
Abstract
We assessed the incidence and clinical significance of the fetal region of homozygosity (ROH) detected using single nucleotide polymorphism (SNP) array by analyzing clinical information and pregnancy outcomes. We collected data on 6176 mid- and late pregnancies. All fetuses were subjected to SNP array analysis. Fetuses with ROH were analyzed by karyotyping, parental SNP array verification, whole-exome sequencing, and/or placental studies. Eighty-seven ROHs met our reporting thresholds. Thirty-four fetuses were detected from noninvasive prenatal testing-positive results, with the most common detection rate (2.03%). Twenty-four cases were diagnosed using ultrasound abnormalities; fetal growth restriction was the indication with the highest diagnostic rate. Fifteen cases of uniparental disomy in mid- and late pregnancy were identified (0.24%). Nine cases were of ROH accompanied by aneuploidy or pathogenic/likely pathogenic copy number variants with an adverse pregnancy outcome rate of 88.9%. Of the remaining 78 cases, 14 carriers had adverse outcomes (including two cases of imprinting syndrome), 63 had normal development after birth, and one was lost to follow-up. ROH is relatively common in mid- and late-term pregnancies; its incidence is higher than that reported previously. SNP array is effective in assessing ROH and should be combined with multiple techniques to evaluate ROH’s clinical relevance.
Similar content being viewed by others

Introduction
The region of homozygosity (ROH), or a long contiguous stretch of homozygosity (LCSH), refers to an uninterrupted tract of homozygous haplotypes with a neutral genomic copy number1. Genome-wide data suggest that ROH is among the most common characteristics of the human genome2. Small ROHs, usually less than 3–5 Mb, are present in all populations, including outbred individuals, reflecting recombination rates and population history2,3,4. ROH is nonrandom and more prevalent in areas of high linkage disequilibrium and low recombination5. Long ROHs, the focus of this study, are relatively common in the general population and can arise from the chance of unions among parents with a common ancestor or from homologous genomic regions, both from one parent each. ROHs involving multiple chromosomes usually represent regions of autozygosity belonging to identity by descent (IBD). Generally, when an isolated ROH (involving only a single chromosome) is detected, particularly when longer than 10 Mb, segmental or whole-chromosome uniparental disomy (UPD) is a likely underlying mechanism. Thus, the presence of large ROHs may be due to parental relatedness or chromosomal aberrations, which may result in abnormal phenotypes associated with an increased risk of homozygous recessive disorders or through gene imprinting in UPD. The detection rates for long ROH are rarely reported and vary with thresholds, populations, and sample types; however, results from four million population screens indicate that UPD for all chromosomes has an overall prevalence of 0.05%6. Nevertheless, not all UPDs have clinically recognizable consequences; only a few chromosomes contain regions with parent-specific gene expression (imprinting), such as separate regions of 6q24, 7p11.2p13, 7q32, 11p15, 14q32, 15q11q13, and 20q137. Most UPD carriers, especially ROH carriers, present with normal phenotypes, and the clinical significance of ROH remains unclear.
ROH can be detected using a chromosomal microarray (CMA). For over a decade, single-nucleotide polymorphism-based CMA (SNP array) analyses have been broadly implemented in prenatal diagnosis to detect genomic imbalances. It detects copy number variants (CNVs), ROH, UPD, and possible consanguinity8. CMA can provide additional clinically pathogenic or likely pathogenic CNVs in about 0.55–6% of prenatal cases versus karyotyping9,10. However, data on prenatally diagnosed ROH are limited, rendering the interpretation of abnormal ROH levels and genetic counseling difficult. The threshold for reporting ROH has been suggested to be greater than 10 Mb for nonimprinted chromosomes, greater than 5 Mb for the terminus, and 10 Mb for interstitial ROH for imprinted chromosomes8,11. In this study, we reviewed cases of ROH detected using an SNP array in amniotic fluid or cord blood samples, tracking the perinatal outcomes of fetuses with ROH. The aim of this study was to establish the overall diagnostic rate of CMA in our setting and obtain more information on the correlation between ROH results and phenotype.
Materials and methods
Ethics approval and consent to participate
This study was reviewed and approved by the ethics committee of Changsha Hospital for Maternal and Child Health Care. All pregnant women received genetic counseling and provided informed consent before the invasive prenatal test. All experiments were performed in accordance with relevant named guidelines and regulations.
