-K) Distribution of SUSD3, SHCBP1, and SIM2 in normal and malignant cells.
Validation of genes in LRS expression levels
The expression levels of SHCBP1, SIM2, VGF, GABRQ, and SUSD3 were elevated in cancer tissues compared to paired paracancer tissues (Fig. 11 A-E). And the expression level of CLIC6 was decreased (Fig. 11F). Box plots show the expression levels of genes in different datasets (Fig. 11G-K). Figure 11L-P shows the expression levels of genes in breast cancer cell lines. In clinical cancer tissues, mRNA expression levels of SHCBP1, SIM2, VGF, GABRQ, and SUSD3 were elevated, and mRNA expression levels of CLIC6 were reduced (Fig. 12A-F).

Validation of genes in LRS across different datasets. (A-F) Expression levels of SHCBP1, SIM2, CLIC6, VGF, GABRQ, and SUSD3 in BC tissues and adjacent normal tissues in the TCGA-BRCA cohort. (G-J) Expression levels of the prognostic signature genes in GSE42568, GSE24124, GSE45827, and GSE57297. (K-P) Expression of the prognostic signature genes in cell lines in the CCLE database. * P < 0.05; ** P < 0.01; *** P < 0.001; **** P < 0.0001.

Experimental validation of genes in the LRS. (A-F) mRNA expression levels of SIM2, SHCBP1, SUSD3,VGF, GABRQ, and CLIC6 in breast cancer tissues and paracancerous tissues. * P < 0.05; ** P < 0.01; *** P < 0.001.
Prediction of potential small-molecule drugs
By CMap analysis, we obtained the top three small molecule complexes targeting VGF, SUSD3, SHCBP1, and GABRQ (Fig. 13A, C, E, G). We selected the drug with the smallest score for molecular docking. Figure 13B, D, F, and H show the molecular docking models for VGF + AH-6809, SUSD3 + W-13, SHCBP1 + arachidonyltrifluoromethane, and GABRQ + clofibrate.

Identification of potential drugs for targeting key genes. (A, C, E, G) Top three potential drugs predicted by CMap analysis. (B, D, F, H) Schematic diagram of the molecular docking model of drugs and proteins.
Discussions
Breast cancer is a complex disease caused by the progressive accumulation of multiple mutations and epigenetic dysregulation of key genes and protein pathways14. Breast cancer progression involves multiple steps, including uncontrolled cell proliferation and aberrant apoptosis, where activation of oncogenes and/or suppression of tumor suppressor genes are key events and are always accompanied by epigenetic changes15,16. Cancer cells can use epigenetic plasticity to adapt to the changing tumor microenvironment, evade the immune system, and develop resistance to therapies17. Epigenetic modifications are thought to be alterable, and drugs targeting these epigenetic modifications will be promising therapeutic targets for cancer treatment.
Numerous studies have demonstrated that lactylation promotes tumor development by regulating gene expression. Hypoxia-induced glycolysis was found to promote β-catenin lactylation and enhance β-catenin stability and expression, thereby aggravating the malignant proliferation of colorectal cancer (CRC) cells18. Another study found that inactivation of von Hippel-Lindau (VHL) triggered a positive feedback loop of histone lactylation-PDGFRβ signaling to promote clear cell renal cell carcinoma (ccRCC) progression19. In addition, other studies have found that lactylation is closely associated with the remodeling of the tumor microenvironment. Lactic acid regulates Treg cell production through lactylation of Lys72 in MOESIN, which enhances TGF-β signalling in the tumor microenvironment and maintains an immunosuppressive tumor microenvironment20. In another study, lactylation enriched in the promoter region of METTL3 was found to drive high expression of METTL3, which effectively induced immunosuppressive functions in myeloid cells in colon cancer via the METTL3/m6A/JAK1/STAT3 axis21. These findings suggest that lactylation plays an important role in tumor progression. The emergence of this new histone modification may provide new perspectives for cancer therapy.
In this study, we obtained 18 lactylation-related genes with prognostic significance by one-way Cox and Kaplan–Meier analysis. Using an unsupervised clustering algorithm, we classified breast cancer patients into two clusters: L1 and L2. Patients in the L1 subgroup had a poorer prognosis and were strongly associated with biological processes that promote cancer development (cell cycle and DNA replication), whereas the L2 subgroup was strongly associated with biological processes that are relatively cancer-suppressive (cell killing and immune activation). This is consistent with its poor prognosis and suggests a potential synergy between lactation-targeted and immune checkpoint blockade therapies in L1 patients. Notably, we also found that biological processes related to protein ubiquitination were activated in the L1 subtype. Ubiquitination of proteins is a dynamic and multi-aspect post-translational modification that involves a variety of cellular processes22. Ubiquitination of tumor suppressor protein factors or deubiquitination of tumor oncogenic protein factors often leads to cancer progression23. This finding suggests a mechanistic link between lactation and ubiquitination in promoting tumor invasiveness. It also suggests that L1 patients characterized by upregulated ubiquitination may benefit from proteasome inhibitors (e.g., bortezomib) or E3 ligase-targeted therapies, whereas L2 patients may respond better to standard therapies. In conclusion, L1/L2 classification is not only a reliable prognostic biomarker, but also provides an important reference for breast cancer patients’ personalized treatment.
In recent years, machine learning techniques have shown great potential in the medical field, especially in cancer research and treatment. With the rapid progress of the large data and computational power, machine learning methods have become an important tool for early diagnosis, prediction of prognosis, and personalized treatment of cancer24,25,26,27. In this study, we successfully screened six core genes by multiple machine learning methods and constructed gene signatures by the GBM algorithm with the highest C-index. The LRS is the independent prognostic predictor and shows robust predictive performance in different datasets. In addition, we found that the LRS outperforms clinical characteristics (stage, T stage, and age) and can well predict the prognosis of BC patients with different subgroups of clinical characteristics. In order to better improve the clinical value of LRS, we combined the clinical characteristics to construct a nomogram that can predict the prognosis of BC patients at 1, 3, and 5 years. The results of ROC curves and calibration curves show that the nomogram we constructed has good predictive performance and can be used as an accurate and stable tool for predicting the prognosis of BC patients.
The LRS consists of six core genes (SHCBP1, SIM2, VGF, GABRQ, SUSD3, and CLIC6), and previous studies have demonstrated that these genes play an important role in cancer progression. SHCBP1 is aberrantly expressed in a wide range of tumors, and through a variety of possible mechanisms, plays an important role in cancer development (cell proliferation, cell cycle, invasion and metastasis)28,29,30. SIM2 was found to be elevated in gliomas, and inhibition of SIM2 reduced the growth, migration, and invasion of glioma cells31. Another study found that overexpression of SIM2 reversed the inhibition of proliferation, migration, and invasion of colorectal cancer cells by TMEM75 (lncRNA) knockdown32. Expression of VGF is an independent prognostic predictor of oral squamous cell carcinoma, and overexpression of VGF promotes the proliferation and migration of oral squamous cell carcinoma cells33. Another study found that VGF in non-small cell lung cancer tissues underwent epigenetic modifications leading to increased gene transcription, which in turn promoted epithelial-mesenchymal transition and resistance to kinase inhibitors34. GABRQ has been demonstrated to serve as a novel prognostic biomarker for clear cell renal cell carcinoma35. In addition, researchers have found that knockdown of GABRQ inhibits the proliferation of hepatocellular carcinoma cells36. SUSD3 was found to be a breast cancer cell proliferation promoter, and knockdown of SUSD3 inhibited cell adhesion, spreading and motility of cancer cells37. In addition, another study found a trend of decreased SUSD3 expression in patients treated with anastrozole endocrine therapy38. CLIC6 has also been found to serve as a potential therapeutic target in gastric and endometrial cancers39,40. We explored the differential expression of these genes in different datasets and verified their expression in clinical tissues by QT-PCR. The results of PCR showed that SHCBP1, SIM2, VGF, GABRQ, and SUSD3 were up-regulated in breast cancer tissues, while CLIC6 was down-regulated in breast cancer tissues. Finally, we predicted potential drugs for highly expressed genes (SHCBP1, VGF, GABRQ, and SUSD3) by the CMap method and molecular docking. The results of molecular docking showed that AH-6809, clofibrate, W-13, and arachidonyltrifluoromethane could serve as potential inhibitors of genes. Previous studies have confirmed the inhibitory effects of these drugs in breast cancer. As an inhibitor of prostaglandin E2, AH 6809 significantly reduces the proliferation and invasion of breast cancer cells by silencing the EP2 receptor41. As an inhibitor of peroxisome proliferators, clofibrate has been shown to inhibit the proliferation of breast cancer cells42. The results of in vitro studies showed that W-13 significantly inhibited the proliferation of breast cancer cell lines43. This study provides valuable insights into potential drug candidates and their targets, but they also have their inherent limitations and require experimental validation. Although the predicted drugs AH-6809, W-13, and clofibrate show promise in cancer therapy, their potential anticancer effects and mechanisms of action require further experimental studies.