Subjects
From August 2017 to September 2023, 6176 consecutive series of samples, including amniotic fluid (6044 samples) and cord blood (132 samples), were collected and analyzed successfully at the center of Changsha Hospital for Maternal and Child Health Care. The mean age of the pregnant women was 31.15 years (range: 16–53 years), and the mean gestational age was 22.63 weeks (range: 15–37 weeks). The indications for prenatal testing include abnormal ultrasound findings, abnormal NIPT results, abnormal result on maternal serum biochemical screening, and advanced maternal age (≥ 35), as shown in Table 1. SNP array analyses were performed for all patients. If the SNP array revealed ROH, the parents’ blood samples were analyzed to determine whether the fetal ROHs were IBD or UPD. WES was used to rule out homozygous recessive disorders if necessary.
SNP array analysis
The SNP-array analysis was conducted on the Affymetrix CytoScan platform (Thermo, USA). Amniocyte or umbilical cord blood genomic DNA was extracted using a TIANamp® Micro DNA Kit (Tiangen, China), after which 250 ng DNA was digested, ligated, PCR-amplified, purified, fragmented, labeled, and hybridized to the Affymetrix CytoScan 750 K array, which includes 550,000 CNV markers and 200,000 SNP markers. The raw data were analyzed using the Chromosome Analysis Suite (ChAS) 4.2. Interpretation and reporting of constitutional CNVs were performed according to the standards and guidelines released by the American College of Medical Genetics (ACMG)12. Herein, we report clinically significant CNVs with deletions > 500 kb or duplications > 1 Mb in size. Pathogenicity was classified as follows: pathogenic (P), likely pathogenic (LP), variants of uncertain significance, likely benign, or benign. ROH was reported for the following four conditions: (1) ROH is over 10 Mb in size on nonimprinted chromosomes; (2) ROH is greater than 5 Mb on termini or greater than 10 Mb on interstitials of imprinted chromosomes (6, 7, 11, 14, 15, and 20), and (3) the percentage of ROH is greater than or equal to 6.25%. The percentage of ROH is suggestive of the coefficient of inbreeding (F) and can be estimated as the sum of all homozygous regions greater than 3 Mb distributed on autosomes divided by the total autosomal genomic length (approximately 2881 Mb for GRCh37/hg19). The percentage was 25%, and parental relatedness was first degree (12.5%, second degree; 6.25%, third degree; and 3.125%, fourth degree)1. (4) Sex chromosomes were excluded unless the entire ROH was on one chromosome because males are always hemizygous. X chromosomes in females are often observed with increased ROH because of limited recombination1,2. If we detected the ROH in prenatal samples using the SNP array, parents were given the choice to undergo parental SNP-array or trio-WES analysis to identify the origin of the ROH. Some databases were used as reference resources, including geneimprint (https://geneimprint.com/), UPD-start (http://upd-tl.com/upd.html), Online Mendelian Inheritance in Man (https://www.omim.org/) and literatures in PubMed (https://pubmed.ncbi.nlm.nih.gov/).
Karyotyping and whole-exome sequencing
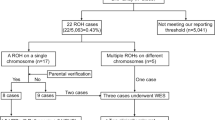
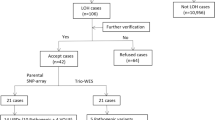
Amniotic fluid or cord blood were collected for 7-14-day amniotic fluid cell culture and 3-day umbilical cord blood cell culture. Cell harvesting involved the termination of culture, hypotonic treatment, fixation, sectioning, and G-banding. Subsequently, scanning was performed using a GSL-120 chromosome automatic analyzer (Leica Biosystems, Germany). Karyotypes were named according to the International System for Human Cytogenetic Nomenclature (2020). To detect potential homozygous recessive pathogenic genes, WES was further performed after obtaining parental consent. Sample DNA was extracted, fragmented randomly, purified and enriched to construct DNA libraries. The DNA libraries were sequenced using the NextSeq500 sequencer according to the manufacturer’s protocols (Illumina, USA). For sequence alignment, variant calling, and annotation, the sequences were mapped to their location with the human genome reference sequence (GRCh37/hg19 build) using Burrows-Wheeler software (version 0.59). All single nucleotide variants and insertion/deletions were annotated with multiple public population frequency databases and in-house database. The annotated variants were filtrated in a stepwise model that included genotype-driven, phenotype-driven, and manual review13,14. Candidate variants were classified according to the ACMG guidelines15 and ClinGen variant curation expert panel guidelines16,17. A multidisciplinary team of clinical and laboratory geneticists, obstetricians and genetic counselors reviewed all the rare phenotype-related variants to identify the reportable variants. The overall flow of the fetal ROH analysis is illustrated in Fig. 1.