The tumor microenvironment (TME) is increasingly being recognized as a key factor in the development, progression, and treatment of cancerous lesions44. The TME is the cellular and molecular environment associated with tumor cells, which can influence the conditions affecting tumor development and progression45. The TME consists of immune cells, stromal cells, the extracellular matrix (ECM), and other secreted factors46,47. And the interaction of these components determines the cancer trends (cancer inhibition and promotion). In this study, we found that the high-LRS group had higher levels of infiltration of Gamma delta T cells, Type II T helper cells, and Regulatory T cells, which have been found to suppress anti-tumor immunity in cancer48,49,50. The results of enrichment analysis showed that biological processes such as Cell cycle, DNA replication, histone phosphorylation, and histone methylation were activated in the high-LRS group, while immune response-related biological processes were suppressed. There is growing evidence that various aberrant histone modifications promote tumor development and metastasis by inducing epigenetic, transcriptional, and phenotypic alterations51,52. Furthermore, histone modifications can interact with each other to the extent that one modification recruits or activates chromatin modification complexes to produce different histone modifications53,54. Thus we speculate that lactylation and other histone modifications crosstalk with each other to jointly influence breast cancer development. In conclusion, these results suggest that the high-LRS group has a tumor microenvironment that promotes tumor development, which may reveal the reason for their poorer prognosis. These results highlight potential therapeutic strategies, including immunotherapies targeting immune-suppressive cells (e.g., Regulatory T cells, Gamma delta T cells) and combination therapies that address both immune evasion and epigenetic alterations in the TME. Such approaches could help restore anti-tumor immunity and improve therapeutic outcomes in patients with a high-LRS.
With the introduction of novel anticancer drugs, significant progress has been made in the treatment of breast cancer patients over the past 15 years55. Recent studies have emphasized the application of novel systemic therapies and the use of radiotherapy combined with cell cycle inhibitors (CDK4/6 inhibitors)56,57. In the present study, we found that patients in the high-LRS group were more sensitive to chemotherapeutic agents, whereas the low-LRS group was more sensitive to CDK4/6 inhibitors. These results suggest that the LRS can serve as potential predictors of personalized treatment for BC patients. By identifying chemotherapy sensitivity in high-LRS patients, it could help avoid unnecessary treatments, while its association with CDK4/6 inhibitor response in low-LRS patients might optimize the use of targeted therapies. The translational potential of LRS in personalizing breast cancer treatment is promising, and further validation in larger cohorts is needed to confirm its clinical utility and integration into current therapeutic strategies.
Although the gene signature we constructed showed good predictive results, we must recognize that it has some limitations. First, this study relies mainly on retrospective data and still needs to be validated in a real cohort. Second, the lack of functional validation of the gene characterization means that the underlying mechanisms, especially the relationship between the identified genes and lactation, remain speculative. Further experimental studies are needed to confirm the biological relevance of these genes. In addition, potential batch effects between datasets may affect the robustness of the results, as differences in sample processing and data collection may influence the results. Future studies should address these limitations by using prospective cohorts and validating the functional role of genes to improve the reliability and applicability of gene characterization.
Notably, future studies should further explore the role of lactonization in drug resistance and metastasis, as the effects of lactonization on gene expression regulation and the tumor microenvironment suggest that lactonization is closely linked to cancer progression. Given the observed association between lactonization and epigenetic plasticity, investigating the interactions between lactonization and other histone modifications could provide more insight into the mechanisms driving tumor aggressiveness and immune evasion. In addition, elucidating the role of lactonization in regulating drug resistance, especially to chemotherapeutic agents and targeted therapies, may help to discover new strategies to overcome treatment failure.
Conclusion
In conclusion, we established a lactylation-associated gene signature in BC through multiple machine learning, which can assess the prognosis of BC patients as well as their response to drugs. In addition, we predicted potential inhibitors of genes, which may provide new insights into targeted therapies for BC patients.
Materials and methods
Data acquisition and processing
Lactation-related genes (LRGs) were obtained from the GeneCards database and from previous studies. By removing duplicated genes, we obtained 402 LRGs (Supplementary Table S2). Gene expression data and clinical data for TCGA-BRCA and METABIC were downloaded from the TCGA database and cBioPortal database. Data on GSE96058, GSE86166, GSE42568, GSE45827, GSE24124, and GSE57297 were downloaded from the GEO database. Basic information on these datasets can be found in Supplementary Table S3. Differentially expressed genes (DEGs) in the TCGA-BRCA cohort were obtained by the ‘limma’ R package58, and the intersection genes of DEGs and LRGs were extracted.
Construction of lactation-associated subtypes
We obtained lactylation-related genes with prognostic significance by one-way Cox analysis. Based on the expression profiles of these genes, we performed unsupervised consensus clustering analysis (K = 2–9) on 1044 tumor samples from the TCGA-BRCA cohort using the ConensusClusterPlus package59. The optimal K values were filtered based on the proportion of ambiguous clustering and the cumulative distribution function (CDF). Differentially expressed genes (LRDEGs) among subtypes were obtained using the ‘limma’ package, with the screening criteria of P < 0.05, log2|FC|≥ 1. In addition, we performed GO and KEGG enrichment analyzes using the ‘ClusterProfiler’ package60 and analyzed the biological processes underlying the differences between the subtypes using the ‘GSVA’ package61.
Screening of genes related to lactation
Prognostically relevant LRDEGs were screened by one-way Cox with a threshold of P < 0.05. Prognostic LRDEGs were further screened by three machine learning methods, including CoxBoost, random survival forest (RSF), and the least absolute shrinkage and selection operator (Lasso)62,63,64. The CoxBoost algorithm was performed with the ‘CoxBoost’ R package (version 1.5). The optimal Penalty was selected by the optimCoxBoostPenalty function, and the genes were screened by the model constructed by the optimal.step. The RSF algorithm was performed with the ‘randomForestSRC’ R package (version 3.3.1) with ntree set to 1000 and nodesize set to 95. The Lasso algorithm is performed with the ‘glmnet’ R package (version 4.1–8), with parameters that include a nlambda of 1000 and a lambda of lambda.min. Finally, the overlapping genes of the 3 algorithms were extracted for constructing gene signature.
Construction of gene signature related to lactation
Referring to the methods in previous studies65, we used 15 machine learning algorithms in multiple datasets (TCGA-BRCA, METABRIC, GSE96058, and GSE86166) to build the machine learning framework. The 15 machine learning algorithms are GBM, SuperPC, Survival-Svm, Ridge, Enet, plsRCox, Lasso, CoxPH, StepCox, SurvReg, CoxBoost, Rpart, GlmBoost, CTree, and ObliqueRSF. The GBM and SuperPC algorithms were implemented using the ‘gbm’ R-package (version 2.2.2) and the ‘superpc’ R-package (version 1.12), and both fit the model with tenfold cross-validation. The Survival-Svm algorithm was implemented using the ‘survivalsvm’ R-package (version 0.0.5), with the gamma.mu value set to 1. The Ridge, Lasso, and Enet algorithms were implemented using the glmnet package, and were executed individually by setting alpha. When alpha is equal to 0, the Ridge algorithm is executed. When alpha is greater than 0 and less than 1, the Enet algorithm is executed. When alpha is equal to 1, the Lasso algorithm is executed. The plsRcox model was constructed using the ‘plsRcox’ R package (version 1.7.7). The CoxPH and StepCox algorithms were performed using the coxph and step functions, respectively, of the ‘survival’ R package (version 3.7–0). The CoxBoost algorithm was performed as previously described. Rpart was implemented with the ‘rpart’ R package (version 4.1.23). glmBoost was implemented with the ' mboost’ R package (version 2.9–11) with sigma set to 0.1. The CTree algorithm was built using the ctree function of the ‘party’ R package (version 1.3–17). The ObliqueRSF algorithm was implemented using the ORSF function of the ‘obliqueRSF’ R package, with the alpha set to 0.5 and the ntree set to 100. We constructed prognostic models by machine learning with the same parameters in all four datasets and calculated the C-index for each machine learning algorithm. The C-index is a metric that evaluates the predictive performance of a model, and the closer it is to 1, the better the predictive performance. We finally selected the machine learning algorithm with the highest average C-index in the four datasets to construct the genetic features (LR score). The samples were divided into high-risk and low-risk groups based on the median LR score, and the survival outcomes of BC patients in the low-risk and high-risk groups were analyzed by Kaplan–Meier curves. In addition, 1-, 3- and 5-year ROC curves for LR score were plotted. We chose the machine learning algorithm with the highest average C-index to construct the gene signature (LRS). The samples were divided into high-LRS and low-LRS groups based on the median of LRS, and the survival outcomes of BC patients in the low-LRS and high-LRS groups were analysed by Kaplan–Meier curves. In addition, 1-, 3- and 5-year ROC curves for LRS were plotted.