Clinical follow-up
The pregnancy outcomes were followed up for six months to one year in all cases, and for two to three years in cases with ROH. Follow-up assessments included measurements of birth weight, length, head circumference, and growth.
Results
ROH in different clinical indications
Eighty-seven cases of ROH from 6176 samples, with a prenatal detection rate of 1.41% (87/6176), were identified. The rate of ROH ≥ 10 Mb was 1.38% (85/6176). ROH detection rates for different prenatal diagnostic indications are shown in Table 1. Overall, the most common detection rate was from abnormal non-invasive prenatal testing (NIPT) results (34/1674 (2.03%)), followed by previous adverse pregnancy (8/430 (1.86%)), high risk for Down’s syndrome (17/1254 (1.36%)), and ultrasound abnormalities (24/2120 (1.13%)). Of the 34 ROH cases with positive NIPT results, 29 were confirmed to be in the chromosome or region with aberrant NIPT results, with a concordance rate of 85.3% (29/34). Fetal growth restriction (FGR) showed the highest detection rate (7/185 (3.78%)) among fetuses with different ultrasound abnormalities.
General distribution of ROH
We divided the 87 cases into different categories: (1) according to the number of chromosomes involved, 11 (12.6%) cases were found on multiple chromosomes, and 76 (87.4%) cases were on a single chromosome, suggesting a UPD frequency of 1.23% (76/6176). In the 11 cases with multiple ROHs, two were from consanguineous marriages close to the third degree (F-value = 0.0815) and fourth degree (F-value = 0.0302); in the other nine, there was no indication of consanguinity. (2) According to the size of the segments involved, 82 (94.3%) cases involved segments of a chromosome (ranging from 9.06 to 74.06 Mb) and 5 (5.7%) involved the entire chromosome, including 1, 2, 3, 7, and X. (3) According to the location of the chromosomes involved, 24 (27.6%) cases were on imprinted chromosomes and 63 (72.4%) were on nonimprinted chromosomes.
When ROH occurred on a single chromosome, its occurrence appeared to be nonrandom, and chromosomes 2 (12/76 (14.8%)), 1 (7/76 (14.8%)), 16 (7/76 (14.8%)), and 18 (7/76 (14.8%)) were involved most often. ROH covering the entire length of the chromosome was considered a complete UPD. In our study, 15 cases of definite UPD (including three cases without parental verification and ROH present on the entire chromosome) were found among the 6176 cases of mid to late pregnancy, with a detection rate of 0.24%.
Outcome and follow-up
Of the 87 ROH fetuses, nine were cases of ROH with chromosomal aneuploidy or P/LP CNVs, with a detection rate of 10.3% (9/87). Of the remaining 78 cases, ROH was located on imprinted chromosomes in 23 cases (23/78 (29.5%)) and on nonimprinted chromosomes in 55 cases (55/78 (70.5%)). Overall, 23 carriers had adverse outcomes (including two cases of imprinting syndrome), 63 carriers had normal development after birth, and one carrier was lost to follow-up.
ROH in fetuses with aneuploidy or P/LP CNVs
The pregnancy outcomes of the nine fetuses with ROH were as follows (Table 2): eight terminations of pregnancies (TOPs) and one term birth (the pregnant woman refused to undergo any further genetic tests, except for ultrasound, and the fetus had a normal ultrasound during pregnancy and a normal phenotype after birth). We followed up on the late ultrasound findings in some cases and found that Case 10 presented with severe FGR and oligohydramnios at 22 weeks. The rate of adverse pregnancy outcomes in fetal ROH with aneuploidy and P/LP CNVs was 88.9% (8/9).