Nomogram construction
The nomogram was composed of the LRS and other clinical characteristics. In addition, we plotted 1-, 3-, and 5-year ROC curves, calibration curves, and 1-, 3-, and 5-year DCA curves for the nomogram.
Immune cell infiltration analysis
The Cibersort, ESTIMATE, and ssGSEA algorithms of the ‘IOBR’ R package66 were used to assess immune cell infiltration in the high- and low-LRS groups. In addition, the correlation of the LRS with immunoinhibitor, immunostimulator, major histocompatibility complex (MHC) molecule, chemokine, and chemokine receptor was analyzed.
Functional enrichment analysis
Differentially expressed genes (log2|FC|≥ 1, P < 0.05) among different LRS groups were obtained by the ‘limma’ R package, and these genes were analyzed by GO and KEGG enrichment analyzis67,68,69. In addition, the relationship between LRS and cancer-related biological processes was analysed.
Drug sensitivity analysis
Drug sensitivity analyses were performed using the ‘oncoPredict’ package70. Half maximum inhibitory concentration (IC50) was used to indicate drug sensitivity.
Single-cell analysis and spatial transcriptome analysis
In addition, the expression levels of CTRGs in different cells in GSE176078 (single-cell dataset) were analyzed using the Sparkle database (https://grswsci.top/analyze), as well as their spatial distribution in GSE203612-GSM6177603-NYU-BRCA2. Cell markers for single-cell analysis can be found in Supplementary Table S4.
Identification of potential drugs
Referring to the methodology used in a previous study71, drug features were downloaded from the Connectivity Map database (CMap, https://clue.io/), and the expression profiles of the top 150 up-regulated and top 150 down-regulated were selected as input data. CMap scores were calculated using the limit sum (XSum) algorithm, and the top 3 drugs with the lowest CMap scores were selected for visualisation. The small-molecule drugs and proteins with the lowest CMap scores were selected for molecular docking. The 3D structures of the proteins were obtained from the Uniprot database (https://www.uniprot.org/). The 3D structures of the small molecule drugs were downloaded from PubChem (https://www.ncbi.nlm.nih.gov/pccompound). Finally, the molecular docking model of protein and small-molecule drug complex was constructed by CB-Dock2 (https://cadd.labshare.cn/cb-dock2/php/index.php).
Real-time fluorescent polymerase chain reaction (RT-PCR)
Tissue samples from breast cancer patients were provided by Shanxi Province Cancer Hospital. This study was conducted in accordance with the Declaration of Helsinki and was approved by the Research Ethics Committee of Shanxi Province Cancer Hospital (Grant No. KY2023163). Subjects included in this study signed an informed consent form. Total RNA was extracted from tissues using a Trizol kit. Reverse transcription (RT) was performed using a Takara kit. The steps of real-time quantitative PCR reaction are as follows: conFig. the reverse transcription system; perform real-time PCR reaction; calculate the amount of mRNA expression of the target gene. The primers for genes are shown in Supplementary Table S5.
Statistical analyses
All data analysis and statistical analyses were performed using R version 4.3.0. The Wilcoxon test was used to compare data between two groups, and the Kruskal–Wallis test was used to compare data between multiple groups. Kaplan–Meier analyses were used to assess differences in overall survival (OS) between groups. P < 0.05 indicates statistical significance.
Data availability
The datasets used in this study can be downloaded from The Cancer Genome Atlas database (https://portal.gdc.cancer.gov/), cBioPortal for Cancer Genomics (https://www.cbioportal.org/), and Gene Expression Omnibus (https://www.ncbi.nlm.nih.gov/geo/).
References
Bray, F. et al. Global cancer statistics 2022: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA A Cancer J. Clinicians 74, 229–263 (2024).
Goel, S. & Chandarlapaty, S. Emerging therapies for breast cancer. Cold Spring Harb. Perspect Med. 13, a041333 (2023).
Pankotai-Bodó, G., Oláh-Németh, O., Sükösd, F. & Pankotai, T. Routine molecular applications and recent advances in breast cancer diagnostics. J. Biotechnol. 380, 20–28 (2024).
Nolan, E., Lindeman, G. J. & Visvader, J. E. Deciphering breast cancer: from biology to the clinic. Cell 186, 1708–1728 (2023).
Sher, G. et al. Epigenetic and breast cancer therapy: Promising diagnostic and therapeutic applications. Semin. Cancer Biol. 83, 152–165 (2022).
Hussain, S. et al. Role of epigenetics in carcinogenesis: Recent advancements in anticancer therapy. Semin. Cancer Biol. 83, 441–451 (2022).
Garcia-Martinez, L., Zhang, Y., Nakata, Y., Chan, H. L. & Morey, L. Epigenetic mechanisms in breast cancer therapy and resistance. Nat. Commun. 12, 1786 (2021).
Karami Fath, M. et al. The role of epigenetic modifications in drug resistance and treatment of breast cancer. Cell Mol. Biol. Lett. 27, 52 (2022).
Zhang, D. et al. Metabolic regulation of gene expression by histone lactylation. Nature 574, 575–580 (2019).
Wang, W., Wang, H., Wang, Q., Yu, X. & Ouyang, L. Lactate-induced protein lactylation in cancer: functions, biomarkers and immunotherapy strategies. Front. Immunol. 15, 1513047 (2025).
Su, J. et al. Functions and mechanisms of lactylation in carcinogenesis and immunosuppression. Front. Immunol. 14, 1253064 (2023).
Yu, J. et al. Histone lactylation drives oncogenesis by facilitating m6A reader protein YTHDF2 expression in ocular melanoma. Genome Biol. 22, 85 (2021).
Pan, L. et al. Demethylzeylasteral targets lactate by inhibiting histone lactylation to suppress the tumorigenicity of liver cancer stem cells. Pharmacol. Res. 181, 106270 (2022).
Olopade, O. I., Grushko, T. A., Nanda, R. & Huo, D. Advances in breast cancer: pathways to personalized medicine. Clin. Cancer Res. 14, 7988–7999 (2008).
Yin, J., Gu, T., Chaudhry, N., Davidson, N. E. & Huang, Y. Epigenetic modulation of antitumor immunity and immunotherapy response in breast cancer: biological mechanisms and clinical implications. Front. Immunol. 14, 1325615 (2024).
Thakur, C. et al. Epigenetics and environment in breast cancer: New paradigms for anti-cancer therapies. Front. Oncol. 12, 971288 (2022).
Trnkova, L. et al. Epigenetic deregulation in breast cancer microenvironment: Implications for tumor progression and therapeutic strategies. Biomed. Pharmacother. 174, 116559 (2024).
Miao, Z., Zhao, X. & Liu, X. Hypoxia induced β-catenin lactylation promotes the cell proliferation and stemness of colorectal cancer through the wnt signaling pathway. Exp. Cell Res. 422, 113439 (2023).
Yang, J. et al. A positive feedback loop between inactive VHL-triggered histone lactylation and PDGFRβ signaling drives clear cell renal cell carcinoma progression. Int. J. Biol. Sci. 18, 3470–3483 (2022).
Gu, J. et al. Tumor metabolite lactate promotes tumorigenesis by modulating MOESIN lactylation and enhancing TGF-β signaling in regulatory T cells. Cell Rep. 39, 110986 (2022).
Xiong, J. et al. Lactylation-driven METTL3-mediated RNA m6A modification promotes immunosuppression of tumor-infiltrating myeloid cells. Mol. Cell 82, 1660-1677.e10 (2022).
Han, S. et al. The role of ubiquitination and deubiquitination in tumor invasion and metastasis. Int. J. Biol. Sci. 18, 2292–2303 (2022).
Ye, Z., Yang, J., Jiang, H. & Zhan, X. The roles of protein ubiquitination in tumorigenesis and targeted drug discovery in lung cancer. Front. Endocrinol. 14, 1220108 (2023).
Swanson, K., Wu, E., Zhang, A., Alizadeh, A. A. & Zou, J. From patterns to patients: Advances in clinical machine learning for cancer diagnosis, prognosis, and treatment. Cell 186, 1772–1791 (2023).
Balkenende, L., Teuwen, J. & Mann, R. M. Application of deep learning in breast cancer imaging. Semin. Nucl. Med. 52, 584–596 (2022).
Yassi, M., Chatterjee, A. & Parry, M. Application of deep learning in cancer epigenetics through DNA methylation analysis. Briefings Bioinform. 24, bbad11 (2023).
Sheehy, J. et al. Gynecological cancer prognosis using machine learning techniques: A systematic review of the last three decades (1990–2022). Artif. Intell. Med. 139, 102536 (2023).
Yang, C. et al. SHCBP1 interacting with EOGT enhances O-GlcNAcylation of NOTCH1 and promotes the development of pancreatic cancer. Genomics 113, 827–842 (2021).
Zhou, M. et al. The dynamic role of nucleoprotein SHCBP1 in the cancer cell cycle and its potential as a synergistic target for DNA-damaging agents in cancer therapy. Cell Commun. Signal 22, 131 (2024).