ROH in fetuses on imprinted chromosomes without aneuploidy or P/LP CNVs
Of the 23 patients, 14 underwent parental SNP array, WES, or prenatal single-WES analysis, and the remaining 10 refused further genetic testing. The pregnancy outcomes of the 23 fetuses with ROH on imprinted chromosomes were as follows (Table 3): five TOPs (of which two UPDs were located in defined imprinted chromosomes (Case 62 and 73), one carried paternal UPD6 with severe FGR (Case 72) and two had abnormal ultrasound phenotypes (Case 55 and 74)), and 18 had a full term birth (one carried paternal UPD7 and homozygous SUGCT mutation with an overweight phenotype after birth; the remaining 17 term births showed normal growth and development). The rate of adverse pregnancy outcomes in fetal ROH on imprinted chromosomes without aneuploidy and P/LP CNVs was 26.1% (6/23).
ROH in fetuses on nonimprinted chromosomes without aneuploidy or P/LP CNVs
Pregnancy outcomes of the 55 fetuses with ROH were as follows (details are shown in Supplementary Table S1, and UPD cases are listed in Table 4): four TOPs (three with FGR and one with positive NIPT showed fetal intrauterine distress and hypercapnia in late pregnancy), three preterm births with trisomy detected by NIPT and combined with ultrasound abnormalities late in pregnancy (one died prematurely, one grew to catch up at 2 years old, and one had anal atresia and heart malformation), one miscarriage after amniotic fluid infusion with insufficient amniotic fluid, one unexplained premature rupture of membranes, one lost to follow-up, and the remaining 45 term births showed normal growth and development. The rate of adverse pregnancy outcomes in fetal ROH on nonimprinted chromosomes without aneuploidy or P/LP CNVs was 14.5% (8/55).
Discussion
SNP array, considered the gold standard, have the advantage of detecting ROH and may reveal instances of UPD, ancestral homozygosity, and consanguinity. The incidence of ROH varies according to the population, threshold, and sample type. The detection rate of ROH in villous abnormalities in miscarriages is reportedly in the range of 2–3%18,19. A retrospective study of 14,574 clinical cases (including mental retardation, developmental delays, and multiple congenital anomalies), primarily from the United States and Mexico, found that 1.32% of cases carried one ROH greater than 10 Mb on a single chromosome and 0.47% of cases carried two or more ROHs on multiple chromosomes20. In this study, we reviewed 6176 fetal samples subjected to SNP array at our center over six years. ROH cases meeting our reporting threshold were further analyzed; the detection rate was 1.41% (87/6176), which was significantly higher than the 0.43% (22/5063)21, 0.96% (106/11062)22, and 0.97% (100/10294)23 reported at other centers in our country. This difference is most likely related to the sample type. In our study, the proportion of invasive diagnoses due to positive NIPT results was as high as 27.1% (1674/6176), with the highest positive rate for ROH (2.03%) among all indications. NIPT uses cell-free fetal DNA released from the placenta in the maternal blood, reflecting placental chromosomal conditions more accurately than fetal abnormalities24. In our study, 85.3% (29/34) of ROH cases with normal copy numbers were confirmed in chromosomes or regions with positive NIPT results. In cases of aberrant NIPT, especially on chromosomes with clinically significant imprinting disorders on chromosomes 6, 7, 11, 14, 15, and 20, prenatal diagnostic techniques that recognize ROH or UPD, such as SNP array, should be prioritized.
UPD occurs when an initial chromosomal imbalance self-corrects into a normal karyotype. The result may be heterodisomy (two homologous but genetically different chromosomes), isodisomy (two identical copies of a single chromosome), or partial hetero/isodisomy, depending on the meiotic recombination events. Trisomy rescue, monosomy rescue, and gamete complementation are the primary mechanisms underlying self-correction25. At least 19% of the reported UPD cases are due to trisomic rescue; in many cases of isodisomy, it is due to monosomy rescue26,27. Incomplete self-correction results in a cytogenetic abnormality in UPD, and approximately 35% of the comprehensively studied UPD cases present with an abnormal karyotype, including chromosome aneuploidy (often mosaic) or a small supernumerary marker chromosome26. In our study, we identified nine cases of ROH with aneuploidy or P/LP CNVs in 87 samples, among which were two interstitial triplications coupled with distal segmental isodisomy/ROH and whose possible mechanism was one-ended DNA break repair coupled with microhomology-mediated break-induced replication (Table 2; Cases 14 and 42)28. In other cases, data from the NIPT suggested a submicroscopic deletion/duplication, parental SNP array studies confirmed the presence of segmental UPD with normal copy number in the same region, and direct placental testing revealed the deletion/duplication consistent with NIPT results (Table 4; Case 87). The mechanism of deletion rescue can explain the formation of segmental UPD/ROH29,30. In conclusion, these cases deepen our understanding of the possible mechanisms underlying ROH formation and provide supporting evidence for genomic rearrangement models.
In our study, we identified 15 cases of UPD from 6176 mid- to late-term pregnancies, with a detection rate of 0.24%, significantly higher than the general population frequency of 0.05%6. The detection rate of UPD in the prenatal period would be much higher than this incidence because more than one-third of UPDs cannot be detected by CMA31, and many of the large ROHs in our study were not verified in parents, resulting in an inconclusive diagnosis of UPD or IBD segments. The medical consequences of UPD include autosomal recessive disease (when isodisomy is present) or aberrant imprinting. Specific syndromes are associated with UPD, including Prader-Willi syndrome (maternal UPD15), Angelman syndrome (paternal UPD15), (transient) neonatal diabetes mellitus (paternal UPD6), Silver-Russell syndrome (maternal UPD7), Beckwith-Wiedemann syndrome (paternal UPD11) and the maternal UPD14 syndrome25. Most of these syndromes are characterized by disordered growth and/or development, particularly prenatal growth. However, the relationship between imprinting disorders and growth in some UPDs, such as UPD16, is controversial, and more evidence is needed. Among the 15 cases of UPD, six presented with FGR in later pregnancy, including two cases of maternal UPD16 screened from positive NIPT results (Table 4; Cases 48 and 80). Both cases showed severe FGR and one of them was born with very low birth weight and catch-up after birth during a long-term intensive follow-up, which supports the view that chromosome 16 does not undergo underlying imprinting. FGR was the most common ultrasound symptom observed in our study, and confined placental mosaicism (CPM) accounts for approximately 10% of the causes of moderate or severe FGR32. It is critical to recognize whether FGR is caused by CPM or imprinting disorders in clinical practice. The combination of genetic counseling, karyotyping, WES, CMA, NIPT, placental studies, and prenatal ultrasonography can provide valuable insights into the identification and management of UPD, primarily when associated with FGR.
Detecting ROH by itself is not diagnostic of any underlying condition and may be clinically benign. The first step in assessing the clinical relevance of observed ROH is to determine whether ROH is combined with an abnormal karyotype or P/LP CNVs; if combined, there is a high risk of adverse pregnancy outcomes after birth because of genomic imbalances. The second step is distinguishing between excessive homozygosity found in multiple regions throughout the genome and ROH restricted to a single chromosome. High frequency of autosomal recessive disorders in consanguineous marriages is well established. For fetuses conceived by consanguineous parents, ultrasound screening and trio-WES should first be performed to rule out autosomal recessive disorders; if ruled out, these cases have better outcomes. We determined whether the ROH was located on an imprinted chromosome. Segmental ROH may be indicative of partial hetero/isodisomy. When an ROH is encountered on chromosomes 6, 7, 11, 14, 15, or 20 and does not seem to involve an imprinted gene region, it may still be necessary to rule out heterodisomy in the adjacent imprinted gene region. Regardless of ROH involvement in the imprinted chromosomes, WES can be used to determine homozygous mutations in recessive pathogenic genes. In our study, further WES testing was performed in only a few cases; thus, a few cases of recessive genetic disorders exposed to ROH regions were found. Many ROHs show uncertain clinical significance, presenting substantial challenges for prenatal genetic counseling. For these ROHs, the main concern is the increased risk of monogenic disorders caused by the exposure of recessive genes within the region. According to the hypothetical risk assessment of Mendelian genetic disorders, for the total homozygous regions less than 180 Mb (indicating a parental relatedness equivalent to third degree)1, the risk of autosomal recessive disorders is similar to that of the general non-consanguineous population. Therefore, it is not advisable to give excessive clinical attention to these ROHs with unclear clinical significance, and the published consensus statements in China do not recommend reporting them in prenatal reports33.
Conclusion
In conclusion, ROH is relatively common in mid- and late-term pregnancies, and its incidence is higher than that reported previously. The SNP array is an effective test for the ROH. The clinical relevance of ROH should be evaluated using a combination of techniques, including karyotyping, parental SNP array verification, WES, systematic ultrasound monitoring, and placental studies.