Lin, Y. & Cai, H. Biological functions and therapeutic potential of SHCBP1 in human cancer. Biomed. Pharmacother. 160, 114362 (2023).
Su, Y. et al. MiR-200a impairs glioma cell growth, migration, and invasion by targeting SIM2-s. NeuroReport 25, 12–17 (2014).
Jin, X., Liu, G., Zhang, X. & Du, N. Long noncoding RNA TMEM75 promotes colorectal cancer progression by activation of SIM2. Gene 675, 80–87 (2018).
Chou, C.-H. et al. The upregulation of VGF enhances the progression of oral squamous carcinoma. Cancer Cell Int. 24, 115 (2024).
Marwitz, S. et al. Epigenetic modifications of the VGF gene in human non-small cell lung cancer tissues pave the way towards enhanced expression. Clin. Epigenet. 9, 123 (2017).
Lee, D. et al. GABRQ expression is a potential prognostic marker for patients with clear cell renal cell carcinoma. Oncol. Lett. https://doi.org/10.3892/ol.2019.10960 (2019).
Li, Y.-H. GABA stimulates human hepatocellular carcinoma growth through overexpressed GABAA receptor theta subunit. WJG 18, 2704 (2012).
Moy, I. et al. Estrogen-dependent sushi domain containing 3 regulates cytoskeleton organization and migration in breast cancer cells. Oncogene 34, 323–333 (2015).
Mello-Grand, M. et al. Gene expression profiling and prediction of response to hormonal neoadjuvant treatment with anastrozole in surgically resectable breast cancer. Breast Cancer Res. Treat 121, 399–411 (2010).
Fu, Z. et al. Construction of miRNA-mRNA-TF regulatory network for diagnosis of gastric cancer. Biomed. Res. Int. 2021, 1–9 (2021).
López-Ozuna, V. M. et al. Identification of predictive biomarkers for lymph node involvement in obese women with endometrial cancer. Front. Oncol. 11, 695404 (2021).
Qian, X., Zhang, J. & Liu, J. Tumor-secreted PGE2 inhibits CCL5 production in activated macrophages through cAMP/PKA signaling pathway. J. Biol. Chem. 286, 2111–2120 (2011).
Penna, F., Bonelli, G., Baccino, F. & Costelli, P. Cytotoxic properties of clofibrate and other peroxisome proliferators: relevance to cancer progression. CMC 17, 309–320 (2010).
Wei, J.-W., Hickie, RobertA. & Klaassen, DavidJ. Inhibition of human breast cancer colony formation by anticalmodulin agents: Trifluoperazine, W-7, and W-13. Cancer Chemother. Pharmacol. 11, 86–90 (1983).
Burgos-Panadero, R. et al. The tumour microenvironment as an integrated framework to understand cancer biology. Cancer Lett. 461, 112–122 (2019).
Rodríguez-Bejarano, O. H., Parra-López, C. & Patarroyo, M. A. A review concerning the breast cancer-related tumour microenvironment. Crit. Rev. Oncol. Hematol. 199, 104389 (2024).
Bejarano, L., Jordāo, M. J. C. & Joyce, J. A. Therapeutic targeting of the tumor microenvironment. Cancer Discov. 11, 933–959 (2021).
Junttila, M. R. & De Sauvage, F. J. Influence of tumour micro-environment heterogeneity on therapeutic response. Nature 501, 346–354 (2013).
Fleming, C., Morrissey, S., Cai, Y. & Yan, J. γδ T cells: unexpected regulators of cancer development and progression. Trends Cancer 3, 561–570 (2017).
Basu, A. et al. Differentiation and regulation of TH cells: a balancing act for cancer immunotherapy. Front. Immunol. 12, 669474 (2021).
Ohue, Y. & Nishikawa, H. Regulatory T (Treg) cells in cancer: Can Treg cells be a new therapeutic target?. Cancer Sci. 110, 2080–2089 (2019).
Neganova, M. E., Klochkov, S. G., Aleksandrova, Y. R. & Aliev, G. Histone modifications in epigenetic regulation of cancer: Perspectives and achieved progress. Semin. Cancer Biol. 83, 452–471 (2022).
Yang, Y., Zhang, M. & Wang, Y. The roles of histone modifications in tumorigenesis and associated inhibitors in cancer therapy. J. Nat. Cancer Center 2, 277–290 (2022).
Lee, J.-S., Smith, E. & Shilatifard, A. The language of histone crosstalk. Cell 142, 682–685 (2010).