Flowchart of fetal ROH analysis in our cohort. ROH, region of homozygosity; SNP array, single nucleotide polymorphism array; *, Parental SNP-array and WES were performed simultaneously in six cases.
Data availability
The raw sequence data reported in this paper have been deposited in the Genome Sequence Archive (Genomics, Proteomics & Bioinformatics 2021) in National Genomics Data Center (Nucleic Acids Res 2024), China National Center for Bioinformation / Beijing Institute of Genomics, Chinese Academy of Sciences (GSA-Human: HRA010642) that are publicly accessible at https://ngdc.cncb.ac.cn/gsa-human.
References
Kearney, H. M., Kearney, J. B. & Conlin, L. K. Diagnostic implications of excessive homozygosity detected by SNP-based microarrays: consanguinity, uniparental disomy, and recessive single-gene mutations. Clin. Lab. Med. 31, 595–613 (2011). ix.
Ceballos, F. C., Joshi, P. K., Clark, D. W., Ramsay, M. & Wilson, J. F. Runs of homozygosity: windows into population history and trait architecture. Nat. Rev. Genet. 19, 220–234 (2018).
Gonzales, P. R. et al. Interpretation and reporting of large regions of homozygosity and suspected consanguinity/uniparental disomy, 2021 revision: A technical standard of the American college of medical genetics and genomics (ACMG). Genet. Med. 24, 255–261 (2022).
McQuillan, R. et al. Runs of homozygosity in European populations. Am. J. Hum. Genet. 83, 359–372 (2008).
Gibson, J., Morton, N. E. & Collins, A. Extended tracts of homozygosity in outbred human populations. Hum. Mol. Genet. 15, 789–795 (2006).
Nakka, P. et al. Characterization of prevalence and health consequences of uniparental disomy in four million individuals from the general population. Am. J. Hum. Genet. 105, 921–932 (2019).
Butler, M. G. Imprinting disorders in humans: a review. Curr. Opin. Pediatr. 32, 719–729 (2020).
Armour, C. M. et al. Practice guideline: joint CCMG-SOGC recommendations for the use of chromosomal microarray analysis for prenatal diagnosis and assessment of fetal loss in Canada. J. Med. Genet. 55, 215–221 (2018).
Wapner, R. J. et al. Chromosomal microarray versus karyotyping for prenatal diagnosis. N Engl. J. Med. 367, 2175–2184 (2012).
Sagi-Dain, L. et al. Chromosomal microarray vs. NIPS: analysis of 5541 low-risk pregnancies. Genet. Med. 21, 2462–2467 (2019).
[Guideline for the application of chromosomal microarray analysis in prenatal diagnosis. ]. Zhonghua Fu Chan Ke Za Zhi 58, 565–575 (2023). (2023).
Riggs, E. R. et al. Technical standards for the interpretation and reporting of constitutional copy-number variants: a joint consensus recommendation of the American college of medical genetics and genomics (ACMG) and the clinical genome resource (ClinGen). Genet. Med. 22, 245–257 (2020).
Huang, Y. et al. Exome sequencing in fetuses with short long bones detected by ultrasonography: A retrospective cohort study. Front. Genet. 14, 1032346 (2023).
Huang, Y. et al. ALG8-CDG: advances in molecular and prenatal phenotyping facilitate prenatal diagnosis and genetic counseling. Qjm (2025).
Richards, S. et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American college of medical genetics and genomics and the association for molecular pathology. 17, 405–424 (2015).
Abou Tayoun, A. N., Pesaran, T., DiStefano, M. T. & Oza, A. Recommendations for interpreting the loss of function PVS1 ACMG/AMP variant criterion. 39, 1517–1524 (2018).
Brnich, S. E. et al. Recommendations for application of the functional evidence PS3/BS3 criterion using the ACMG/AMP sequence variant interpretation framework. Genome Med. 12, 3 (2019).
Xu, Z., Liu, N., Gao, L. & Yu, D. Application of chromosomal microarray analysis in genetic reasons of miscarriage tissues. 17, 85–93 (2024).
Wang, Y. et al. Clinical application of SNP array analysis in first-trimester pregnancy loss: a prospective study. Clin. Genet. 91, 849–858 (2017).
Wang, J. C. et al. Regions of homozygosity identified by oligonucleotide SNP arrays: evaluating the incidence and clinical utility. Eur. J. Hum. Genet. 23, 663–671 (2015).
Liang, B. et al. Prenatal diagnosis of fetuses with region of homozygosity detected by single nucleotide polymorphism array: a retrospective cohort study. J. Hum. Genet. 67, 629–638 (2022).
Xue, H. et al. Genetic testing for fetal loss of heterozygosity using single nucleotide polymorphism array and whole-exome sequencing. Sci. Rep. 14, 2190 (2024).
Liu, J. et al. Absence of heterozygosity detected by single-nucleotide polymorphism array in prenatal diagnosis. 57, 314–323 (2021).
Chiu, R. W. K. & Lo, Y. M. D. Cell-free fetal DNA coming in all sizes and shapes. 41, 1193–1201 (2021).
Del Gaudio, D. et al. Diagnostic testing for uniparental disomy: a points to consider statement from the American college of medical genetics and genomics (ACMG). Genet. Med. 22, 1133–1141 (2020).
Liehr, T. Uniparental Disomy (UPD) in Clinical Genetics. A Guide for Clinicians and Patients, (Uniparental Disomy (UPD) in Clinical Genetics (A Guide for Clinicians and Patients, 2014).
Papenhausen, P. et al. UPD detection using homozygosity profiling with a SNP genotyping microarray. Am. J. Med. Genet. A. 155a, 757–768 (2011).
Carvalho, C. M. et al. Absence of heterozygosity due to template switching during replicative rearrangements. Am. J. Hum. Genet. 96, 555–564 (2015).
Caldwell, S. et al. Deletion rescue resulting in segmental homozygosity: A mechanism underlying discordant NIPT results. Am. J. Med. Genet. A. 182, 2666–2670 (2020).
Johnson, J. P. et al. Deletion rescue’ by mitotic 11q uniparental disomy in a family with recurrence of 11q deletion Jacobsen syndrome. Clin. Genet. 85, 376–380 (2014).
Hoppman, N., Rumilla, K., Lauer, E., Kearney, H. & Thorland, E. Patterns of homozygosity in patients with uniparental disomy: detection rate and suggested reporting thresholds for SNP microarrays. Genet. Med. 20, 1522–1527 (2018).
Miyagami, K. et al. Prenatal identification of confined placental mosaicism in pregnant women with fetal growth restriction. 29, 896–903 (2022).
Liu, W. et al. [A consensus recommendation for the interpretation and reporting of copy number variation and regions of homozygosity in prenatal genetic diagnosis]. Zhonghua Yi Xue Yi Chuan Xue Za Zhi. 37, 701–708 (2020).
Bu, X. et al. Case report: paternal uniparental disomy on chromosome 7 and homozygous SUGCT mutation in a fetus with overweight after birth. Front. Genet. 14, 1272028 (2023).
Bu, X. et al. Prenatal diagnosis of complete paternal uniparental isodisomy for chromosome 3: a case report. Mol. Cytogenet. 14, 50 (2021).
Funding
This research was funded by Program of Health Commission of Hunan Province (No. 202301036352 and 202305026432), Natural Science Foundation of Hunan Province (No. 2025JJ50517 and 2023JJ60398), and Natural Science Foundation of Changsha (No. kq2208479 and kq2403191).
Author information
Authors and Affiliations
Contributions
X.B. and X.Y. wrote the original draft; L.Z., G.Z. and R.T. carried out all the molecular genetic analysis and participated in the design of the work; C.P. and S.Z. collected all clinical data and participated in conceiving the work; S.L. and J.L. designed the work and drafted and revised the manuscript. All authors reviewed the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethical approval
The studies involving humans were approved by the Changsha Hospital for Maternal & Child Healthcare. The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent for participation in this study was provided by the participants’ legal guardians/next of kin. Written informed consent was obtained from the minor(s)’ legal guardian/next of kin for the publication of any potentially identifiable images or data included in this article.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Bu, X., Yu, X., Zeng, L. et al. A retrospective single center analysis of fetuses with region of homozygosity detected by single nucleotide polymorphism array. Sci Rep 15, 13623 (2025). https://doi.org/10.1038/s41598-025-98497-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-025-98497-9