Suganuma, T. & Workman, J. L. Crosstalk among histone modifications. Cell 135, 604–607 (2008).
Meattini, I. et al. International multidisciplinary consensus on the integration of radiotherapy with new systemic treatments for breast cancer: european society for radiotherapy and oncology (ESTRO)-endorsed recommendations. Lancet Oncol. 25, e73–e83 (2024).
Burstein, H. J. Systemic therapy for estrogen receptor-positive, HER2-negative breast cancer. N. Engl. J. Med. 383, 2557–2570 (2020).
Morrison, L., Loibl, S. & Turner, N. C. The CDK4/6 inhibitor revolution — a game-changing era for breast cancer treatment. Nat. Rev. Clin. Oncol. 21, 89–105 (2024).
Ritchie, M. E. et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 43, e47–e47 (2015).
Wilkerson, M. D. & Hayes, D. N. ConsensusClusterPlus: a class discovery tool with confidence assessments and item tracking. Bioinformatics 26, 1572–1573 (2010).
Yu, G., Wang, L.-G., Han, Y. & He, Q.-Y. clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS: J. Integrative Biol. 16, 284–287 (2012).
Hänzelmann, S., Castelo, R. & Guinney, J. GSVA: gene set variation analysis for microarray and RNA-Seq data. BMC Bioinformatics 14, 7 (2013).
LeRoy, G. Requirement of RSF and FACT for transcription of chromatin templates in vitro. Science 282, 1900–1904 (1998).
Friedman, J., Hastie, T. & Tibshirani, R. Regularization paths for generalized linear models via coordinate descent. J. Stat. Soft. https://doi.org/10.18637/jss.v033.i01 (2010).
Gonzalez-Angulo, A. M. et al. Functional proteomics characterization of residual breast cancer after neoadjuvant systemic chemotherapy. Ann. Oncol. 24, 909–916 (2013).
Zhang, N. et al. An artificial intelligence network-guided signature for predicting outcome and immunotherapy response in lung adenocarcinoma patients based on 26 machine learning algorithms. Cell Prolif. 56, e13409 (2023).
Zeng, D. et al. IOBR: Multi-Omics Immuno-Oncology Biological Research to Decode Tumor Microenvironment and Signatures. Front. Immunol. 12, 687975 (2021).
Kanehisa, M., Furumichi, M., Sato, Y., Kawashima, M. & Ishiguro-Watanabe, M. KEGG for taxonomy-based analysis of pathways and genomes. Nucleic Acids Res. 51, D587–D592 (2023).
Kanehisa, M. & Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 28, 27–30 (2000).
Kanehisa, M. Toward understanding the origin and evolution of cellular organisms. Protein Sci. 28, 1947–1951 (2019).
Maeser, D., Gruener, R. F. & Huang, R. S. oncoPredict: an R package for predicting in vivo or cancer patient drug response and biomarkers from cell line screening data. Briefings Bioinform. 22, bbab260 (2021).
Yang, C. et al. A survey of optimal strategy for signature-based drug repositioning and an application to liver cancer. Elife 11, e71880 (2022).
Funding
This work was supported by the grants from the Basic Research Program of Shanxi Province (Free Exploration Category) [Grant number: 202103021224022] and the Basic Research Program of Shanxi Province (Free Exploration Category) [Grant number: 202103021224428].
Author information
Authors and Affiliations
Contributions
Conceptualization: J.F.Z., Y.X.W. and Y.C.; Methodology: L.P.L., J.Y.H., J.R.W., and H.W.X.; Formal analysis and investigation: J.F.Z., L.P.L., and Y.X.W.; Writing—original draft preparation: L.P.L. and J.F.Z.; Writing—review and editing: L.P.L., J.F.Z., J.R.W. and H.W.X.; Funding acquisition: J.F.Z. and Y.C.; Resources: J.F.Z. and Y.C.; Supervision: J.F.Z. and Y.X.W. All authors contributed to the article and approved the submitted version.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethical approval
This study was conducted in accordance with the Declaration of Helsinki and was approved by the Research Ethics Committee of Shanxi Province Cancer Hospital (Grant No. KY2023163). Subjects included in this study signed an informed consent form. It is necessary to respect the privacy of the individuals involved in the study.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Zhao, J., Li, L., Wang, Y. et al. Identification of gene signatures associated with lactation for predicting prognosis and treatment response in breast cancer patients through machine learning. Sci Rep 15, 13575 (2025). https://doi.org/10.1038/s41598-025-98255-x
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-98255-x
